
Abstract
N6-methyladenosine (m6A) is the most prevalent modification found in eukaryotic RNA and played a significant role in various cancers. However, the mechanism by which m6A modification influences cervical cancer (CC) tumorigenesis remains unclear. Therefore, we aim to elucidate the role and mechanism of METTL3 in CC progression. In the present study, we observed a significant upregulation of METTL3 in CC tissues and cell lines. Knockdown of METTL3 resulted in reduced growth, migration, and invasion of CC cells, as well as affected apoptosis, while overexpression of METTL3 reversed these effects. Through a combined analysis of meRIP-seq and Ribo-seq data following METTL3 knockdown, NEK2 was identified as a key target of METTL3 in CC cells. Correlation analysis, MeRIP-qPCR, and luciferase reporter assay suggested that METTL3 regulates NEK2 expression through m6A modification. NEK2 synergized with METTL3 to mediate the malignant phenotype of CC cells. The METTL3-NEK2 axis promoted CC progression by activating the Wnt/β-catenin pathway and inhibiting the apoptosis pathway. In conclusion, METTL3 facilitated the malignant progression of CC and contributed to the formation of the METTL3-NEK2 regulatory axis in an m6A-dependent manner, which represented a potential target for CC therapy.
Similar content being viewed by others
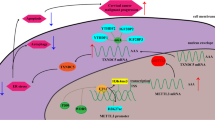


Introduction
Cervical cancer (CC) remains the fourth most common gynecological malignant tumor, with approximately 341,831 deaths and 604,127 new cases each year in worldwide1. The morbidity rate of CC is steadily declining in developed countries, attributed to the widespread implementation of the HPV vaccine and cervical cancer screening2. However, in developing countries, the mortality rate from CC has not decreased to the desired level, primarily due to poor public awareness, limited screening coverage, and challenges faced by women living in remote areas3. Patients diagnosed with CC at an early stage who receive prompt treatment exhibit a better prognosis; conversely, the prognosis for recurrent and metastatic CC remains unsatisfactory4,5. Consequently, it is essential to identify novel therapeutic targets to reduce CC mortality and enhance the survival of patients with this disease.
N6-methyladenosine (m6A) modification plays a crucial role in RNA stability, splicing, translocation, and nuclear localization, all of which are vital for tumor proliferation, invasion, and metastasis6. The m6A modification process is primarily mediated by methyltransferases (writers), demethylases (erasers), and reading proteins (readers)7. Recent studies have revealed that the demethylase ALKBH5 is associated with distant metastasis and lymph node metastasis in gastric cancer8. METTL3, a methyltransferase, catalyzes the formation of m6A modifications9. In recent years, the potential of METTL3 as a tumor biomarker has been identified in various malignant tumors. For example, METTL3 inhibits the expression of SOCS2 through the reading protein YTHDF2, thereby promoting the proliferation and metastasis of hepatocellular carcinoma10. Unfortunately, the mechanism by which METTL3 mediates m6A modification in CC remains unclear.
NEK2, a member of the Never in Mitosis A (NIMA)-related kinases family, is involved in various cellular functions, including cell cycle control, centrosome organization, DNA damage response, and RNA splicing11. Increasing evidence indicates that NEK2 is overexpressed in various malignant tumors, particularly in CC12. Nevertheless, there has been no report exploring the role of NEK2 and METTL3 in CC carcinogenesis. In this study, we aim to investigate that NEK2 is identified as the key target of METTL3 and promotes the development of CC through the WNT pathway and apoptosis pathway.
Materials and methods
RNA-seq and microarray datasets and bioinformatics analysis
The TCGA-CESC database was acquired from the cancer genome atlas genomic data commons (TCGA-GDC) Data Portal (https://portal.gdc.cancer.gov/, accessed on November 30, 2022). The databases GSE63514, GSE52903, GSE63591, GSE85324, and GSE112795 were sourced from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo, accessed on December 1, 2022). The TCGA-CESC database comprises 306 CC and 3 normal cervical tissues. The GSE63514 database contains 28 CC and 24 normal cervical tissues, and the GSE52903 database includes 55 CC and 17 normal cervical tissues. The GSE63591 database comprises ribosome sequencing (Ribo-seq) profiling of METTL3 knockdown and control samples. Lastly, the GSE85324 and GSE112795 databases encompass MeRIP-seq data from HeLa cells.
Clinical samples collection
Between 2020 and 2022, a total of 22 CC and 11 normal cervical tissues were collected from patients who underwent hysterectomy and biopsy prior to receiving chemotherapy and radiotherapy at The Second Affiliated Hospital of Zhengzhou University. All tissues were pathologically examined and subsequently preserved at -80 °C. This study received approval from the ethical committee of The Second Affiliated Hospital of Zhengzhou University (2022131).
