
Abstract
Objective
Previous observational studies have indicated associations between various inflammatory cytokines and diabetic nephropathy (DN) caused by type 2 diabetes mellitus (T2DM). However, the causality remains unclear. We aimed to further evaluate the causal association between 91 inflammatory cytokines and DN using bidirectional two-sample Mendelian randomization (MR) analysis.
Method
Summary statistics for DN were obtained from a publicly available genome-wide association study (GWAS) analysis. Data pertaining to inflammatory cytokines were derived from a GWAS protein quantitative trait locus (pQTL) study. The primary analytical approach employed the inverse variance weighted (IVW) method, complemented by MR-Egger regression, weighted mode (WM), and weighted median (WME) methods to evaluate the causal association between inflammatory cytokines and DN. Sensitivity analyses were conducted to validate the robustness of the findings.
Result
Among individuals of European ancestry, the IVW method results revealed a positive causal association between the gene expression of tumor necrosis factor ligand superfamily member 14 (TNFSF14), and TNF-related activation-induced cytokine (TRANCE) with DN. Conversely, a negative causal association was observed between the gene expression of interleukin-1-alpha (IL-1α), and transforming growth factor-alpha (TGF-α) with DN. Among individuals of East Asian ancestry, the IVW method results indicated a negative causal association between the gene expression of glial cell line-derived neurotrophic factor (GDNF) and DN. Notably, these findings persisted without evidence of horizontal pleiotropy or heterogeneity, ensuring their robustness and reliability.
Conclusion
The MR analysis underscores a causal association between inflammatory cytokines and DN, providing an important reference and evidence for the study of DN.
Similar content being viewed by others

Introduction
Diabetic nephropathy (DN) emerges as a prevalent complication of type 2 diabetes mellitus (T2DM)1, and SNPs confer genetic susceptibility for T2DM2. Studies have highlighted the complex interaction between glucose metabolism issues and abnormal kidney blood flow, which triggers inflammation and leads to changes in kidney blood vessels. These changes are key to the onset and progression of DN3,4,5. Inflammatory cytokines are critical in DN’s development and progression6. For instance, Zhang demonstrated that IL-17C contributes to DN and that inhibiting IL-17C may offer a therapeutic approach7. Murakoshi found that IL-6 is crucial for the proliferation of glomerular mesangial cells and the activation and expansion of B cells. As a pleiotropic cytokine, IL-6 has various effects on the body and directly influences the inflammatory response in DN8. Nakamura identified MCP-1 as an early marker of kidney function changes in DN, with its levels reflecting the disease’s progression9. Furthermore, investigations have indicated that DN progression may be influenced by the activation of different signal transduction pathways mediated by inflammatory cytokines10 and other biomarkers11,12. However, the precise impact of these cytokines on DN at the genetic level is still unknown, highlighting the need for further research.
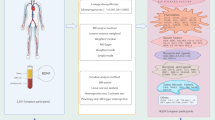
Mendelian randomization (MR) analysis stands as a potent method for elucidating causal associations between exposures and outcomes. It uses single nucleotide polymorphisms (SNPs) as instrumental variables (IVs)13. In recent years, MR has become a valuable tool for assessing these causal associations14. In our study, we used a two-sample bidirectional MR analysis to examine the causal association between inflammatory cytokines and DN caused by T2DM. This provided genetic evidence for their association. The procedural framework of our study is depicted in Fig. 1.

The protocol of our study procedure.
Material and method
Exposure and outcome data sources
For the estimation of SNP effects associated with inflammatory cytokines, we utilized GWAS summary statistics (GSCT90274758-GSCT90274848) provided by Zhao15, encompassing 91 inflammatory cytokines and 14,824 individuals of European ancestry. Summary statistics for DN were retrieved from a publicly available GWAS analysis, incorporating 1,032 cases and 451,248 controls of European ancestry (ebi-a-GCST90018832), along with 220 cases and 132,764 controls of East Asian ancestry (ebi-a-GCST90018612), sourced from the IEU OpenGWAS Project website (gwas.mrcieu.ac.uk). The European ancestry dataset comprised 452,280 individuals and 24,190,738 SNPs16, while the East Asian ancestry dataset included 132,984 individuals and 12,447,074 SNPs16.
As this study relies on publicly available data, no additional ethical approval or consent was required. To mitigate the influence of race-related confounding factors, the study population’s genetic background was predominantly of European ancestry. Additionally, data from individuals of East Asian genetic background were incorporated to enhance the robustness and generalizability of the findings.
