
Abstract
Gray’s grenadier anchovy, Coilia grayii, is an important anadromous fish species with economic value in near ocean ecosystems. Despite its significance, the lack of genomic resources has constrained our understanding of its genetic foundation, phylogenetic relationships, and adaptive evolution strategies. In this study, we assembled a chromosome-level reference genome for C. grayii by integrating PacBio HiFi long-reads, MGI short-reads, and Hi-C sequencing data. The resulting genome is 920.64 Mb in size, with a contig N50 of 36.45 Mb. The genome contains 324.19 Mb of repetitive sequences, and 29,496 protein-coding genes were predicted, with 29,395 functionally annotated. BUSCO analysis revealed that 95.2% of the 3,640 benchmarking genes were complete, underscoring the high quality of the assembly. This high-quality genome will provide crucial insights into the phylogeny, evolutionary history, and genetic basis of adaptive traits in Coilia species.
Similar content being viewed by others


Background & Summary
Coilia grayii, commonly known as Gray’s grenadier anchovy, is a small anadromous fish species belonging to the family Engraulidae1. This species is predominantly found in the coastal waters and estuaries of Southeast Asia, ranging from the Bay of Bengal to the South China Sea2. C. grayii plays a crucial role in the ecosystem dynamics of these regions, serving as a vital link in the aquatic food web. In its natural habitat, C. grayii occupies a niche that bridges marine and freshwater ecosystems3. It is typically found in brackish waters of estuaries and coastal areas, where it feeds on zooplankton and small invertebrates. As a prey species, it supports various predators, including larger fish, seabirds, and marine mammals, thus playing a significant role in energy transfer within the ecosystem4. The economic importance of C. grayii is multifaceted. In wild fisheries, it is a target species for small-scale commercial and subsistence fishing operations throughout its range. The fish is often consumed fresh, dried, or processed into fish sauce, contributing to local food security and livelihoods1. Given the adaptability of C. grayii to varying salinity levels and its nutritional value, and with the successful aquaculture of another species Coilia nasus within the same genus, there is growing interest in the potential for its controlled cultivation5,6.
Previous studies on C. grayii have primarily focused on its unique migratory behaviors, which are crucial for understanding its life cycle and population dynamics. Research demonstrated that C. grayii engages in anadromous migration, ascending rivers from estuarine areas to spawn in freshwater environments during its reproductive season, yet predominantly resides in the higher salinity waters of estuaries and adjacent marine areas7. This migratory pattern not only influences the species’ distribution but also has implications for its genetic structure and local adaptations4. The adaptive traits of C. grayii in the context of changing environmental conditions have also been a subject of scientific interest. Recent studies have shown that the species possesses remarkable osmoregulatory capabilities, allowing it to thrive in environments with fluctuating salinity levels8. Furthermore, ongoing research suggests that C. grayii may serve as a valuable bioindicator for assessing the health of estuarine ecosystems, given its sensitivity to changes in water quality and environment9,10.
Despite the ecological and economic significance of C. grayii, genomic resources for this species remain limited. To date, only fragmented genomic data, such as mitochondrial DNA sequences and a handful of nuclear markers, have been available2,11,12. These resources, while valuable for preliminary phylogenetic and population genetic studies, have been insufficient for in-depth investigations into the genetic basis of the species’ unique adaptations and migratory behaviors. The lack of a high-quality, chromosome-level genome assembly for C. grayii represents a significant gap in our ability to comprehensively study this species. Such a resource would be invaluable for enhancing our understanding of the genetic mechanisms underlying migration, osmoregulation, and adaptation to varying environmental conditions. A complete genome assembly would also facilitate comparative genomic analyses with other anchovy species, potentially revealing insights into the evolution of catadromous life histories in this group of fishes.