Cell culture and transfection
The human CC cell lines SiHa and HeLa were purchased from the Procell Life Science & Technology Co., Ltd. (Wuhan, China) and cultured in DMEM (Solarbio, Beijing) with 10%FBS (Hyclone, Logan, Utah) in a humid atmosphere with 5% CO2 at 37 °C. Small interfering RNA (siRNA) against METTL3 (siMETTL3), NEK2 (siNEK2), WNT1(siWNT1) and negative control (siNC) were designed and synthesized by GeneCreate Bioengineering Co., Ltd. (Wuhan, China). The designated plasmids of pCDNA3.1, pCDNA3.1- METTL3 (oeMETTL3), and pCDNA3.1- NEK2 (oeNEK2) were transfected into SiHa and HeLa cells using Lipofectamine 3000 (Invitrogen, USA). The sequences of siRNA were displayed in Supplementary Table 1.
RNA extraction and quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNAs were extracted from human cells and tissues using the Trizol reagent (Invitrogen, USA) and processed with RQ1 DNase (Promega, USA) to remove DNA. All RNA samples were kept at -80 °C for future usage. Reverse transcription reactions were carried out using the ReverTra Ace qPCR RT Kit (TOYOBO Life Science, China). qRT-PCR was developed with SYBR GREEN (Solarbio, China) and 2×Taq PCR MasterMix (Solarbio, China). PCR conditions consisted of denaturing at 95 °C for 1 min, and 40 cycles of denaturing at 95 °C for 15 s followed by annealing and extension at 60 °C for 30 s. The relative expression of the target genes was calculated using the 2−ΔΔCt method, normalized with the reference gene of GAPDH13. Primer sequences were displayed in (Supplementary Table 2).
Western blot
Human cells and tissues were lysed in RIPA buffer containing 1 nM PMSF, and protein concentrations were measured using a BCA Protein Assay kit (Biosharp, China). The proteins were subsequently separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Following separation, the proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore, USA) using a transfer buffer composed of 25 mM Tris, 0.2 M glycine, and 25% methanol. After blocking with 5% skimmed milk, the membranes were incubated with primary antibodies: METTL3 (1:1000, ab195352, Abcam, UK), NEK2 (1:1000, ab227958, Abcam, UK), β-catenin (1:1000, R22820, ZenBio, China), Bax (1:1000, 200958, ZenBio, China), BCL2 (1:1000, 381702, ZenBio, China), WNT1 (1:500, E1A0392C-1, EnoGene, China) and GAPDH (1:2500, ab9485, Abcam, UK) overnight at 4℃. The membrane was then incubated with appropriate HRP-conjugated affinipure goat anti-Rabbit IgG (1:5000, SA00001-2, Proteintech, USA).
Immunohistochemistry (IHC)
The tissues sections were deparaffinized in xylene and subsequently rehydrated using a gradient of ethanol concentrations. Citrate buffer was prepared to facilitate antigen retrieval. BSA was added as the blocking buffer. The tissue sections were incubated overnight at 4℃ with primary antibodies METTL3 (1:1000, ab195352, Abcam, UK) and NEK2 (1:1000, ab227958, Abcam, UK). Following this, the sections were incubated with HRP-labeled goat anti-rabbit IgG (1:200, D110058, Sangon Biotech, China) and subsequently treated with DAB solution (Solarbio, China). The immunolabeled slides were counterstained with hematoxylin, dehydrated, and mounted. Finally, the tissue sections were photographed using an inverted microscope (Olympus, Japan).
Cell proliferation assay
The SiHa and HeLa cells were planted at a density of 5 × 103 cells per well in 96-well plates with different transfections. 10 µL cell Counting Kit-8 (CCK-8, Solarbio, China) reagent was added at the indicated time points after transfection (24, 48, and 72 h) for another 30 min. The absorbance at 450 nm was measured by a microplate reader (Heales, China).
Wound healing and transwell assays
For cell migration assay, the SiHa and HeLa cells were inoculated at a density of 5 × 105 cells per well in 6-well plates. After cells adhesion, a 200 µL pipette was used to draw a horizontal line on the cell surface. Images were captured immediately at the indicated time points (0, 24, and 48 h). The migration distances and migration rates were calculated by the Image J program (version 1.8.0). For cell transwell assay, transwell chambers were placed into 24-well plates with diluted matrigel gel (Becton, USA). The SiHa and HeLa cells were added in the upper chamber containing a 100µL serum-free medium, and the bottom chamber was filled with DMEM medium containing 10% FBS. After 24 h, following removal of the cells on the top surface of the upper chambers, the remaining cells were fixed in 4% paraformaldehyde, stained with 1% crystal violet, and then washed with PBS. Cell staining were photographed with inverted microscope.