Instrumental variables selection
The selection of genetic variants as instrumental variables (IVs) was associated with the exposure of inflammatory cytokines. The selection of IVs in this study adhered to the three-hub hypothesis (Fig. 2).

The hub hypothesis of the MR analysis.
Firstly, in screening the GWAS data, only SNPs meeting the criterion of P < 5e− 06 were considered. A linkage disequilibrium test was subsequently conducted on the included SNPs to ensure compliance with the independence hypothesis. During SNP selection, parameters were meticulously controlled, with a threshold of R2 < 0.001 and a maximum distance of 10,000 kb, aiming to mitigate linkage disequilibrium and identify independent SNPs17. Secondly, the PhenoScanner database was utilized to further validate whether the identified SNP loci exhibited an association with other potential confounding factors18. Lastly, to assess the susceptibility of the included SNPs to weak IV bias, F statistics were employed, with a threshold set at F > 10 (calculated using the formula F = β^2/SE^2, where β represents the effect on the exposure and SE represents the standard error). SNPs with F < 10 were considered susceptible to weak IV bias and were consequently excluded to prevent their influence on the results19.
Statistical analysis
MR analysis
To unveil the causal association between inflammatory cytokines and DN, a comprehensive array of analytical methodologies was employed, encompassing the IVW, WME, WM, MR-Egger regression, and forest plot visualization20. The utilization of multiple analytical approaches aimed at fortifying the robustness and reliability of the findings. A significance threshold of P < 0.05 was employed to infer a causal association between exposure (inflammatory cytokines) and outcome (DN)21.
To attenuate the potential for a specific type of error and evaluate the impact of multiple testing on our findings, we additionally employed the q value to rectify the false discovery rate (FDR). The q value < 0.1 signifies a significant association. The P < 0.05 yet the q value ≥ 0.1, an association between inflammatory cytokines and DN is considered suggestive22.
Sensitivity analysis
The P-value derived from Cochran’s Q statistics of the IVW was utilized to assess the heterogeneity of IVs. A P-value ≥ 0.05 signifies the absence of heterogeneity in causal analysis23. Furthermore, the funnel plot was employed as a visual tool to discern heterogeneity, with a symmetrical distribution of SNPs indicative of homogeneity in the results24. Depending on the presence or absence of heterogeneity, either a random-effects model or a fixed-effects model was selected. The presence of pleiotropy was meticulously assessed via MR-Egger regression, with analysis conducted by examining the intercept of MR-Egger regression in a scatter plot25. Moreover, LOO analysis was conducted to evaluate stability, identifying SNPs with significant influence when individually removed26. The risk association between inflammatory cytokines and DN was quantified using OR and 95%CI, with statistical significance denoted by P < 0.05.
Bidirectional MR analysis
A two-sample bidirectional MR analysis was undertaken to investigate the potential reverse causal association between DN (exposure) and inflammatory cytokines (outcome). The procedural steps for bidirectional MR analysis closely mirrored those of the MR analysis delineated previously.
Statistical software
All MR analyses were executed utilizing R (version 4.3.1) and the TwoSampleMR package.
Results
Instrumental variables selection
Among individuals of European ancestry, a total of 14, 14, 26, and 29 SNPs were extracted from TNFSF14, TRANCE, IL-1α, and TGF-α, respectively (Fig. 3A). Among individuals of East Asian ancestry, 8 SNPs were extracted from GDNF (Fig. 3B). Notably, the F statistics of the IVs enlisted in this study exceeded 10, indicative of minimal bias from weak IVs, thereby ensuring the robustness of the results (Supplementary Table 1).

Plot of MR analysis results. (A) Forest plot of the MR analysis results in European ancestry. (B) Exposure represents the 4 inflammatory cytokines, where id.exposure represents the GWAS ID of the inflammatory cytokines in the IEU OpenGWAS Project website. nsnp represents the number of SNPs, and pval represents the P value. (C) Bubble chart of the MR analysis results in East Asian ancestry. The abscissa represents β value, and the ordinate represents the P value, where red represents β value > 0, and blue represents β value < 0. The bigger the bubble, the darker the color, which means the greater the P value.