Several key gaps exist in our current understanding of C. grayii’s genetics, particularly in relation to its migratory and adaptive behaviors. First, the genetic basis of its catadromous migration remains poorly understood. While it is clear that this behavior is likely controlled by a complex interplay of genes, the specific loci involved and their regulatory mechanisms have yet to be identified. Second, the genetic underpinnings of C. grayii’s remarkable osmoregulatory capabilities are not well-characterized, limiting our ability to predict how the species might respond to changing salinity regimes in the face of climate change. Moreover, there is a lack of information on the population genetic structure of C. grayii across its range. This knowledge gap hampers efforts to effectively manage and conserve the species, especially considering the increasing pressures from overfishing and habitat degradation. Understanding the genetic connectivity among populations is crucial for developing informed conservation strategies and sustainable fisheries management plans.
The primary objective of this research is to assemble a high-quality, chromosome-level genome for C. grayii. This genomic resource will serve as a foundation for investigating the genetic loci associated with migration, osmoregulation, and environmental adaptation in this species. We expect that this study will provide insights into the genetic architecture underlying C. grayii’s unique life history and adaptive traits. By identifying specific genes and regulatory regions associated with migration and osmoregulation, we anticipate gaining a deeper understanding of how this species has evolved to thrive in its dynamic environment. In conclusion, this study on the genomics of C. grayii represents a significant step forward in our understanding of this ecologically and economically important species. By bridging the current knowledge gaps, we anticipate that our research will not only advance the field of fish genetics but also contribute to the conservation and sustainable utilization of this valuable resource in the face of ongoing environmental changes.
Methods
Sample collection and sequencing
A healthy male specimen of C. grayii (body weight: 17.1 g, body length: 16 cm, Fig. 1) was collected from the Pearl River Estuary (latitude: 22°48′10.915″N, longitude: 113°36′28.192″E, Dongguan, China). High-quality genomic DNA was extracted from its muscle tissue for comprehensive sequencing, including MGI short-read sequencing, long-read single-molecule real-time (SMRT) sequencing, and Hi-C sequencing. Additionally, samples were collected from six distinct tissues: muscle, brain, gills, heart, liver, and stomach. RNA was extracted from these tissues, pooled, and used to prepare total RNA for RNA sequencing. To preserve sample integrity, all tissues were rapidly frozen in liquid nitrogen and stored at −80 °C in an ultra-low temperature freezer.

A picture of the C. grayii for sequencing and assembly.
High-molecular-weight (HMW) genomic DNA was extracted from the collected samples using the CTAB method and purified with the Grandomics Genomic Kit for regular sequencing, following the manufacturer’s standard procedure. DNA degradation and contamination were assessed on 1% agarose gels, and DNA purity was evaluated using a NanoDrop™ One UV-Vis spectrophotometer (Thermo Fisher Scientific, USA), with OD260/280 ratios of 1.87 and OD260/230 ratios of 2.32. DNA concentration was measured using a Qubit® 4.0 Fluorometer (Invitrogen, USA), with a final concentration of 337.0 ng/µL. High-quality DNA was subsequently used for library preparation and high-throughput sequencing.
For short-read sequencing, 1–1.5 μg of genomic DNA (gDNA) was randomly fragmented using a Covaris ultrasonicator, and fragments were size-selected to an average length of 200–400 bp using the Agencourt AMPure XP-Medium Kit (Beckman Coulter, USA). The selected DNA fragments underwent end-repair, 3′ adenylation, adapter ligation, and PCR amplification, followed by purification with the AxyPrep Mag PCR Clean-up Kit (Axygen, USA). The resulting double-stranded PCR products were heat-denatured and circularized using a splint oligo sequence to generate single-stranded circular DNA (ssCir DNA), which was used as the final library. Library quality was assessed by QC, and sequencing was performed on the MGISEQ-2000 platform, generating a total of 50.38 Gb of raw reads with an average depth of 44.58× (Table 1).For PacBio sequencing, SMRTbell libraries were prepared using the SMRTbell prep kit 3.0 (Pacific Biosciences, USA; Product PN: 102-141-700) according to the kit manual. The preparation steps included DNA shearing with the Megaruptor 3 system, followed by repair, A-tailing, adapter ligation, nuclease treatment, and size selection to enrich for long DNA fragments. Sequencing was performed on the PacBio Revio platform in Circular Consensus Sequencing (CCS) mode. A single SMRT Cell produced 41.41 Gb of high-quality CCS long reads, filtered using the CCS software (https://github.com/PacificBiosciences/ccs) with stringent parameters (min-passes 3, min-rq 0.99, and min-length 100). Notably, 100% of the filtered reads achieved Q20 (≥99% base accuracy), ensuring data reliability for genome assembly. The final HiFi reads represented a genome depth of 36.64× with an average read length of 38,121 bp (Table 1)13.