Apoptosis assay
The SiHa and HeLa cells apoptosis were detected with FITC-Annexin V Apoptosis Detection Kit (Solarbio, China). Cells were rinsed with PBS and stained with FITC-Annexin V and PI in the dark for 5 min at room temperature. The stained cells were subjected to FACScalibur flow cytometry (Becton Dickinson, USA).
Quantification of mRNA m6A and methylated RNA immune-precipitation qPCR (MeRIP-qPCR)
For m6A quantification, 100–200 ng of mRNA was digested by nuclease P1 in 25 µl of buffer containing NH4OAc at 42 °C for 2 h, followed by the addition of alkaline phosphatase with incubation. The sample was enzymatically hydrolyzed and extracted with chloroform. The total amount of m6A in RNA was measured using LC-MS/MS by AB Sciex 6500 + QTRAP mass spectrometer. The ratio of m6A to A was calculated based on the determined concentrations.
MeRIP-qPCR assay14 was performed as previously described to identify the m6A modification of NEK2, following the recommendations of the GenSeq® m6A MeRIP Kit (Cloudseq, China). After RNA isolation, total RNA was fragmented and immunoprecipitated with an anti-m6A antibody or IgG using protein A/G magnetic beads. Bound RNA was subjected to reverse transcription followed by RT-qPCR analysis.
Luciferase reporter assay
The sequences of METTL3 containing either the wild-type (WT) or mutant (MUT) binding site of NEK2 were designed and synthesized by GenePharma (Shanghai, China). SiHa cells were co-transfected with the corresponding plasmids using Lipofectamine 3000 (Invitrogen, USA). Following a 48-hour incubation period, the activities of Firefly and Renilla luciferase were measured using the Dual Luciferase Reporter Assay Kit (Promega, USA), in accordance with the manufacturer’s instructions.
Statistical analysis
Data analyses were conducted using GraphPad Prism (version 8.0.1) and R (version 4.1.2). Student’s t-test, Chi square test and Fisher’s precision probability test were used to assess the significant difference between two groups. Pearson’s correlation analysis was employed to evaluate the relationship between METTL3 and NEK2. P-values were set at less than 0.05 to indicate statistical significance.
Results
METTL3 is highly expressed in CC
According to data acquired from the TCGA-CESC database, we first investigated the expression levels of METTL3 in CC tissues. The results demonstrated that METTL3 was significantly upregulated in CC (Fig. 1A). Similar findings were obtained from the GSE63514 and GSE52903 databases, which also indicated higher levels of METTL3 in CC (Fig. 1B,C). To validate these results, qRT-PCR analysis showed that CC tissues exhibited a higher mRNA expression level of METTL3 compared to normal cervical tissues (Fig. 1D). Based on the qRT-PCR results, we categorized the tissues into METTL3-low-expression and METTL3-high-expression groups and conducted a correlation analysis between METTL3 expression and clinicopathological characteristics. We found that FIGO stage (P = 0.004), lymph vascular space involvement (LVSI) (P = 0.049), and lymph node metastasis (P = 0.013) were significantly associated with METTL3 expression, as detailed in (Supplementary Table 3). Furthermore, Kaplan-Meier analysis indicated that patients with higher METTL3 expression had lower overall survival (OS) rates (Fig. 1E). Additionally, we randomly selected three pairs of tissues, revealing that METTL3 protein expression was higher in CC tissues than in normal tissues (Fig. 1F), and this trend was further confirmed by immunohistochemistry (IHC) assay (Fig. 1G).

METTL3 displayed higher expression level in CC. (A) The mRNA expression level of METTL3 in TCGA-CESC database. (B–C) The mRNA expression level of METTL3 in different CC databases from GEO (GSE63514 and GSE52903). (D) The mRNA expression level of METTL3 in 22 CC and 11 normal cervical tissues was detected by qRT-PCR. (E) Kaplan-Meier analysis of CC patients with high or low METTL3 expression in TCGA-CESC database. (F) The protein expression of METTL3 in 3 CC and 3 normal cervical tissues was measured by western blot. (G) The protein expression of METTL3 in CC and normal cervical tissues was measured by IHC. *P < 0.05. ***P < 0.001.