MR analysis
Among individuals of European ancestry, the IVW analysis unveiled a significant positive causal association between the gene expression of TNFSF14 (OR = 1.249, 95%CI 1.018–1.532, P = 0.033) and TRANCE (OR = 1.287, 95%CI 1.051–1.577, P = 0.015) with DN. Conversely, the gene expression of IL-1α (OR = 0.712, 95%CI 0.514–0.984, P = 0.040) and TGF-α (OR = 0.701, 95%CI 0.493–0.998, P = 0.049) elucidated a significant negative causal association with DN (Fig. 4A, B).


MR estimates of the causal effects on DN. (A) Forest plot of MR estimates of the causal effects of 4 inflammatory cytokines on DN in European ancestry. The green diamond, red square, grey circle, and purple triangle symbols denote the WME method, MR-Egger method, IVW method, and WM method, respectively. (B) Circular graph of estimates of the causal effects of 4 inflammatory cytokines on DN in European ancestry. The redder the color, the greater the P value. (C) Forest plot of MR estimates of the causal effects of 1 inflammatory cytokines on DN in East Asian ancestry. (D) The green diamond, red square, grey circle, and purple triangle symbols denote the WME method, MR-Egger method, IVW method, and WM method, respectively. (E) Circular graph of estimates of the causal effects of 1 inflammatory cytokines on DN in East Asian ancestry. The redder the color, the greater the P value.
Among individuals of East Asian ancestry, the gene expression of GDNF (OR = 0.391, 95%CI 0.163–0.931, P = 0.034) elucidated a significant negative causal association with DN (Fig. 4C, D). The forest plot depicting single SNP MR results is provided in Supplementary Fig. 1. Although the results of the WME, WM, and MR-Egger regression analyses did not attain statistical significance, their directional consistency with the IVW analysis was evident (Fig. 5A, E, Supplementary Table 2). Under the premise that all SNPs function as effective IVs, devoid of horizontal pleiotropy, and the outcomes derived from the IVW analysis remained unbiased, the efficacy of the IVW analysis in furnishing dependable effect estimates surpasses that of alternative methodologies27, it concluded that inflammatory cytokines exhibit a causal association with DN.

Scatter plots of SNP analysis. (A) Gene expression of IL-1α; (B) Gene expression of TGF-α; (C) Gene expression of TNFSF14; (D) Gene expression of TRANCE; (E) Gene expression of GDNF; The X-axis denotes the impact of the SNP on the inflammatory cytokine, while the Y-axis represents the SNP’s influence on DN. Each black dot signifies a single SNP, with the line segment depicting the 95%CI. The slope of the straight line reflects the causal estimation derived from the MR method. In this visualization, the light blue line corresponds to the IVW method, the blue line represents the MR Egger method, the reseda line signifies the WME method and the dark green line represents the WM method.
However, after FDR correction, it was found that only the gene expression of IL-1α had a negative causal association with DN (q value = 0.05). The other inflammatory cytokines were considered to be suggestive of DN.
Sensitivity analysis
In both individuals of European ancestry and East Asian ancestry, Cochran’s Q test revealed no evidence of heterogeneity among the included IVs (P > 0.05). Moreover, the intercept test of MR-Egger regression indicated that pleiotropy did not introduce bias to the results (P > 0.05) (Supplementary Table 3). Funnel plots visually depicted that potential confounders were unlikely to affect causality (Fig. 6A–E). LOO sensitivity analysis demonstrated that the remaining SNPs did not significantly alter the analysis results upon individual SNP removal (Fig. 7A–E).

Funnel plots of sensitivity analysis. (A) Gene expression of IL-1α; (B) Gene expression of TGF-α; (C) Gene expression of TNFSF14; (D) Gene expression of TRANCE; (E) Gene expression of GDNF; The light blue represents the IVW method, and the dark blue represents the MR Egger method.


Forest plots of LOO analysis. (A) Gene expression of IL-1α; (B) Gene expression of TGF-α; (C) Gene expression of TNFSF14; (D) Gene expression of TRANCE; (E) Gene expression of GDNF; In this representation, each black dot symbolizes the DN with increased standard deviation (SD) in the inflammatory cytokine, generated by utilizing each SNP as an individual IV. Conversely, the red dot denotes the causal estimation derived from all SNP combinations using various MR methods. The horizontal line segment represents the 95%CI. Specifically, the IVW causal estimate illustrates how the overall estimate (depicted by the red horizontal line) might be disproportionately influenced by the removal of a single variant (indicated by the black horizontal line).