For Hi-C sequencing, fresh muscle tissue was treated with 2% formaldehyde to create DNA-protein crosslinks. The library preparation involved the digestion of crosslinked DNA, biotin labeling, proximity ligation, and subsequent DNA purification14. The resulting Hi-C libraries were sequenced on the MGISEQ-2000 platform using 150 bp paired-end reads, enabling the detection of spatial interactions between chromosomal regions. This process generated 90.83 Gb of Hi-C data, with an average sequencing depth of 80.38× (Table 1).
Total RNA was extracted from pulverized tissue under cryogenic conditions using TRIzol reagent (Tiangen Biotech, Beijing, China; Catalog No. GDP424). RNA was pooled from muscle, brain, gills, heart, liver, and stomach for RNA sequencing on the MGISEQ-2000 platform. This approach yielded 15.76 Gb of RNA-seq data, which was subsequently used for genome-wide prediction of protein-coding genes (Table 1).
De novo assembly and Hi-C assembly
K-mer analysis was performed using Jellyfish (v2.2.10) and GenomeScope v1.015, applying the parameters “k = 21, p = 2”. This analysis estimated the genome size to be 1135.14 Mb, with a heterozygosity rate of 2.22% and the model predicted a duplicated sequence content (dup) of 1.6% (Fig. 2).

The K-mer distribution of the C. grayii genome.
The de novo genome assembly was constructed from a 41.41 Gb PacBio long-read dataset (Table 1) using Hifiasm (v0.19.8)16. This assembly resulted in a 1,666 Mb genome, consisting of 4,664 contigs with an N50 contig length of 1.90 Mb (Table 2).
Following the initial genome assembly, further refinement was carried out using Purge_Dups(v1.2.5)17 in combination with Minimap2(v2.22)18. Minimap2 was employed to align reads against the assembled contigs, allowing for the assessment of coverage across various regions and the identification of repetitive elements through self-alignments. Purge_Dups then utilized this data to classify and filter out repetitive sequences, effectively distinguishing primary assembly sequences from potential haplotypes. This process led to the removal of redundancies and the resolution of haplotypes, resulting in a deduplicated genome of 921 Mb, comprising 1153 contigs with an N50 length of 2.90 Mb (Table 2).
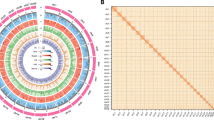
Hi-C data was subsequently utilized to anchor and orient the draft genome contigs into chromosome-scale assemblies. The deduplicated genome assembly was first indexed with BWA (v0.7.17)19 and SAMtools (v1.7)20. Hi-C reads were aligned to the genome using BWA-MEM, and the resulting alignment files were sorted with SAMtools sort. PCR duplicates, which are reads artificially replicated during PCR amplification from the same DNA fragment, were removed using bammarkduplicates2 in biobambam2 (v2.0.87)21. Refinement of the assembly was further achieved with Yahs (v1.1)22, which employed Hi-C data to improve scaffold ordering, producing an updated assembly. Gap and telomere analyses were performed using PretextMap (v0.1.9, https://github.com/sanger-tol/PretextMap), followed by manual curation with PretextView (v0.2.5, https://github.com/sanger-tol/PretextView). Scaffolds exhibiting strong interaction signals were clustered, facilitating the delineation of chromosomal boundaries. The Hi-C data were utilized to further anchor and orient the assembled sequences onto 24 chromosomes, spanning a total of 872.88 Mb with a contig N50 length of 36.45 Mb and covering approximately 94.80% of the scaffold-level genome (Table 3, Fig. 3A,B). The chromosomes ranged in length from 27.48 to 45.69 Mb (Table 3).