METTL3 regulates the proliferation, migration, invasion and apoptosis of CC
To investigate the role of METTL3 in CC, we downregulated METTL3 (siMETTL3) and upregulated METTL3 (oeMETTL3) in SiHa and HeLa cells. qRT-PCR and western blot analyses were employed to confirm transfection efficiency (Fig. 2A-C). The proliferation of SiHa and HeLa cells was significantly enhanced in the oeMETTL3 group compared to the normal control group, while this trend was reversed in the siMETTL3 group (Fig. 2D). Results from wound healing and transwell assays demonstrated that inhibition of METTL3 led to reduced migration and invasion, whereas the migratory and invasive capacities of SiHa and HeLa cells were activated under METTL3 overexpression (Fig. 2E-F). Flow cytometry analysis of apoptosis indicated that METTL3 knockdown enhanced apoptosis in CC cells (Fig. 2G). Furthermore, qRT-PCR results revealed that METTL3 overexpression increased the expression of migration and invasion-related genes, Twist and Snail, while downregulating the epithelial marker E-cadherin (Fig. 2H). Collectively, these findings indicate that METTL3 inhibits apoptosis while promoting the migration, invasion, and proliferation of CC cells.

METTL3 regulates the proliferation, migration, invasion and apoptosis of CC cells in vitro. (A) The expression of METTL3 in CC cells transfected with normal control (siNC), METTL3-targeted (siMETTL3-1#, siMETTL3-2#, and siMETTL3-3#) siRNAs was detected by qRT-PCR. (B) The mRNA expression level of METTL3 in METTL3 up- or down-regulated CC cells was confirmed by qRT-PCR. (C) The protein expression level of METTL3 in METTL3 up- or down-regulated CC cells was confirmed by western blot analysis. (D) The effect of METTL3 on CC cells proliferation at different time points was measured by CCK-8 assay. (E) The effect of METTL3 on CC cells migration was detected by wound healing assay. (F) The effect of METTL3 on CC cells invasion was tested by transwell assay. (G) The effect of METTL3 on CC cells apoptosis was detected by flow cytometry. (H) qRT-PCR indicated the Twist, Snail, and E-cadherin expression in the transfection of METTL3 overexpression. *P < 0.05. **P < 0.01. ***P < 0.001.
NEK2 acts as the target of METTL3 in CC
To further investigate the downstream targets and underlying mechanisms of METTL3 in CC, we analyzed the characteristics of m6A modification in five CC and three normal cervical tissues. Our findings revealed that the level of m6A was significantly upregulated in the CC tissues (Fig. 3A). A schematic diagram illustrated the m6A motif associated with METTL3 (Fig. 3C). Analysis of Ribo-seq data (GSE63591) identified 1,651 upregulated genes and 1,350 downregulated genes in METTL3 knockdown HeLa cells compared to normal control HeLa cells (Fig. 3D- E). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis indicated that METTL3 knockdown in CC cells led to significant alterations in the cell cycle, apoptosis, DNA replication, and the p53 signaling pathway (Fig. 3F). Additionally, we analyzed MeRIP-seq data (GSE85324 and GSE112795) from HeLa cells. By overlapping the results of Ribo-seq and meRIP-seq, NEK2 was identified as a candidate target of METTL3. The SRAMP database confirmed that NEK2 contained m6A sites with high reliability (Fig. 3B). To validate NEK2 as a candidate target of METTL3, we first examined the correlation between NEK2 and METTL3 expression in CC tissues. Correlation analysis demonstrated a positive relationship between METTL3 and NEK2 expression (Fig. 3-H). Subsequently, we assessed NEK2 expression following METTL3 overexpression and knockdown in SiHa and HeLa cells using qRT-PCR and western blot analysis. METTL3 inhibition significantly reduced both mRNA and protein levels of NEK2, whereas METTL3 overexpression resulted in increased expression levels of NEK2 (Fig. 3I-K). MeRIP-qPCR analysis showed marked enrichment of NEK2 m6A modification. Notably, METTL3 inhibition diminished NEK2 m6A modification, while METTL3 overexpression enhanced the modification level in SiHa cells (Fig. 3L). To investigate whether METTL3 directly bound to the m6A modification sites of NEK2 mRNA, a luciferase reporter assay was conducted. Wild-type NEK2 (NEK2-WT) and m6A binding domain mutated NEK2 (NEK2-MUT) were ectopically expressed in SiHa cells. As shown in Fig. 3M, the overexpression of METTL3 led to an increase in luciferase activity in the NEK2-WT group, while these changes were abolished in the NEK2-MUT group. These results suggest that METTL3 regulates NEK2 expression through m6A modification.

