
Abstract
Copy number variation (CNV) tends to occur in genetically enriched regions and is likely associated with a number of complex diseases such as skin aging. In this study, we investigated the genome-wide CNVs in 20 wrinkled skin cases (WSC) of Xiang pigs and 63 controls, and identified 7893 copy number variable regions (CNVRs). We estimated the F-statistic (Fst) at each locus and identified that 93 case-controls stratified CNVRs (Fst > = 0.15) overlapped with 87 known genes. Functional enrichment analysis showed that most of these genes were predominantly enriched in pathways and terms related to the extracellular matrix. Finally, we found that some CNVs were predicted to have high effects on genes such as VCAN, TIMP1 and FOXO1 through transcriptional amplification, transcript ablation and so on. Most of the genes overlapped with those CNVRs have been reported to be related to aging in human or animals. The copy numbers presented the positive correlations with the transcript level of the genes in skins between the cases and controls. Our results suggested that those 22 CNVRs, including 19 CNV losses and 3 CNV gains, were putatively associated with the skin wrinkle of Xiang pigs.
Similar content being viewed by others

Introduction
Genetic variation in human and animal genomes occurs in many forms, including copy number variations (CNVs), structural variations (SVs), insertions/deletions (INDELs), and single nucleotide polymorphisms (SNPs). Copy Number variation (CNV) is a complex multi-allelic variation (conventionally > 1 Kb), which represent an important source of genomic variation in mammalian genomes1. Unlike SNPs and SVs, CNVs corresponds to relatively large regions in the genome, containing more nucleotide sequences and having higher mutation probability2. This affects gene expression, phenotypic difference and phenotypic adaptation by changing gene structure and dose3. A typical example is the white coat phenotype produced by duplication of the KIT gene in pigs4.
In recent years, the biological role of copy number variants (CNVs) has been closely linked to disease. CNVs have been identified as factors that can increase susceptibility to the disease5, affect host-microbiome interactions6, and contribute to the development of both common and rare genetic diseases7 and syndromes8. CNVs can serve as biomarkers for certain pathological processes, such as cancer9, or even as potential confounders when evaluating the results of certain genetic diagnostic tests. CNVs have also been implicated in the formation of specific phenotypes in animals. For example, hereditary periodic fever syndrome in Shar-Pei dogs is caused by a high copy number of 16.1 Kb repeats upstream of the HAS2 gene10. This mutation results in periodic fever in Shar-Pei dogs, which leads to a phenotype of thick skin and heavily folding. Moreover, it has been strongly linked that CNV is associated with ageing. As observed in the elderly, the number of copies of mtDNA reduce11. And it has been approximated that the number of copies decreases by a few per cent for every passing decade12. The copy number of the human satellites III (1q12) has increased in a skin fibroblast cell line undergoing senescence13. Recurrent microdeletions and duplications in the CNVs of the 15q11.2 human genomic region is associated with neurodevelopmental disorders14. The correlation between aging and the CNTNAP4 gene, alongside its polymorphic variants in the CNVR6782.1del/del intron, has been established15. It appears that Copy Number Variations (CNVs) are associated with aging and complex diseases.
The Xiang pig is a local mini-pig breed in Guizhou Province, China. A few diseased individuals were found in Xiang pigs, as indicated by generalized skin wrinkles (see Supplementary Fig. S1). The earliest discovery was made at 2 months of age, which we believe is a premature aging phenomenon similar to HGPS syndrome in humans16. Individuals presenting with wrinkled skin typically exhibit a reduction in growth rate and an elevated risk of developing disease. Anatomical findings showed that affected individuals had thickened skin and a thinner subcutaneous fat layer. Histologically, the skin of diseased individuals exhibited epidermal hyperplasia and structural dermal disorders. In a previous study17, we identified and validated 65 small fragments of structural variants (SVs) associated with skin ageing in Xiang pigs by comparing the genomic SVs in cases with systemic wrinkled skin and four controls. In this study, we eliminated the superfluous controls and directly utilized skin wrinkled cases and normal Xiang pigs as research subjects to detect copy number variation (CNV) and selection signatures on the genome. Subsequently, a validation was conducted in a hybridisation pedigree constructed with wrinkled skin cases as dams to elucidate the aetiology of skin wrinkling/ageing in Xiang pigs.
Results
Detection of CNVs in wrinkled skin cases and controls
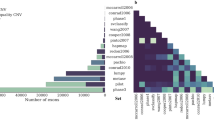
Based on the Illumina HiSeq2500 platform, we performed whole-genome resequencing and genome-wide CNVs detection on 20 cases of wrinkled skin (WSC) and 63 controls from the Xiang pig population. The mapped read depth ranged from 9.76 to 18.45, with an average depth of 13.75 per sample (Supplementary Table S1). The CNVs were called by CNVnator and CNVcaller software from the NGS data based on the Read Depth method. A total of 7893 CNVRs were identified in all samples, including 5753 losses and 2140 gains (Supplementary Tables S2-3). The CNVRs covered 47.34% of the porcine genome (Sscrofa 11.1) in all populations. The group of wrinkled skin cases had 5562 CNVRs, with 4559 losses and 1003 gains. These CNVs were located on all chromosomes except for Y chromosome with a mean size of 147,229.16 bp ranging from 600 to 1,996,600 bp (Table 1; Fig. 1). All the CNVR maps for 83 pigs were showed in Fig. 2. In addition, sequence coverage did not correlate with the number of CNVs identified in each individual (Fig. 3).