Hi-C chromatin interaction map and circos plot of the genome assembly. (A) Circos plot of the C.grayii genome assembly. (a) GC ratio; (b) Gene density in heatmap; (c) GC skewness; (d) See the N ratio in the point chart; (e) Basic chromosome skeleton. (B) Hi-C intra-chromosomal contact map of the C.grayii genome assembly.
Repetitive sequence annotation
For the annotation of repetitive sequences, we employed RepeatMasker (v4.1.6)23 using the Dfam database, which leverages advanced HMMs to identify known repeats, and RepBase for comprehensive family representation. To identify species-specific repeats not present in public databases, RepeatModeler (v2.0.5)24 was utilized to generate de novo repeat libraries through iterative clustering and sequence refinement. The annotations from Dfam, RepBase, and RepeatModeler were then consolidated into a single dataset, with overlapping annotations merged and redundancies eliminated. Based on these analyses, we identified a total of 324.31 Mb (35.22% of the assembled C. grayii genome) of repeats in the C. grayii genome (Table 4).
Protein-coding gene prediction and annotation
Gene predictions were conducted using a combination of homology-based, transcriptome-based, and de novo methods. For homology-based prediction, full-genome protein sequences from closely related species, including Sardina (Sardina pilchardus, GCF_963854185.1), European anchovy (Engraulis encrasicolus, GCF_034702125.1), American shad (Alosa sapidissima, GCF_018492685.1), and Japanese grenadier anchovy (Coilia nasus, GCA_027475355.1) were sourced from GenBank. MMseqs. 2 (v15-6f452)25 was used to align these homologous protein sequences against the target genome, with hits filtered by “identity > 0.1, evalue < 1e-3”. Overlapping High Scoring Segment Pairs (HSPs) resulting from alternative splicing were merged, followed by additional filtering using “identity > 0.2, evalue < 1e-9, query coverage > 0.3”. Subsequently, Genewise (v2.4.1)26, Gth (v1.7.3)27 and Exonerate (v2.2.0)28 were employed for precise spliced alignments of matched proteins to homologous sequences, facilitating the prediction of gene structures. RNA-Seq datasets from six tissues were processed using Trimmomatic (v0.39)29 for quality control, and the trimmed reads were aligned to the reference genome using HISAT2 (v2.1.0)30. Open Reading Frames (ORFs) were identified from assembled transcripts with TransDecoder (v5.5.0, https://github.com/TransDecoder/TransDecoder). Additionally, de novo gene prediction was performed using Augustus (v3.5.0)31. The results from these three methods were integrated to form a non-redundant reference gene set, comprising 29,496 protein-coding genes (Table 2).
For annotation, genomic sequences were aligned against the NT database using BLAST+ (v2.13.0)32 with an e-value threshold of 1e-10. Predicted proteins were further compared against the NR and UniProt databases33 using DIAMOND (v2.1.8)34. Ultimately, 29,395 genes (99.66%) were successfully annotated by at least one database (Table 2). The number of predicted genes exceeds that found in other Coilia species, including Coilia nasus. Additionally, to annotate Gene Ontology (GO) terms, KEGG pathways, and identify protein families, we utilized 12ggnog-mapper (v2.1.12)35 for functional annotation. This tool enables the mapping of predicted gene models to several public databases, including KEGG, GO, and PFAM. The default parameters were used for gene annotations across these databases. This resulted in a total of 27,512 genes (93.27%) were successfully annotated. Specifically, 26,017 genes (88.21%) were mapped to the PFAM database, and 18,367 genes (62.27%) were assigned GO terms, while 18,253 genes (61.88%) were annotated with KEGG Orthology (KO) terms. The detailed results are presented in Table 5.