NEK2 acts as the target of METTL3 in CC. (A) m6A quantification analysis in the CC and normal cervical tissues. (B) The SRAMP database revealed the NEK2 m6A sites. (C) Schematic diagram demonstrated the m6A motif of METTL3. (D) Volcano map showed differentially expressed genes in Ribo-seq upon METTL3 knockdown. (E) Heatmap showed differentially expressed genes in Ribo-seq upon METTL3 knockdown. (F) KEGG pathway analysis of differentially expressed genes in Ribo-seq upon METTL3 knockdown. (G) The correlation between NEK2 and METTL3 expression in TCGA-CESC database. (H) The correlation between NEK2 and METTL3 expression in CC tissues. (I) qRT-PCR indicated the NEK2 mRNA expression in the transfection of METTL3 knockdown. (J) qRT-PCR indicated the NEK2 mRNA expression in the transfection of METTL3 overexpression. (K) Western blot indicated the NEK2 protein expression in the transfection of METTL3 knockdown or METTL3 overexpression. (L) MeRIP-qPCR indicated the NEK2 mRNA enrichment precipitated by m6A antibody (M) Luciferase reporter assay was performed to investigate METTL3 directly bound the m6A modification sites of NEK2 mRNA. *P < 0.05. **P < 0.01. ***P < 0.001.
NEK2 plays an oncogenic role in CC
Through the analysis of transcriptome data from the TCGA-CESC and GEO databases (GSE63514 and GSE52903), we observed that NEK2 mRNA expression was higher in CC tissues compared to normal cervical tissues (Fig. 4A). This finding was corroborated by qRT-PCR, which confirmed the differential expression in 22 CC and 11 normal cervical tissues (Fig. 4B). We further explored the clinical significance of NEK2 expression as determined by qRT-PCR results. The data indicated that CC tissues with adenocarcinoma and LVSI exhibited higher NEK2 expression levels than those with squamous cell carcinoma and without LVSI (Supplementary Table 4). Kaplan-Meier analysis revealed that CC patients with low NEK2 expression did not experience any survival benefits (P = 0.301, Fig. 4C) the TCGA-CESC database. Subsequently, we assessed NEK2 protein abundance in CC and normal cervical tissues using IHC assay, which demonstrated a similar trend of increased NEK2 expression in CC tissues compared to normal cervical tissuess at the protein level (Fig. 4D). Collectively, these results suggest that NEK2 is highly expressed at both the mRNA and protein levels in CC.

NEK2 displayed higher expression level in CC. (A) The mRNA expression level of NEK2 in TCGA-CESC database, and GEO (GSE63514 and GSE52903). (B) The mRNA expression level of NEK2 in 22 CC and 11 normal cervical tissues was detected by qRT-PCR. (C) Kaplan-Meier analysis of CC patients with high or low NEK2 expression in TCGA-CESC database. (D) The protein expression of NEK2 in CC and normal cervical tissues was measured by IHC. **P < 0.01. ***P < 0.001.
To further investigate the role of NEK2 in CC, we conducted gain- and loss-of-function assays. qRT-PCR and western blot analyses confirmed the efficiency of NEK2 knockdown and overexpression following transfection (Fig. 5A-C). CCK-8 assays indicated that NEK2 deletion significantly suppressed cell proliferation, whereas NEK2 overexpression enhanced proliferation in SiHa and HeLa cells (Fig. 5D). The wound healing assay demonstrated that the migration ability was significantly inhibited upon NEK2 knockdown, while the opposite effect was observed with NEK2 upregulation in CC cells (Fig. 5E). Furthermore, transwell assays revealed that NEK2 knockdown markedly hindered the invasive capabilities of SiHa and HeLa cells, whereas NEK2 overexpression increased the invasiveness of CC cells (Fig. 5F). Furthermore, the apoptosis ability of NEK2-overexpressing in SiHa and HeLa cells were significantly decreased, whereas knockdown of NEK2 significantly increased cell apoptosis (Fig. 5G). Results from qRT-PCR demonstrated that NEK2 overexpression enhanced the expression of migration and invasion-related genes, Twist and Snail, while concurrently reducing the expression of E-cadherin, indicating the occurrence of epithelial-mesenchymal transition (Fig. 5H).

NEK2 regulates the proliferation, migration, invasion and apoptosis of CC cells in vitro. (A) The expression of NEK2 in CC cells transfected with normal control (siNC), NEK2-targeted (siNEK2-1#, siNEK2-2#, and siNEK2-3#) siRNAs was detected by qRT-PCR. (B) The mRNA expression level of NEK2 in NEK2 up- or down-regulated CC cells was confirmed by qRT-PCR. (C) The protein expression level of NEK2 in NEK2 up- or down-regulated CC cells was confirmed by western blot analysis. (D) The effect of NEK2 on CC cells proliferation at different time points was measured by CCK-8 assay. (E) The effect of NEK2 on CC cells migration was detected by wound healing assay. (F) The effect of NEK2 on CC cells invasion was tested by transwell assay. (G) The effect of NEK2 on CC cells apoptosis was detected by flow cytometry. (H) qRT-PCR indicated the Twist, Snail, and E-cadherin expression in the transfection of NEK2 overexpression. *P < 0.05. **P < 0.01. ***P < 0.001.