Size distribution of CNVRs. The distribution of CNV fragment size in the groups with wrinkled skin (cases) and those without (controls) is presented herewith. A consistent trend is evident in the distribution of CNV size between the two groups, with small fragments of CNV being more prevalent in the loss type and large fragments of CNV being more prevalent in the gain type.

A comprehensive chromosome mapping of all CNVRs. Two types of CNVR were identified: gain (green) and loss (grey). The appearance of vertical bars on a chromosome indicates the position of discrete types of CNVs.

The relationship between the number of variants and the sequence coverage for each sample. No correlation was observed between sequence coverage and the number of CNVRs identified in each individual.
Comparison with CNVRs identified in previous reports
Comparing our CNV data to those reported previously in several other studies for pigs, and the results showed that our data has a 27.26% overlap in CNV events with other researchers (Table 2). Comparison showed that the average overlap rate was is 9.91%, which was is higher than the average overlap rate of 2.13% found in previous studies18. The comparison also showed that the overlapping CNV events accounted for 61.93% of the total events, while the newly identified CNVRs accounted for 38.07%. These results suggest that the detected CNV events were highly reliable. Here we adopted the principle that two CNVRs share at least one base to identify overlapping CNV events.
Population stratification analysis
To discover CNVR that were differentiated between WSC and controls, the pairwise F-statistic (Fst) between wrinkled skin and cases controls samples were calculated by using the NGS data at each locus. As shown in Supplementary Table S3 (Fig. 4), pairwise Fst values showed generally large (Fst = 0.151–0.248) to very large (Fst = 0.25–0.44) genetic divergence between WSC and controls. About 86.87% (n = 6819) of the CNVRs showed nonsignificant genetic differentiation (Fst = 0.0–0.05), while 11.96% (n = 944) of the CNVRs showed moderate genetic differentiation (Fst = 0.05–0.15), 0.988% (n = 77) and 0.203% (n = 16) of the CNVRs showed large (Fst = 0.15–0.248) to very large (Fst = 0.25–0.44) genetic differentiation, respectively. A Fst value of 0.15 was used as a threshold for identifying potentially selective CNVR. In total, we identified 93 WSC-controls stratified CNVRs, including 17 gains and 76 losses, which had a length ≥ 0.8 kb, and the largest event was CNVR_4865, a 1.715 Mb gain in 11 chromosome. In order to study the potential function of the population-stratified CNVRs, the CNV coverage area were annotated. As shown in Table S4, there were 87 known genes (such as VCAN, YKT6, CAMK2B, GCK, WNT9A, FOXO1, THSD1), 57 novel genes, 1 pseudogene, and 45 noncoding RNA genes overlapped with 62 CNVRs. A total of 31 of the 93 CNVRs were located in the intergenic region, without covering any genes.

Identification of regions with differentiation in WSC compared to controls. A total of 93 differential CNVRs were identified using a threshold of Fst > = 0.15 (indicated by the red line).
We further investigated the impact of population-stratified CNVRs on covering genes by Variant Effect Predictor (VEP). CNVR_1233 deletion (loss) indicated a high impact predicted to be loss-of-function, including feature truncation, transcript ablation, transcript amplification and frame shift variants on host genes. Total of 7 DUPs (gains) were characterized to be whole-gene duplication which generally caused the copy gain of an entire gene. CNVR_4865, for example, overlaped 11 ensembl genes, including SOX11, FOXO1, MRPS31, SLC25A15, THSD1, CKAP2, NEK3, NEK5, ALG11, CCDC70 and one novel gene. Whole-gene duplication results in transcript amplification, which affects gene expression in a dose-dependent manner.
GO and KEGG analysis of the genes covered by population-stratified CNVRs
In order to gain a deeper understanding of the biological functions of the genes overlapped with the population-stratified CNVRs and the mechanism of the relationship between CNVRs and disease, we conducted gene ontology (GO) and KEGG enrichment analysis on the 87 known genes using the KOBAS tool. There were 47 genes that mapped to 149 KEGG pathways (Supplementary Table S5), including 54 significant pathways (P < 0.05), which mainly enriched in Insulin and Insulin resistance, FoxO, Notch, N-Glycan biosynthesis, Amino sugar and nucleotide sugar metabolism, Mitophagy – animal, Cancer, Th1 and Th2 cell differentiation, Endocrine resistance, AGE-RAGE signaling pathway in diabetic complications, and ErbB signaling pathway. There were 85 genes mainly enriched in 63 significant GO terms (P < 0.01), including protein N-linked glycosylation, extracellular matrix, metalloendopeptidase activity, mitophagy, elastic fiber assembly, negative regulation of apoptotic process, acetylglucosaminyltransferase activity, glutathione peroxidase activity, response to mitochondrial depolarization, extracellular matrix organization, hyaluronic acid binding and others.