Bidirectional MR analysis
Among individuals of European ancestry, the reverse MR analysis revealed a negative causal association between the gene expression of Axin-1, C–C motif chemokine 28 (CCL28), Leukemia inhibitory factor receptor (LIFR), TNF-related apoptosis-inducing ligand (TRAIL), TNF-beta (TNF-β), and Vascular endothelial growth factor A (VEGFA) with DN. Among individuals of East Asian ancestry, the reverse MR analysis unveiled a negative causal association between the gene expression of Interleukin-10 (IL-10), and Programmed cell death 1 ligand 1 (PD-L1) with DN (Supplementary Fig. 2A–F).
Conversely, no causal association was observed between DN and the inflammatory cytokines identified as having a positive association (TNFSF14, TRANCE, IL-1α, TGF-α, and GDNF) (Fig. 8A–E).

Forest plot of the bidirectional MR analysis of the positively identified inflammatory cytokines. (A) Gene expression of IL-1α; (B) Gene expression of TGF-α; (C) Gene expression of TNFSF14; (D) Gene expression of TRANCE; (E) Gene expression of GDNF; Outcome represents the inflammatory cytokines, OR represents the odds ratio, CI represents the confidence interval, and pval represents the P value.
Discussion
In this study, we conducted a meticulous analysis using large-scale GWAS data to explore the causal association between inflammatory cytokines and DN. In order to satisfy the three hypotheses. Firstly, only SNPs meeting the criterion of P < 5e-06, R2 < 0.001, and kb = 10,000 were considered. Secondly, the PhenoScanner database was utilized to exclude potential confounding factors. Lastly, SNPs with F > 10 were finally included in the study. The IVW analysis revealed that the gene expression of TNFSF14 and TRANCE were associated with a higher risk of DN, while the gene expression of IL-1α, TGF-α, and GDNF were associated with a lower risk of DN. Subsequent sensitivity analyses confirmed the robustness and consistency of these findings. Additionally, reverse MR analysis showed no discernible causal association between DN and the inflammatory cytokines identified as having a positive association.
TNFSF14, predominantly expressed in activated T lymphocytes and other immune cells28, comprises 254 amino acids and functions as both a membrane-bound and soluble molecule. Its key roles include tumor immunity, inflammatory responses, autoimmune disease pathogenesis, and thymic negative selection29. These functions are primarily mediated through interactions with LTβR (TNFRSF3) and HVEM (TNFRSF14). Studies using TNFSF14-deficient mice (TNFSF14 KO mice) have highlighted its importance in regulating immune responses and inflammatory processes, despite the mice having normal peripheral lymphoid organs30. Notably, TNFSF14 is implicated in promoting renal fibrosis31. TNF-related activation-induced cytokines facilitate ICAM-1 and VCAM-1 induction via protein kinase C-dependent NF-kB activation in endothelial cells32.
IL-1 plays a crucial role in immune regulation and the orchestration of inflammatory responses. Elevated serum levels and gene expression of IL-1 have been consistently observed in DN patients. This aligns with studies showing an association between IL-1 gene polymorphisms and the risk of end-stage DN33. Functionally, IL-1 upregulates ICAM-1, VCAM-1, and E-cadherin, promoting leukocyte adhesion to endothelial cells, mononuclear macrophage infiltration into the glomerulus, and accelerating glomerulosclerosis34. In DN patients, renal tubular cells show increased expression of IL-1α, with hyperglycemia triggering its local production, thereby exacerbating renal fibrosis35.
TGF-α, a prominent member of the epidermal growth factor family, acts as a specific ligand for the epidermal growth factor receptor (EGFR). Its precursor, translated by mRNA, consists of 160 amino acids. This precursor undergoes excision and glycosylation by various proteolytic enzymes, producing a mature TGF-α of 50 amino acids, which is then released into the cell36. Clinical studies have shown that blocking EGFR with TGF-α monoclonal antibodies significantly inhibits the onset and progression of DN37. TGF-α, as a secreted protein, is implicated in the pathogenesis of numerous diseases. In cell models exposed to high glucose levels, circFTO has been shown to upregulate TGF-α gene expression, thus modulating the injury of ARPE-19 cells induced by high glucose38.