Over expressed NEK2 reverses the silencing METTL3 effects on the CC apoptosis and migration
Next, we provided additional evidence that the m6A modification regulates the expression of NEK2. A low expression of METTL3 led to a marked decrease in the mRNA levels of both METTL3 and NEK2, however, these effects were significantly mitigated upon the reintroduction of NEK2 (Fig. 6A). Furthermore, the knockdown of METTL3 significantly inhibited cell migration and enhanced apoptosis in CC cells. Conversely, when NEK2 was reintroduced into these cells, the aggressive cancer progression that had been inhibited by METTL3 knockdown was substantially restored (Fig. 6B- C). Collectively, our results suggest that NEK2 synergizes with METTL3 to mediate the malignant phenotype of CC cells.

Over expressed NEK2 reversed the silencing METTL3 effects on the CC apoptosis and migration. (A) qRT-PCR indicated the NEK2 and METTL3 mRNA expression in the transfection of METTL3 knockdown with or without NEK2 overexpression. (B) The effect of METTL3 knockdown with or without NEK2 overexpression on CC cells apoptosis was detected by flow cytometry. (C) The effect of METTL3 knockdown with or without NEK2 overexpression on CC cells migration was detected by wound healing assay. **P < 0.01. ***P < 0.001.
METTL3 regulates the expression of NEK2 via Wnt/β-catenin signaling and apoptosis pathway
We next investigated the underlying mechanisms of the METTL3-NEK2 axis in regulating the tumorigenic potential of CC cells. The results confirmed that the mRNA and protein levels of Wnt1, β-catenin, and Bcl2 were significantly increased, while Bax levels were decreased following the overexpression of NEK2 (Fig. 7A- B). Conversely, the knockdown of NEK2 resulted in opposite effects in CC cells. Specifically, METTL3 knockdown led to a decrease in the protein levels of Wnt1 and Bcl2, whereas METTL3 overexpression enhanced the protein levels of these factors (Fig. 7C). Furthermore, interference with Wnt1 downregulated the expression of Wnt1 and β-catenin in Siha cells. Notably, Wnt1 knockdown, when combined with the overexpression of NEK2 or METTL3, promoted the expression of Wnt1 and β-catenin, effectively reversing the effects of Wnt1 interference (Fig. 7D). In summary, these findings reveal that the METTL3-NEK2 axis regulates the Wnt/β-catenin signaling and the apoptosis pathway.

METTL3-NEK2 axis regulates the Wnt/β-catenin signaling and apoptosis pathway. (A) qRT-PCR indicated the Wnt1, β-catenin, Bcl2, and Bax mRNA expression in the transfection of NEK2 overexpression or NEK2 knockdown (B) Western blot indicated the Wnt1, β-catenin, Bcl2, and Bax protein expression in the transfection of NEK2 overexpression or NEK2 knockdown. (C) Western blot indicated the Wnt1, and Bcl2 protein expression in the transfection of METTL3 overexpression or METTL3 knockdown. (D) qRT-PCR and Western blot indicated the Wnt1 and β-catenin expression in the transfection of Wnt1 knockdown, Wnt1 knockdown combined with overexpression of NEK2 or METTL3. *P < 0.05. **P < 0.01. ***P < 0.001.
Discussion
For patients with CC, surgery, radiation, and chemotherapy have long been the mainstays of treatment15. Recently, novel therapeutics, such as immunotherapy16 and antibody-drug conjugates17 have emerged, providing new treatment opportunities for patients with CC. However, there remains a risk of recurrence and metastasis even after these interventions. Recurrence and metastasis are currently the leading causes of death among patients with CC18. Hence, understanding the underlying mechanisms involved in the development of CC is of significant clinical importance.
m6A, which is located in the conserved RRACH sequence (where R = G or A; H = A, C, or U), is the most abundant modification found in eukaryotic mRNA, as well as in non-coding RNAs, including miRNA, lncRNA, and circRNA19. The m6A modification process is dynamic and reversible. Advances in enzymology have led to the discovery of m6A methyltransferases, such as METTL3, METTL14, and WTAP, as well as m6A demethylases, including FTO and ALKBH5. Additionally, reading proteins, such as the YTH protein family (YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2) and the insulin-like growth factor 2 mRNA binding protein family (IGF2BP1, IGF2BP2, and IGF2BP3), have also been identified20. It has been reported that m6A is closely associated with CC21. Du et al.22. found that YTHDF3 promoted the translation of RAD51D in an m6A-dependent manner, contributing to radio-resistance in CC. Zhen et al.23. demonstrated that ALKBH5 demethylates and destabilizes SIRT3 in an m6A-IGF2BP1-dependent manner, which repressed CC growth, lipid metabolism, and tumorigenesis by downregulating ACC1. Furthermore, RANBP2 has been shown to promote the growth, migration, and invasion of CC cells, while YTHDF1 regulates RANBP2 translation in an m6A-dependent manner, highlighting it as a potential target for CC treatment24.