Screening of candidate CNVRs related with wrinkled skin cases
We then investigated the CNVR-covering genes that involved in the biological processes and pathways of skin aging, such as apoptosis, survival, endocytosis, cell differentiation, oxidative stress, DNA damage and repair, extracellular matrix, immune and metabolism, and combined with the previously reported genes and databases (https://www.ncbi.nlm.nih.gov/gene/?term=aging) related to aging, we identified 22 CNVRs putatively associated with wrinkled skin cases, containing 61 known genes (Table 3). Three CNVRs (CNVR_1195, CNVR_1854, CNVR_4865) were predicted to cause transcript amplification effects on genes (such as FOXO1, ADAMTS2, MAPK9, SQSTM1, NEK5, NEK3). Ten known genes, including VCAN, CAMK2B, TIMP1, and LGALS2, were affected by 9 deletion variants resulting in transcript ablation, stop lost, feature truncation. Ten other CNVRs may impact the intronic regions of the AGK, SDR1, DMD, LPGAT1, and ADAMTS18 genes.
CNV assay
Six RNA-seq datasets were used to investigate the impact of candidate CNVRs on the expression of overlapping genes in case and control skin samples. Of the 61 known genes overlapping 22 candidate CNVRs, 56 were expressed in the skin (Supplementary Table S6). Wrinkled skin cases and controls showed differential expression of twelve genes (Fig. 5), including TIMP1, FOXO1 and ARAF. The expression levels of TIMP1, FOXO1, and ARAF genes were 1.7–3 times lower in the case group compared to the control group.

The expression trends of 12 CNVR-overlapping differential genes in cases of wrinkled skin (WSC) and control skin (XP).
To further clarify the functional significance of the candidate CNVRs in skin wrinkling traits in aromatic pigs, we validated CNV_VCAN (CNVR_1233), CNV_TIMP1 (CNVR_7451). We evaluated the copy numbers of CNV_VCAN, CNV_TIMP1 in F2 populations of the WSC × LW family using quantitative qPCR. All individuals were divided into three categories according to 2−ΔΔCT values (Supplementary Table S7): those with 2−ΔΔCT values ranging from 1.5 to 2.5 as normal type and those with values less 1.5 or more than 2.5 represented loss and gain, separately. The types of normal, loss and gain were detected in the 58 samples from the F2 population. The frequency of copy number variation of the three CNV genes in the F2 population was inconsistent. The proportion of losses accounted for by CNV_TIMP1 was 39.66%, followed by CNV_VCAN at 58.62%. CNVs may affect the phenotype by altering the transcription of genes through a dosage effect. We evaluated the relationship between CNV types of CNV_VCAN, CNV_TIMP1 and skin wrinkles traits in the F2 population using a general linear model (Table 4). The skin wrinkles trait was expressed as wrinkled fraction, which is equal to the wrinkle value divided by the profile value. The study suggests that skin wrinkling was affected by different CNV genotypes, specifically CNV_TIMP1. Increasing the copy number of CNV_TIMP1 resulted in more wrinkles on the F2 population (P < 0.05). RNA-seq data also showed a significant increase in the expression of gained CNV_TIMP1 copy number phenotypes at 6 and 12 months of age, particularly at 12 months (P < 0.05). However, the correlation between CNV_VCAN copy number and skin wrinkles was not strong. However, at 12 months of age, the skin wrinkle index was significantly higher in the loss type compared to the normal type.
Discussion
In this study, we identified 7893 CNVRs from 83 individuals by whole-genome resequencing. And the CNVRs accounted for 1157.34 Mb or 47.34% of the pig genome, which was higher than the CNV detection rate in previous studies18,19,20. In our research, more CNVR was detected. The reason for the difference in genomic CNV coverage may be due to differences in sample size, sequencing depth, and detection methods. At the same time, we found that the copy number deletion event (5753) was more common than the copy number duplication event, which was consistent with previous reports21,22. Different CNV formation mechanisms have different preferences for the formation of copy number variation types. It could be partially explained by Non-allelic Homologous Recombination (NAHR) because non-allelic homologous recombination tends to create more deletions than duplications3, and partially by technical bias.
There was limited concordance between our research and previous CNV studies. However, the average overlap rate of 9.91% was higher than in previous studies18. The underlying reason for this difference may be due to the difference in population size and genetic background between different studies, as well as the difference in sequencing depth and the algorithm called by CNV. Moreover, the results of researches are also inconsistent based on the assembly of different versions of the genome. After all, there were dramatically differences between Sscrofa 11.1 and Sscrofa 10.223.