TRANCE, a novel member of the TNF family, is predominantly expressed in T cells. It acts as a dendritic cell (DC)-specific survival factor and activates JNK when it interacts with its receptor. TRANCE significantly enhances DC function by increasing the survival rate of DCs in vivo39. TNF-α, a transmembrane protein made up of 233 amino acids, is produced by mononuclear macrophages, glomerular mesangial cells, and renal tubular epithelial cells. With a molecular weight of 26 kDa, TNF-α is a primary regulator of immune and inflammatory responses. Experimental evidence indicates that TNF-α can induce the synthesis of inflammatory mediators such as prostaglandin, leukotriene, and IL-1 in cultured human mesangial cells. These inflammatory mediators are implicated in the pathogenesis of DN40.
GDNF, a glycosylated disulfide-bonded homodimeric protein, is derived from the serum-free medium of the rat glial cell line B49. It is widely expressed throughout the body, with varying levels in tissues at different developmental stages and after different degrees of nerve injury. Initially identified as a neurotrophic factor for midbrain dopaminergic neurons, GDNF has potential therapeutic applications in Parkinson’s disease41,42. Subsequent studies have highlighted its neuroprotective and significant analgesic properties43. GDNF and its receptor are also expressed in the pancreas, where they promote pancreatic cell proliferation, reduce apoptosis, enhance cell function, and improve glucose tolerance44. Diabetic mice show decreased GDNF concentrations; however, local administration of GDNF or overexpression of the GDNF gene in mice has been shown to reduce apoptosis, enhance β-cell quality and proliferation, promote insulin secretion, and improve local tissue function. Despite these benefits, the efficacy of GDNF in controlling blood sugar levels remains debated45.
In summary, TNFSF14 and TRANCE are hypothesized to contribute to the onset and progression of DN through abnormal infiltration into renal tissues. Monitoring inflammatory cytokine levels in patients’ serum could help assess their condition and provide a basis for the clinical diagnosis and treatment of DN.
The study possesses several strengths. Firstly, its substantial sample size helps mitigate the influence of confounding factors on the results. Secondly, employing MR analysis allows for a robust estimation of the causal association between exposures and disease, thereby circumventing the issue of reverse causality inherent in traditional observational studies. Thirdly, it represents the first attempt to uncover the association between inflammatory cytokines and DN at the genetic level. However, certain limitations need to be acknowledged. Firstly, the outcome data originate from European and East Asian populations, limiting the generalizability of the findings, the generalizability of our findings to other populations should be validated using local data. Secondly, the lack of detailed information, such as age and gender, hinders the possibility of conducting further subgroup analyses. Therefore, they must be validated in future large-sample clinical trials. Thirdly, while MR facilitates the evaluation of the enduring impacts of genetically predisposed inflammatory cytokines throughout an individual’s lifespan, it may not directly encapsulate the attenuation of these factors in adulthood due to the influence of diverse unreported regulators.
Conclusion
This study provides an initial insight into the genetic aspect of the causal association between inflammatory cytokines and DN. It revealed that the gene expression of TNFSF14 and TRANCE exhibit a positive causal association with DN, whereas the gene expression of IL-1α, TGF-α, and GDNF show a negative causal association with DN. These findings imply that targeting and modulating specific inflammatory factors could potentially serve as an effective strategy for the future treatment and prevention of DN.
Abbreviations
- CCL28:
-
C–C motif chemokine 28
- CI:
-
Confidence interval
- DN:
-
Diabetic nephropathy
- IL-1α:
-
Interleukin-1-alpha
- IL-10:
-
Interleukin-10
- IVW:
-
Inverse variance weighted
- IVs:
-
Instrumental variables
- GDNF:
-
Glial cell line-derived neurotrophic factor
- GWAS:
-
Genome-wide association study
- LIFR:
-
Leukemia inhibitory factor receptor
- LOO:
-
Leave-one-out
- MR:
-
Mendelian randomization
- OR:
-
Odds ratio
- PD-L1:
-
Programmed cell death 1 ligand 1
- SNPs:
-
Single nucleotide polymorphisms
- TGF-α:
-
Transforming growth factor-alpha
- TNF-β:
-
TNF-beta
- TNFSF14:
-
Tumor necrosis factor ligand superfamily member 14
- TRANCE:
-
TNF-related activation-induced cytokine
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- VEGFA:
-
Vascular endothelial growth factor A
- WM:
-
Weighted mode
- WME:
-
Weighted median
References
Zhang, S. et al. The role of the microbiome in diabetes mellitus. Diabetes Res. Clin. Pract. 172, 108645 (2021).