In this study, we investigated the upregulation of METTL3 expression in CC and its association with poor prognosis, which aligns with previous research findings9,25. Importantly, the overexpression of METTL3 significantly induced proliferation, migration, and invasion of CC cells, while inhibiting apoptosis in vitro. However, the roles of METTL3 in the molecular mechanisms underlying the development and progression of CC remain infrequently reported in existing literature. For instance, METTL3 enhances the stability of FOXD2-AS1, thereby maintaining its expression. FOXD2-AS1 is recruited to the promoter region of p21, leading to the suppression of its expression and the acceleration of CC progression26. To further investigate the regulatory effects of METTL3 on downstream target genes, we conducted Ribo-seq and MeRIP-seq analyses in HeLa cells. Our findings suggest that NEK2 may be a potential target gene of METTL3. METTL3 can bind to NEK2 mRNA and influence its expression in an m6A-dependent manner, thereby promoting the malignant progression of CC.
NEK2 plays a crucial role in the mitotic process by regulating centrosomes, spindles, chromosomes, and various other cellular functions27. It is highly expressed in numerous malignant tumors, including CC and shows promise as a novel tumor marker28. In this study, we investigated the downstream signaling pathways of the METTL3-NEK2 axis and observed significant alterations in the apoptosis signaling pathway in CC cells following METTL3 knockdown. Furthermore, previous studies have demonstrated the association of METTL3 with the expression and membrane localization of β-catenin29. Consequently, we examined the expression changes of Wnt1 and β-catenin, which are associated with the Wnt/β-catenin pathway, as well as the apoptosis-related molecules Bcl2 and Bax, using qRT-PCR and western blot analysis after overexpressing or knocking down NEK2 in CC cells. Our findings suggest that the METTL3-NEK2 axis promotes the progression of CC by activating the Wnt/β-catenin pathway and inhibiting the apoptosis pathway.
However, it is important to acknowledge certain limitations of this study. Firstly, the sample size was relatively small. Additionally, this study did not incorporate any animal experiments. Therefore, future research will explore the potential application of the METTL3-NEK2 axis as a target for targeted therapy in CC.
Conclusion
In summary, our study demonstrated that METTL3 facilitated the malignant progression of CC and led to the formation of the METTL3-NEK2 regulatory axis in an m6A-dependent manner, which might serve as a potential therapeutic target for CC.
Data availability
The datasets generated and/or analysed during the current study are not publicly available due [REASON WHY DATA ARE NOT PUBLIC] but are available from the corresponding author on reasonable request.
References
Sung, H. et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71 (3), 209–249 (2021).
Singh, D. et al. Global estimates of incidence and mortality of cervical cancer in 2020: a baseline analysis of the WHO global cervical cancer elimination initiative. Lancet Glob. Health 11 (2), e197–e206 (2023).
Mezei, A. K. et al. Cost-effectiveness of cervical cancer screening methods in low- and middle-income countries: a systematic review. Int. J. Cancer 141 (3), 437–446 (2017).
Adiga, D., Eswaran, S., Pandey, D., Sharan, K. & Kabekkodu, S. P. Molecular landscape of recurrent cervical cancer. Crit. Rev. Oncol. Hematol. 157, 103178 (2021).
Cohen, P. A., Jhingran, A., Oaknin, A. & Denny, L. Cervical cancer. Lancet 393 (10167), 169–182 (2019).
He, L. et al. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 18 (1), 176 (2019).
Wang, T., Kong, S., Tao, M. & Ju, S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 19 (1), 88 (2020).
Hu, Y. et al. Demethylase ALKBH5 suppresses invasion of gastric cancer via PKMYT1 m6A modification. Mol. Cancer 21 (1), 34 (2022).
Liu, S. et al. METTL3 plays multiple functions in biological processes. Am. J. cancer Res. 10 (6), 1631–1646 (2020).
Chen, M. et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 67 (6), 2254–2270 (2018).
Panchal, N. K. & Evan Prince, S. The NEK family of serine/threonine kinases as a biomarker for cancer. Clin. Exp. Med. 23 (1), 17–30 (2023).