The F statistic has been used to characterize selection for various traits in animals24,25,26. To compare the CNV between individuals with wrinkled skin cases and the control group, we compiled a thorough inventory of population-specific CNV-gene. Large to very large genetic differences between WSC and control were found by F-statistical analysis. The CNVR_5619 (Fst = 0.44) was identified as a divergent differential maximal locus, which overlaped with a novel gene (EGFEM1) and four lncRNA. The EGFEM1 gene encodes a protein that contains EGF-like and EMI domains and involve in calcium binding. Maintaining normal skin stability requires proper calcium homeostasis27. Deletion variants in the EGFEM1 gene may impact cutaneous calcium homeostasis. A total of 93 WSC-controls stratified CNVRs were identified using a threshold of 0.15. For instance, CNVR_1233 (Fst = 0.29), which was a 255.8 kb loss and overlaps with VCAN and two novel genes. The deletion of CNVR_1233 had an impact on four transcripts of VCAN, particularly transcript VCAN-209. The transcript VCAN-209 encodes the protein F1REZ2, which can bind hyaluronic acid (HA) at its N-terminus. The HA through its N-terminal G1 structural domain, leading to the formation of an HA-rich matrix28. Mouse mutants deleted the N-terminal G1 structural domain of versican exhibited reduced binding capacity of HA in the skin and were unable to accumulate in the ECM29. Additionally, fibroblasts showed a significant decrease in collagen biosynthesis29. The loss of the copy number of the N-terminal of the VCAN gene may affect its capacity to bind to HA. In addition, copy number variations (CNVR_7451 and CNVR_4865) were found in the TIMP1 and FOXO1 genes. TIMP1, tissue inhibitors of metalloproteinases 1, inhibit metalloproteinase (MMP) activity. The expression of the TIMP1 gene was decreased in an intrinsic aging model of in vitro skin fibroblasts (HDF) when compared to young HDF donors30. FOXO1, a recognized anti-proliferative and pro-apoptotic factor, has been extensively studied and found to inhibits fibroblast activation and subsequent ECM generation31,32. Partial knockdown of FOXO1 during mouse skin wound healing accelerates the process, while also decreasing the density of collagen33. Each of these genes had varying effects on the proliferation or senescence of fibroblasts. Modifications in their copy number may impact gene expression via a dose effect, thus resulting in a connection to skin wrinkled cases in Xiang pig.
CNV overlapping genes have a wide range of molecular functions. In this study, we analyses the function of CNV overlapping genes identified in population pairs. The analysis of gene enrichment and KEGG pathways revealed that genes overlapping with population-stratified CNVRs were involved in various biological processes. These include extracellular matrix structure formation, skin growth and development (such as FoxO, Notch), and protein metabolism. Degradation of the extracellular matrix of the dermis and the formation of aged fibroblast phenotype are important molecular mechanisms of human skin aging34. The genes that overlap with CNVR are involved in various aspects of the extracellular matrix, such as N-glycan biosynthesis, HA binding, metalloendopeptidase activity, and elastic fibre assembly. Changes in the extracellular matrix components, such as collagen and elastin, have an impact on the skin’s structure and organization35. Skin growth and development are complex processes that involve multiple signaling pathways. These findings may provide insights into aging or wrinkling of the skin of Xiang pigs.
In the end, in our study, we identified 22 CNVRs that may be associated with the wrinkled skin cases of the Xiang pig. These CNVRs overlapped 61 known genes, including FOXO1, ADAMTS2, MAPK9, NEK5, NEK3, VCAN, YKT6, ARAF, TIMP1, GCK, ADAMTS18. FOXO133, NEK536, NEK337, YKT638, ARAF39 and MAPK940 are often associated with cell proliferation, apoptosis and autophagy. Their role may be to regulate skin cell fate, including fibroblasts, which can affect the process of skin ageing and wrinkling. VCAN28, TIMP130, ADAMTS1841, ADAMTS242 genes are associated with extracellular matrix structure. CNVR may affect the expression of these genes through transcript amplification, ablation, or truncation, which could significantly impact skin ageing/wrinkling. Further investigation is required to determine whether the loss of CN affects the proliferation of skin cells through a dose effect.
To gain a better understanding of the role of CN events in WSC, we analyzed the expression of candidate CNVRs overlapping genes using RNA-seq data from both WSC affected and control skin. The majority of the overlapped-genes were expressed in the skin. Only 12 genes, including TIMP1, FOXO1, and ARAF, were differentially expressed in WSC and controls. Differences in gene expression may contribute to the development of wrinkled or aged skin in the Xiang pig. Furthermore, we observed distinct copy numbers of the CNV_TIMP1 and CNV_VCAN genes in the F2 generation of the WSC×LW pedigree. An association was found between an increased copy number of the TIMP1 gene and the skin wrinkle index. The skin appeared more wrinkled when the TIMP1 genotype was gain. TIMP1 is a regulator of the extracellular matrix that inhibits the activity of matrix metalloproteinases. The study revealed that individuals with decreased skin elasticity had higher levels of TIMP143. The skin of diabetic patients exhibits disturbed collagen disorder and loose alignment due to an imbalance of MMP-9/TIMP-1 and MMP-2/TIMP-244. RNA-seq analysis also showed that the expression of TIMP1 was higher in skin samples with copy number gains compared to those with losses at 12 months of age. The CNV of VCAN did not show a significant correlation with either the skin wrinkle index or expression. However, a trend can be observed between a high skin wrinkle index and low expression in loss type. VCAN is associated with the accumulation of HA in the extracellular matrix. Shar-Pei exhibit a distinctive thick and heavily folded skin when there is an over-accumulation of HA in the skin10. Taken together, the results suggest that changes in CN of candidate CNVRs overlapping genes can be used to understand the cause of wrinkled skin cases in Xiang pigs. However, further investigation is required to determine the molecular basis.
In conclusion, the CNVs found in wrinkled skin cases were mapped using second-generation sequencing technology, with normal Xiang pigs as controls. We identified 22 candidate CNVRs associated with skin wrinkling cases in Xiang pigs. These CNVRs are predicted to affect the expression of genes such as VCAN, TIMP1, FOXO1, ADAMTS2, ADAMTS18, AGK, and DMD, through transcript amplification, transcript ablation, stop lost, feature truncation, splicing variant. The transcript levels of genes in case and control skin positively correlate with copy number. We believe that our study provides information to clarify the molecular mechanism of wrinkled skin cases in Xiang pigs. However, the molecular mechanism needs further confirmation.