Chromosomal synteny analysis
We conducted whole-genome sequence alignment between Coilia grayii and Coilia nasus using Minimap2 with the –asm5 parameter, optimized for highly accurate genome assembly comparisons. This alignment provided a comprehensive view of sequence synteny, revealing conserved genomic regions and structural variations. For gene-level synteny analysis, using the MCScanX tool36, we conducted a collinearity analysis between Coilia grayii and Coilia nasus under stringent parameters (MATCH_SCORE: 50, MATCH_SIZE: 5, GAP_PENALTY: -1, OVERLAP_WINDOW: 5, E_VALUE: 1e-05, and a maximum gap of 25).
The gene synteny analysis highlighted several well-conserved regions (Fig. 4), reflecting evolutionary conservation, while scattered and non-linear alignments from sequence synteny suggested structural rearrangements, such as inversions and translocations (Fig. 5). Together, these analyses provide valuable insights into the genomic architecture and evolutionary divergence of Coilia grayii and Coilia nasus.

Genome collinearity between the Coilia grayii and Coilia nasus.

Dotplot showing the whole genome alignment between the Coilia nasus genome assembly and the Coilia grayii genome assembly from this study.
Data Records
The raw sequencing data reported in this paper have been deposited in the NCBI Sequence Read Archive (SRA) under the project accession number SRP53248837. The DNA sequencing data from the PacBio HiFi library are available under the SRA accession number SRR3065632038, the Hi-C library data under SRR3066530739, the MGI short-read genomic sequencing data under SRR3067453140, and the RNA-seq data under SRR3066652841. The assembled genome sequences have been deposited in the NCBI GenBank with the accession number GCA_042479465.142. The genome annotation results have been deposited in the figshare database43.
Technical Validation
For DNA samples designated for MGI and PacBio Revio sequencing, quality and purity were assessed using 0.75% agarose gel electrophoresis and a NanoDrop One UV-Vis spectrophotometer (Thermo Fisher Scientific, USA). The final DNA concentration was determined as 337.0 ng/µL via a Qubit Fluorometer (Invitrogen, USA). RNA integrity was evaluated with an Agilent 2100 Bioanalyzer (Agilent Technologies) alongside agarose gel electrophoresis, while RNA purity and concentration were measured using both NanoDrop and Qubit instruments. These stringent quality control procedures ensured the use of high-quality DNA and RNA for subsequent library preparation and high-throughput sequencing.
Genomic sequences were aligned to the NT database using BLAST+ (v2.13.0), facilitating the annotation of protein-coding genes and the assessment of potential genomic contamination. An e-value threshold of 1e-10 was applied to maintain strict standards. This analysis confirmed that the assembled genome was devoid of artificial or bacterial contaminants.
Genomic integrity was further evaluated using BUSCO (v5.4.7)44 with the Actinopterygii reference dataset. Of the 3,640 benchmarking genes, 3,463 (95.2%) were identified as complete, reflecting the high quality of the genome assembly. The analysis indicated minimal fragmentation, with 57 (1.6%) fragmented and 120 (3.2%) missing BUSCOs. The assembly demonstrated remarkable continuity, with inter-sequence gaps constituting only 0.014%, confirming a highly contiguous and accurate genomic structure (Table 6).
Code availability
No custom code was developed for this study. All software utilized is publicly available, with detailed descriptions of the versions and parameters provided in the Methods section. Where specific parameters are not mentioned, default settings recommended by the developers were applied.
References
Whitehead, P. J. P.; N. FAO species catalogue. Vol.7. Clupeoid fishes of the world (Suborder Clupeoidei). An annotated and illustrated catalogue of the herrings, sardines, pilchards, sprats, shads, anchovies, and wolf-herrings. Part 2. Engraulididae. (1988).
Zhang, Z., Zhang, N., Liu, M. & Gao, T. The complete mitochondrial genome of Coilia grayii (Clupeiformes: Engraulidae). Mitochondrial DNA Part A (2016).
Zhang, L. et al. Fishery Stock Assessments in the Min River Estuary and Its Adjacent Waters in Southern China Using the Length-Based Bayesian Estimation (LBB) Method. Front. Mar. Sci. 7, (2020).
Wang, G. et al. Otolith Microchemistry and Demographic History Provide New Insight into the Migratory Behavior and Heterogeneous Genetic Divergence of Coilia grayii in the Pearl River. Fishes 7, 23 (2022).