Zhang, Y. R. & Zheng, P. S. NEK2 inactivates the Hippo pathway to advance the proliferation of cervical cancer cells by cooperating with STRIPAK complexes. Cancer Lett. 549, 215917 (2022).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25 (4), 402–408 (2001).
Wu, M., Chen, G., Liao, X., Xiao, L. & Zheng, J. YTHDF2 interference suppresses the EMT of cervical cancer cells and enhances cisplatin chemosensitivity by regulating AXIN1. Drug Dev. Res. 83 (5), 1190–1200 (2022).
Podwika, S. E. & Duska, L. R. Top advances of the year: cervical cancer. Cancer 129 (5), 657–663 (2023).
Grau, J. F., Farinas-Madrid, L., Garcia-Duran, C., Garcia-Illescas, D. & Oaknin, A. Advances in immunotherapy in cervical cancer. Int. J. Gynecol. Cancer 33 (3), 403–413 (2023).
Tolcher, A., Hamilton, E. & Coleman, R. L. The evolving landscape of antibody-drug conjugates in gynecologic cancers. Cancer Treat. Rev. 116, 102546 (2023).
Gennigens, C. et al. Recurrent or primary metastatic cervical cancer: current and future treatments. ESMO Open 7 (5), 100579 (2022).
Chen, D. H., Zhang, J. G., Wu, C. X. & Li, Q. Non-coding RNA m6A modification in Cancer: mechanisms and therapeutic targets. Front. Cell. Dev. Biol. 9, 778582 (2021).
Wang, Y. et al. Epigenetic modification of m(6)a regulator proteins in cancer. Mol. Cancer 22 (1), 102 (2023).
Mao, Z., Wang, B., Zhang, T. & Cui, B. The roles of m6A methylation in cervical cancer: functions, molecular mechanisms, and clinical applications. Cell. Death Dis. 14 (11), 734 (2023).
Du, H., Zou, N. Y., Zuo, H. L., Zhang, X. Y. & Zhu, S. C. YTHDF3 mediates HNF1α regulation of cervical cancer radio-resistance by promoting RAD51D translation in an m6A-dependent manner. FEBS J. 290 (7), 1920–1935 (2023).
Zhen, L. & Pan, W. ALKBH5 inhibits the SIRT3/ACC1 axis to regulate fatty acid metabolism via an m6A-IGF2BP1-dependent manner in cervical squamous cell carcinoma. Clin. Exp. Pharmacol. Physiol. 50 (5), 380–392 (2023).
Wang, H. et al. YTHDF1 aggravates the progression of cervical cancer through m(6)A-mediated up-regulation of RANBP2. Front. Oncol. 11, 650383 (2021).
Wang, Q. et al. N(6)-methyladenosine METTL3 promotes cervical cancer tumorigenesis and Warburg effect through YTHDF1/HK2 modification. Cell. Death Dis. 11 (10), 911 (2020).
Ji, F. et al. M(6)a methyltransferase METTL3-mediated lncRNA FOXD2-AS1 promotes the tumorigenesis of cervical cancer. Mol. Therapy Oncolytics 22, 574–581 (2021).
Fang, Y. & Zhang, X. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell. Cycle 15 (7), 895–907 (2016).
Xu, T. et al. Targeting NEK2 impairs oncogenesis and radioresistance via inhibiting the Wnt1/β-catenin signaling pathway in cervical cancer. J. Exp. Clin. Cancer Res. 39 (1), 183 (2020).
Li, J. et al. RNA m(6)a methylation regulates dissemination of cancer cells by modulating expression and membrane localization of β-catenin. Mol. Ther. 30 (4), 1578–1596 (2022).
Funding
This work was sponsored by Henan Province Key Research and Development and Promotion (Science and Technology) Project (222102310651, 242102310405), Henan Province Medical Science and Technology Research Program Joint Construction Project (LHGJ20220476), Key Scientific Research Project of Henan Provincial Education Department (24B320029).
Author information
Authors and Affiliations
Contributions
Yilin Guo, Zhen Xu and Hu Zhao contributed conception and designed this study. Yilin Guo, Yangyang Bai, Lu Wang, Nan Zhang and Wuliang Wang prepared the materials, performed the experiments, and analyzed the data. Yilin Guo wrote the first draft of the manuscript. Zhen Xu and Hu Zhao revised manuscript. All authors contributed to manuscript read, and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, Y., Bai, Y., Wang, L. et al. METTL3 facilitates the progression of cervical cancer by m6A modification-mediated up-regulation of NEK2. Sci Rep 14, 24469 (2024). https://doi.org/10.1038/s41598-024-73601-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-73601-7