Methods
Animal ethics and sample collection
The Guizhou University Subcommittee of Experimental Animal Ethics (EAE-GIU-2020-E015) approved all animal procedures. All methods were performed in accordance with the relevant guidelines and regulations. The study is also in accordance with ARRIVE guidelines. Next-generation sequencing was used to analyze a total of 83 Guizhou Xiang pigs from a local farm, including 20 cases of wrinkled skin (WSC) and 63 individuals of healthy skin (controls) from the Xiang pig populations (Supplementary Table S1).
DNA extraction, libraries construction and sequencing
Genomic DNA was extracted from blood or ear tissue for quality testing. The qualified DNA would be used for the construction of next-generation sequencing libraries. The library was sequenced with Illumina HiSeq2500 instrument (Illumina, USA). The raw sequencing reads were filtered using the default parameters of the NGSQC Toolkit. Alignment default parameters mapped the clean reads to the pig (Sscrofa 11.1) reference genome. SAMtools was used to convert files in SAM format to BAM format. Then, the GATK4.0 software package was used to remove PCR duplication and sort. The recalibration data would be used for CNV detection.
CNV identification and CNVR determination
Bioinformatics detection of genomic variation was performed on the 114 BAM files by CNVnator45 and CNVcaller46 software. Two software is based on the read-depth (RD) method. CNVnator captured RD signals in the genome, genotyped of deletions and duplications, and corrected for GC bias. CNVcaller improved the computational efficiency of complex genomes based on the RD algorithm. The default parameters were used for both programs according to population levels.
To obtain high quality CNV, all CNVs of each sample detected by CNVnator and CNVcaller were merged by bedtools, according to the principle of overlapping at least 1 bp. Then, overlapped CNVs between individuals were merged to generate highly reliable copy number variation regions (CNVRs). CNVs identified in three or more individuals with an overlap of at least 1 bp were defined as CNVRs.
F-statistics analysis of CNV frequencies
To evaluate differences in CNV differentiation within populations, we used F-statistics (Fst) based on genetic polymorphism to reflect signatures of diversifying selection. Fst = (Ht − Hs)/Ht26,47, where Ht represents the expected heterozygosity for the overall total population, which can be expressed as follows: Ht = 1 − ∑(p2− q2). In this context, p and q represent the frequencies of alleles A and a, respectively, within the total population. The Hs value is indicative of the anticipated heterozygosity in subpopulations. The calculation is performed as follows: Hs=\(\:{\sum\:}_{i=1}^{n}\left({H}_{{exp}i}*{n}_{i}\right)\)/NTotal. In this equation, “\(\:{H}_{{exp}i}\)” represents the expected heterozygosity, while “\(\:{n}_{i}\)” denotes the sample size of subpopulation “i”. Fst values must vary between 0 and 1, with closer to 1 indicating greater differences in genetic differentiation between populations. A statistical cut-off value of 0.15 Fst or greater was established, considering the overall distribution.
Gene functional annotation
Based on the Sscrofa11.1 Ensembl GFF file, annotated genes overlapping with the identified CNVRs were retrieved using bedtools (intersect). GO enrichment and functional analysis was performed with KOBAS-i (http://kobas.cbi.pku.edu.cn/). We retained enriched terms with a statistically P < 0.05 and False Discovery Rate (FDR) corrected.
CNV type assay
A CNV type assay was conducted using real-time quantitative PCR (qPCR) and 2−ΔΔCt methods. CNV genotyping was performed on 58 F2 individuals from a pedigree of WSC × Large white pig(LW). The primers used for qPCR amplification were designed using Primer Premier 5.0 software and synthesized by Invitrogen (Shanghai, China). These primers sequences are shown in Supplemen tary Table S8. Real time qPCR assays were performed using SYBR Premix Ex TaqTM (Tli RNaseH Plus) according to the manufacture's instructions. The total volume of PCR amplifications was 20 µL, consisting of the following reagents: 1 µL DNA (around 50 ng), 1 µL (20 pmol/µL) of both forward primer and reverse primer, 10 µL Blue-SYBR-Green mix (2×) and 8 µL water. The PCRs were run as follows: 5 min at 95 °C, followed by 40 cycles of 10 s at 95 °C and 10s at 72 °C; dissolution curve (95 °C, 15 s, 55 °C, 15 s, and 95 °C, 15 s). All reactions were performed on 96-well clear reaction plates, and each sample was amplified in triplicate. We estimated relative expression levels using the cycle threshold (2−ΔΔCt) method, which compares the ΔCt value (Ct of target gene − Ct of GCG in test sample) to that of the reference sample. Definition of CNV type of the target gene: a value equal to approximately 2 was considered a normal, and less or more than 2 represented loss and gain. We evaluated the skin wrinkling and thickness in some individuals of the F2 populations. The quantification of skin wrinkles was expressed as the wrinkle fraction, which is calculated by dividing the wrinkle value by the contour value. The phenotypic data of the skin will be analysed in correlation with CNV typing using SPSS 26.0.