Xu, G.-C., Xu, P., Gu, R.-B., Zhang, C.-X. & Zheng, J.-L. Feeding habits and growth characteristics of pond-cultured Coilia nasus fingerlings. Chinese Journal of Ecology 30, 2014–2018 (2011).
Ma, F. et al. Gap-free genome assembly of anadromous Coilia nasus. Sci Data 10, 360 (2023).
Jiang, T., Liu, H., Huang, H. & Yang, J. Otolith microchemistry of coilia grayii from the pearl river estuary, china. Acta Hydrobiol. Sin 39, 816–821 (2015).
Duan, J. et al. Ecological stoichiometric of C, N and P of Coilia species. Aquaculture and Fisheries https://doi.org/10.1016/j.aaf.2024.03.005 (2024).
Gang, X., Long, S. & Dang, A. Nutrients Affecting the Characteristics of Food-Web Structure in Aquatic Ecosystem of Pearl River. Pol. J. Environ. Stud. 31, 4641–4658 (2022).
Tang, J., Zhang, J., Su, L., Jia, Y. & Yang, Y. Bioavailability and trophic magnification of antibiotics in aquatic food webs of Pearl River, China: Influence of physicochemical characteristics and biotransformation. Science of The Total Environment 820, 153285 (2022).
Yang, Q. et al. Genetics and phylogeny of genus Coilia in China based on AFLP markers. Chin. J. Ocean. Limnol. 28, 795–801 (2010).
Zhou, X., Yang, J.-Q., Tang, W.-Q. & Liu, D. Species validities analyses of Chinese Coilia fishes based on mtDNA COI barcoding. Acta Zoo. Taxon. Sin. 35, 819–826 (2010).
Eid, J. et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 323, 133–138 (2009).
Rao, S. S. P. et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680 (2014).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Tischler, G. & Leonard, S. biobambam: tools for read pair collation based algorithms on BAM files. Source Code for Biology and Medicine 9, 13 (2014).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, btac808 (2023).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Steinegger, M. & Söding, J. MMseqs. 2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol 35, 1026–1028 (2017).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res 14, 988–995 (2004).
Gremme, G., Brendel, V., Sparks, M. E. & Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Information and Software Technology 47, 965–978 (2005).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915 (2019).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34, W435–W439 (2006).
McGinnis, S. & Madden, T. L. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Research 32, W20–W25 (2004).
UniProt Consortium, T. UniProt: the universal protein knowledgebase. Nucleic Acids Research 46, 2699–2699 (2018).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Molecular Biology and Evolution 38, 5825–5829 (2021).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40, e49 (2012).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP532488 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR30656320 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR30665307 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR30674531 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR30666528 (2024).
NCBI GenBank. NCBI https://identifiers.org/ncbi/insdc.gca:GCA_042479465.1 (2024).
A chromosome-level genome assembly of Gray’s grenadier anchovy, Coilia grayii. figshare https://doi.org/10.6084/m9.figshare.27019786.v2 (2024).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
This project was supported by the project ‘Research on Breeding Technology of Candidate Species for Guangdong Modern Marine Ranching’ (Project: 2024-MRB-00-001).
Author information
Authors and Affiliations
Contributions
Jianguo Lu: Conceptualization, Project administration, Supervision, Funding acquisition, Writing - review & editing. Zhenqiang Fu: Conceptualization, Methodology, Investigation, Formal analysis, Visualization, Writing - original draft. Junrou Huang: Resources, Methodology, Software, Data curation. Li Wang: Writing-review & editing. Xuanguang Liang: Methodology, Investigation, Validation. Qinglong Chen: Investigation, Data curation. Yan Hu: Validation, Software. Jia Liu: Validation, Methodology.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fu, Z., Huang, J., Wang, L. et al. A chromosome-level genome assembly of Gray’s grenadier anchovy, Coilia grayii. Sci Data 12, 656 (2025). https://doi.org/10.1038/s41597-025-04834-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04834-y