Tu, Y. et al. Assessment of type 2 diabetes risk conferred by SNPs rs2241766 and rs1501299 in the ADIPOQ gene, a case/control study combined with meta-analyses. Mol. Cell Endocrinol. 396(1–2), 1–9 (2014).
Ni, W. J., Tang, L. Q. & Wei, W. Research progress in signalling pathway in diabetic nephropathy. Diabetes Metab. Res. Rev. 31(3), 221–233 (2015).
Gurley, S. B. et al. Inflammation and immunity pathways regulate genetic susceptibility to diabetic nephropathy. Diabetes 67(10), 2096–2106 (2018).
Tu, Y., Fan, G., Wu, N., Wu, H. & Xiao, H. Association of plasma lead, cadmium and selenium levels with hearing loss in adults: National Health and Nutrition Examination Survey (NHANES) 2011–2012. Br. J. Nutr. 128(6), 1100–1107 (2022).
Alicic, R. Z., Rooney, M. T. & Tuttle, K. R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 12(12), 2032–2045 (2017).
Zhang, F. et al. IL-17C neutralization protects the kidney against acute injury and chronic injury. EBioMedicine 92, 104607 (2023).
Murakoshi, M., Gohda, T. & Suzuki, Y. Circulating tumor necrosis factor receptors: A potential biomarker for the progression of diabetic kidney disease. Int. J. Mol. Sci. 21(6), 1957 (2020).
Nakamura, A. et al. Combination therapy with an angiotensin-converting-enzyme inhibitor and an angiotensin II receptor antagonist ameliorates microinflammation and oxidative stress in patients with diabetic nephropathy. J. Diabetes Investig. 4(2), 195–201 (2013).
Huang, G. et al. Notoginsenoside R1 ameliorates podocyte injury in rats with diabetic nephropathy by activating the PI3K/Akt signaling pathway. Int. J. Mol. Med. 38(4), 1179–1189 (2016).
Tu, Y. et al. Paclitaxel-loaded ROS-responsive nanoparticles for head and neck cancer therapy. Drug Deliv. 30(1), 2189106 (2023).
Tu, Y., Chen, C. & Fan, G. Association between the expression of secreted phosphoprotein - related genes and prognosis of human cancer. BMC Cancer 19(1), 1230 (2019).
Lawlor, D. A., Harbord, R. M., Sterne, J. A., Timpson, N. & Davey Smith, G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27(8), 1133–1163 (2008).
Chang, L., Zhou, G. & Xia, J. mGWAS-Explorer 2.0: Causal analysis and interpretation of metabolite-phenotype associations. Metabolites 13(7), 826 (2023).
Zhao, J. H. et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat. Immunol. 24(9), 1540–1551 (2023).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53(10), 1415–1424 (2021).
Xiang, M. et al. Exploring causal correlations between inflammatory cytokines and systemic lupus erythematosus: A Mendelian randomization. Front. Immunol. 13, 985729 (2022).
Liu, B. et al. Associations of the circulating levels of cytokines with risk of amyotrophic lateral sclerosis: A Mendelian randomization study. BMC Med. 21(1), 39 (2023).
Zou, M., Zhang, W., Shen, L., Xu, Y. & Zhu, Y. Causal association between inflammatory bowel disease and herpes virus infections: a two-sample bidirectional Mendelian randomization study. Front. Immunol. 14, 1203707 (2023).
Burgess, S. & Thompson, S. G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 32(5), 377–389 (2017).
Bowden, J. et al. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: The role of the I2 statistic. Int. J. Epidemiol. 45(6), 1961–1974 (2016).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U S A 100(16), 9440–9445 (2003).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44(2), 512–525 (2015).
Hayreh, S. S. Ocular vascular occlusive disorders: Natural history of visual outcome. Prog. Retin. Eye Res. 41, 1–25 (2014).
Pierce, B. L. & Burgess, S. Efficient design for Mendelian randomization studies: Subsample and 2-sample instrumental variable estimators. Am. J. Epidemiol. 178(7), 1177–1184 (2013).
Li, L. et al. Causal associations between gastroesophageal reflux disease and lung cancer risk: A Mendelian randomization study. Cancer Med. 12(6), 7552–7559 (2023).
Power, G. M. et al. Methodological approaches, challenges, and opportunities in the application of Mendelian randomisation to lifecourse epidemiology: A systematic literature review. Eur. J. Epidemiol. 32(2), 489 (2023).
Mauri, D. N. et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 8(1), 21–30 (1998).