RNA-seq analysis was performed on skin RNA samples taken from three wrinkled cases and three normal controls of Xiang pig. The RNA-seq analysis was performed using a 150-pair-end sequencing protocol on an Illumina Hiseq X10 from Shenzhen Huada Genentech. For sequence comparison, Sus scrofa 11.1 was used as the reference genome, and Subread 2.0.0 featurecounts were used to count gene expression. The expression of samples was normalized by calculating Counts Per Million (CPM)48. The R package DESeq2 was used to analyses the differences in gene expression between WSC and control skins. In addition, skin RNA-seq analyses were performed on 19 F2 hybrid progeny using the same method. The CNV gene expression was extracted and used to analyze its association with CNV genotyping.
Data availability
Sequence data that support the findings of this study have been deposited in the NCBI Sequence Read Archive (SRA) with the primary accession code PRJNA1057488.
References
Scherer, S. W. et al. Challenges and standards in integrating surveys of structural variation. Nat. Genet. 39, S7-15. https://doi.org/10.1038/ng2093 (2007).
Cooper, G. M., Nickerson, D. A. & Eichler, E. E. Mutational and selective effects on copy-number variants in the human genome. Nat. Genet. 39, S22–S29. https://doi.org/10.1038/ng2054 (2007).
Schiavo, G. et al. Copy number variants in Italian large white pigs detected using high-density single nucleotide polymorphisms and their association with back fat thickness. Anim. Genet. 45, 745–749. https://doi.org/10.1111/age.12180 (2014).
Liang, X. et al. Impact of KIT editing on coat pigmentation and fresh meat color in Yorkshire pigs. CRISPR J. 5, 825–842. https://doi.org/10.1089/crispr.2022.0039 (2022).
Kim, Y. H. et al. CCL3L3-null status is associated with susceptibility to systemic lupus erythematosus. Sci. Rep. 11, 19172. https://doi.org/10.1038/s41598-021-98531-6 (2021).
Poole, A. C. et al. Human salivary amylase gene copy number impacts oral and gut nicrobiomes. Cell. Host Microbe 25, 553-564e557. https://doi.org/10.1016/j.chom.2019.03.001 (2019).
Prasad, A. et al. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 (Bethesda) 2, 1665–1685. https://doi.org/10.1534/g3.112.004689 (2012).
Wetzel, A. S. & Darbro, B. W. A comprehensive list of human microdeletion and microduplication syndromes. BMC Genom Data 23, 82. https://doi.org/10.1186/s12863-022-01093-3 (2022).
Zhong, H. et al. Pan-cancer Analysis Reveals Potential of FAM110A as a Prognostic and Immunological Biomarker in Human cancer. Front. Immunol.14, 1058627. https://doi.org/10.3389/fimmu.2023.1058627 (2023).
Olsson, M. et al. A novel unstable duplication upstream of HAS2 predisposes to a breed-defining skin phenotype and a periodic fever syndrome in Chinese Shar-Pei dogs. PLoS Genet. 7, e1001332. https://doi.org/10.1371/journal.pgen.1001332 (2011).
Gibson, G. et al. Assessing mitochondrial DNA variation and copy number in lymphocytes of ~ 2000 sardinians using tailored sequencing analysis tools. PLoS Genet. https://doi.org/10.1371/journal.pgen.1005306 (2015).
Zhang, R., Wang, Y., Ye, K., Picard, M. & Gu, Z. Independent impacts of aging on mitochondrial DNA quantity and quality in humans. BMC Genom. 18, 890. https://doi.org/10.1186/s12864-017-4287-0 (2017).
Ershova, E. S. et al. Copy number variation of human satellite III (1q12) with aging. Front. Genet. 10, 704. https://doi.org/10.3389/fgene.2019.00704 (2019).
van der Meer, D. et al. Association of copy number variation of the 15q11.2 BP1-BP2 region with cortical and subcortical morphology and cognition. JAMA Psychiatry 77, 420–430. https://doi.org/10.1001/jamapsychiatry.2019.3779 (2020).
Dewan, A. et al. A common copy number variation (CNV) polymorphism in the CNTNAP4 gene: Association with aging in females. PLoS ONE https://doi.org/10.1371/journal.pone.0079790 (2013).
Lamis, A. et al. Hutchinson-Gilford progeria syndrome: A literature review. Cureus https://doi.org/10.7759/cureus.28629 (2022).
Xiaoli, L. et al. Detection of genomic structure variants associated with wrinkled skin in Xiang pig by next generation sequencing. Aging 13, 24710–24739. https://doi.org/10.18632/aging.203711 (2021).
Zheng, X. et al. CNV analysis of meishan pig by next-generation sequencing and effects of AHR gene CNV on pig reproductive traits. J. Anim. Sci. Biotechnol. 11, 42. https://doi.org/10.1186/s40104-020-00442-5 (2020).
Ding, R. et al. A composite strategy of genome-wide association study and copy number variation analysis for carcass traits in a Duroc pig population. BMC Genom. 23, 590. https://doi.org/10.1186/s12864-022-08804-1 (2022).
Stafuzza, N. B. et al. A genome-wide single nucleotide polymorphism and copy number variation analysis for number of piglets born alive. BMC Genom. 20, 321. https://doi.org/10.1186/s12864-019-5687-0 (2019).
Chen, C. et al. A comprehensive survey of copy number variation in 18 diverse pig populations and identification of candidate copy number variable genes associated with complex traits. BMC Genom. 13, 733. https://doi.org/10.1186/1471-2164-13-733 (2012).
Li, Y. et al. Identification of genome-wide copy number variations among diverse pig breeds by array CGH. BMC Genom. 13, 725. https://doi.org/10.1186/1471-2164-13-725 (2012).
Warr, A. et al. An improved pig reference genome sequence to enable pig genetics and genomics research. LID https://doi.org/10.1093/gigascience/giaa051 (2020).
E, G. X. et al. Comparative and selection sweep analysis of CNV was associated to litter size in Dazu Black Goats. Anim. Biotechnol. 32, 792–797. https://doi.org/10.1080/10495398.2020.1753756 (2021).
Zhang, W. et al. Population structure and selection signatures underlying domestication inferred from genome-wide copy number variations in Chinese Indigenous Pigs. Genes (Basel) https://doi.org/10.3390/genes13112026 (2022).
Zhu, C., Li, M., Qin, S., Zhao, F. & Fang, S. Detection of copy number variation and selection signatures on the X chromosome in chinese indigenous sheep with different types of tail. Asian-Australas J. Anim. Sci. 33, 1378–1386. https://doi.org/10.5713/ajas.18.0661 (2020).
Lansdown, A. B. Calcium: A potential central regulator in wound healing in the skin. Wound Repair. Regen. 10, 271–285. https://doi.org/10.1046/j.1524-475x.2002.10502.x (2002).
Watanabe, H. Aggrecan and versican: Two brothers close or apart. Am. J. Physiol. Cell Physiol. 322, C967–C976. https://doi.org/10.1152/ajpcell.00081.2022 (2022).
Hatano, S. et al. Versican A-subdomain is required for its adequate function in dermal development. Connect. Tissue Res. 59, 178–190. https://doi.org/10.1080/03008207.2017.1324432 (2018).
Lago, J. C. & Puzzi, M. B. The effect of aging in primary human dermal fibroblasts. PLoS One 14, e0219165. https://doi.org/10.1371/journal.pone.0219165 (2019).
Martins, R., Lithgow, G. J. & Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell. 15, 196–207. https://doi.org/10.1111/acel.12427 (2015).
Xin, Z. et al. FOXO1/3: Potential suppressors of fibrosis. Ageing Res. Rev. 41, 42–52. https://doi.org/10.1016/j.arr.2017.11.002 (2018).
Mori, R. et al. Reduced FOXO1 expression accelerates skin wound healing and attenuates scarring. Am. J. Pathol. 184, 2465–2479. https://doi.org/10.1016/j.ajpath.2014.05.012 (2014).
Fisher, G. J. et al. Skin aging from the perspective of dermal fibroblasts: The interplay between the adaptation to the extracellular matrix microenvironment and cell autonomous processes. J. Cell. Commun. Signal. https://doi.org/10.1007/s12079-023-00743-0 (2023).
Bonté, F., Girard, D., Archambault, J. C. & Desmoulière, A. Skin changes during ageing. Subcell. Biochem. 91, 249–280. https://doi.org/10.1007/978-981-13-3681-2_10 (2019).
Melo-Hanchuk, T. D., Slepicka, P. F., Pelegrini, A. L., Menck, C. F. M. & Kobarg, J. NEK5 interacts with topoisomerase IIβ and is involved in the DNA damage response induced by etoposide. J. Cell. Biochem. 120, 16853–16866. https://doi.org/10.1002/jcb.28943 (2019).
Panchal, N. K. & Evan Prince, S. The NEK family of serine/threonine kinases as a biomarker for cancer. Clin. Exp. Med. 23, 17–30. https://doi.org/10.1007/s10238-021-00782-0 (2023).
Sánchez-Martín, P. et al. ULK1-mediated phosphorylation regulates the conserved role of YKT6 in autophagy. J. Cell Sci. (2023). https://doi.org/10.1242/jcs.260546
An, S. et al. A-Raf: A new star of the family of raf kinases. Crit. Rev. Biochem. Mol. Biol. 50, 520–531. https://doi.org/10.3109/10409238.2015.1102858 (2015).
Sun, Y. et al. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 35, 600–604. https://doi.org/10.3109/10799893.2015.1030412 (2015).
Yang, N. et al. Adamts18 deficiency causes spontaneous SMG fibrogenesis in adult mice. J. Dent. Res. 101, 226–234. https://doi.org/10.1177/00220345211029270 (2021).
Simon, R. et al. Identification of an ADAMTS2 frameshift variant in a cat family with Ehlers–Danlos syndrome. G3 Genes Genomes Genet. https://doi.org/10.1093/g3journal/jkad152 (2023).
O’Brien, M. E. et al. Loss of skin elasticity is associated with pulmonary emphysema, biomarkers of inflammation, and matrix metalloproteinase activity in smokers. Respir. Res. (2019).https://doi.org/10.1186/s12931-019-1098-7
Zhou, P. et al. The imbalance of MMP-2/TIMP-2 and MMP-9/TIMP-1 contributes to collagen deposition disorder in diabetic non-injured skin. Front. Endocrinol. https://doi.org/10.3389/fendo.2021.734485 (2021).