Jungbeck, M. et al. Neutralization of LIGHT ameliorates acute dextran sodium sulphate-induced intestinal inflammation. Immunology 128(3), 451–458 (2009).
Tamada, K. et al. Cutting edge: selective impairment of CD8+ T cell function in mice lacking the TNF superfamily member LIGHT. J Immunol 168(10), 4832–4835 (2002).
Li, Y. et al. Tumor necrosis factor superfamily 14 is critical for the development of renal fibrosis. Aging (Albany NY) 12(24), 25469–25486 (2020).
Min, J. K. et al. TNF-related activation-induced cytokine enhances leukocyte adhesiveness: Induction of ICAM-1 and VCAM-1 via TNF receptor-associated factor and protein kinase C-dependent NF-kappaB activation in endothelial cells. J Immunol 175(1), 531–540 (2005).
Lee, S. H. et al. Polymorphisms in interleukin-1 beta and Interleukin-1 receptor antagonist genes are associated with kidney failure in Korean patients with type 2 diabetes mellitus. Am. J. Nephrol. 24(4), 410–414 (2004).
Yaribeygi, H., Atkin, S. L. & Sahebkar, A. Interleukin-18 and diabetic nephropathy: A review. J. Cell. Physiol. 234(5), 5674–5682 (2019).
Salti, T. et al. Glucose induces IL-1alpha-dependent inflammation and extracellular matrix proteins expression and deposition in renal tubular epithelial cells in diabetic kidney disease. Front. Immunol. 11, 1270 (2020).
Ohyama, T. et al. Transforming growth factor-alpha attenuates hepatic fibrosis: Possible involvement of matrix metalloproteinase-1. Liver Int. 31(4), 572–584 (2011).
Habib, H. A., Heeba, G. H. & Khalifa, M. M. A. Comparative effects of incretin-based therapy on early-onset diabetic nephropathy in rats: Role of TNF-alpha, TGF-beta and c-caspase-3. Life Sci. 278, 119624 (2021).
Huang, Y. et al. circFTO upregulates transforming growth factor-alpha through sponging miR-148a-3p to regulate high glucose-induced ARPE-19 cells injury. Bioengineered 13(5), 11489–11502 (2022).
Wong, B. R. et al. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J. Exp. Med. 186(12), 2075–2080 (1997).
Yang, H. et al. Tim-3 aggravates podocyte injury in diabetic nephropathy by promoting macrophage activation via the NF-kappaB/TNF-alpha pathway. Mol. Metab. 23, 24–36 (2019).
Lin, L. F., Doherty, D. H., Lile, J. D., Bektesh, S. & Collins, F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260(5111), 1130–1132 (1993).
Love, S. et al. Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat. Med. 11(7), 703–704 (2005).
Sah, D. W., Ossipov, M. H., Rossomando, A., Silvian, L. & Porreca, F. New approaches for the treatment of pain: the GDNF family of neurotrophic growth factors. Curr. Top. Med. Chem. 5(6), 577–583 (2005).
Mwangi, S. et al. Glial cell line-derived neurotrophic factor increases beta-cell mass and improves glucose tolerance. Gastroenterology 134(3), 727–737 (2008).
Zhang, L. et al. Repeated systemic administration of human adipose-derived stem cells attenuates overt diabetic nephropathy in rats. Stem Cells Dev. 22(23), 3074–3086 (2013).
Acknowledgements
We thank all contributors to the IEU OpenGWAS Project.
Funding
This study was supported by grants from the National Natural Science Foundation of China (Grant No. 82174293 and 82374355), Science and Technology Support Program of Jiangsu Province (ZD202208), Postgraduate Research and Practice Innovation Program of Jiangsu Province (SJCX24_0967).
Author information
Authors and Affiliations
Contributions
The research was conceptualized by Jiangyi Yu and Qianhua Yan. Siyuan Song conducted the data analysis and contributed to the writing of the paper. All authors reviewed and approved the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
The datasets generated and analysed during the current study are available in the IEU OpenGWAS Project repository, [PERSISTENT WEB LINK is https://gwas.mrcieu.ac.uk/], accession number is ebi-a-GCST90018832 and ebi-a-GCST90018612.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, S., Yan, Q. & Yu, J. Bidirectional two-sample Mendelian randomization analysis unveils causal association between inflammatory cytokines and the risk of diabetic nephropathy. Sci Rep 14, 25425 (2024). https://doi.org/10.1038/s41598-024-73800-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-73800-2