Abyzov, A., Urban, A. E., Snyder, M. & Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984. https://doi.org/10.1101/gr.114876.110 (2011).
Wang, X. et al. CNVcaller: Highly efficient and widely applicable software for detecting copy number variations in large populations. GigaScience 6, 1–12. https://doi.org/10.1093/gigascience/gix115 (2017).
MacEachern, S., Hayes, B., McEwan, J. & Goddard, M. An examination of positive selection and changing effective population size in angus and holstein cattle populations (Bos taurus) Using a high density SNP genotyping platform and the contribution of ancient polymorphism to genomic diversity in domestic cattle. BMC Genom. 10, 181. https://doi.org/10.1186/1471-2164-10-181 (2009).
Corley, S. M., MacKenzie, K. L., Beverdam, A., Roddam, L. F. & Wilkins, M. R. Differentially expressed genes from RNA-Seq and functional enrichment results are affected by the choice of single-end versus paired-end reads and stranded versus non-stranded protocols. BMC Genom. 18, 399. https://doi.org/10.1186/s12864-017-3797-0 (2017).
Keel, B. N., Nonneman, D. J., Lindholm-Perry, A. K., Oliver, W. T. & Rohrer, G. A. A survey of copy number variation in the porcine genome detected from whole-genome sequence. Front. Genet. 10, 737. https://doi.org/10.3389/fgene.2019.00737 (2019).
Bovo, S. et al. Genome-wide detection of copy number variants in european autochthonous and commercial pig breeds by whole-genome sequencing of DNA pools identified breed-characterising copy number states. Anim. Genet. 51, 541–556. https://doi.org/10.1111/age.12954 (2020).
Paudel, Y. et al. Evolutionary dynamics of copy number variation in pig genomes in the context of adaptation and domestication. BMC Genom. 14, 449. https://doi.org/10.1186/1471-2164-14-449 (2013).
Jiang, J. et al. Global copy number analyses by next generation sequencing provide insight into pig genome variation. BMC Genom. 15, 593. https://doi.org/10.1186/1471-2164-15-593 (2014).
Revilla, M. et al. A global analysis of CNVs in swine using whole genome sequence data and association analysis with fatty acid composition and growth traits. PLoS One 12, e0177014. https://doi.org/10.1371/journal.pone.0177014 (2017).
Wang, J. et al. Enhancing genome-wide copy number variation identification by high density array CGH Using diverse resources of pig breeds. PLoS One 9, e87571. https://doi.org/10.1371/journal.pone.0087571 (2014).
Wang, J. et al. Improved detection and characterization of copy number variations among diverse pig breeds by array CGH. G3 (Bethesda) 5, 1253–1261. https://doi.org/10.1534/g3.115.018473 (2015).
Wang, Y. et al. Analysis of genome-wide copy number variations in Chinese indigenous and western pig breeds by 60 K SNP genotyping arrays. PLoS One 9, e106780. https://doi.org/10.1371/journal.pone.0106780 (2014).
Zhao, S., Wiedmann, R. T., Nonneman, D. J. & Rohrer, G. A. Genome-wide copy number variations using SNP genotyping in a mixed breed swine population. Plos One https://doi.org/10.1371/journal.pone.0133529 (2015).
Qiu, Y. et al. Genome-wide detection of CNV Regions and their potential association with growth and fatness traits in Duroc pigs. BMC Genom. 22, 332. https://doi.org/10.1186/s12864-021-07654-7 (2021).
Wang, Y., Zhang, T. & Wang, C. Detection and analysis of genome-wide copy number variation in the pig genome using an 80 K SNP beadchip. J. Anim. Breed. Genet. 137, 166–176. https://doi.org/10.1111/jbg.12435 (2020).
Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 31960641, 32360810, 31672390), the Guizhou provincial High Quality Development Project of Pig Industry in 2022[HQDPPI-2022], the Guizhou Provincial Science and Technology Projects (QKHPTRC[2019]5615, QKHRC[2016]4012), and the Guizhou University DXSCXJJ-[202210657165].
Author information
Authors and Affiliations
Contributions
Conceptualization, J.W. and X.R.; Methodology, X.L, Z.W., and F.H.; Investigation, X.L, F.H., Z.W., W.W., and X.C.; Resources, X.N and S.H.; Writing – Original Draft, X.L.; Writing –Review & Editing, J.W. and X.R.; Funding Acquisition, X.R.; Supervision, J.W. and X.R.All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, X., Hu, F., Wang, W. et al. Genome-wide identification of copy number variations in wrinkled skin cases of Xiang pigs. Sci Rep 14, 19695 (2024). https://doi.org/10.1038/s41598-024-70732-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-70732-9
Keywords
This article is cited by
-
Multi-tool copy number detection highlights common body size-associated variants in miniature pig breeds from different geographical regions
BMC Genomics (2025)
-
Identification of the putative regulatory regions and candidate genes associated with backfat thickness and intramuscular fat content traits in Xiang pigs
BMC Genomics (2025)
-
The systemic wrinkled skin phenotype involves aberrant expression and variation of genes related to the oxidative stress and extracellular matrix in Xiang pigs
BMC Genomics (2025)
