
Abstract
The bacterial group of the phylum Bacteroidota greatly contributes to the global carbon cycle in marine ecosystems through its specialized ability to degrade marine polysaccharides. In this study, it is proposed that two novel facultative anaerobic strains, DS1-an-13321T and DS1-an-2312T, which were isolated from a sea squirt, represent a novel genus, Halosquirtibacter, with two novel species in the family Prolixibacteraceae. The 16S rRNA sequence similarities of these two strains were 91.26% and 91.37%, respectively, against Puteibacter caeruleilacunae JC036T, which is the closest recognized neighbor. The complete genomes of strains DS1-an-13321T and DS1-an-2312T each consisted of a single circular chromosome with a size of 4.47 and 5.19 Mb, respectively. The average amino acid identity and the percentage of conserved proteins against the type species of the genera in the family Prolixibacteraceae ranged from 48.33 to 52.35% and 28.34–37.37%, respectively, which are lower than the threshold for genus demarcation. Strains DS1-an-13321T and DS1-an-2312T could grow on galactose, glucose, maltose, lactose, sucrose, laminarin, and starch, and only DS1-an-2312T could grow on xylose and xylan under fermentation conditions. These strains produced acetic acid and propionic acid as the major fermentation products. Genome mining of the genomes of the two strains revealed 27 and 34 polysaccharide utilization loci, which included 155 and 249 carbohydrate-active enzymes (CAZymes), covering 57 and 65 CAZymes families, respectively. The laminarin-degrading enzymes in both strains were cell-associated, and showed exo-hydrolytic activity releasing glucose as a major product. The xylan-degrading enzymes of strain DS1-an-2312T was also cell-associated, and had endo-hydrolytic activities, releasing xylotriose and xylotetraose as major products. The evidence from phenotypic, biochemical, chemotaxonomic, and genomic characteristics supported the proposal of a novel genus with two novel species in the family Prolixibacteraceae, for which the names Halosquirtibacter laminarini gen. nov., sp. nov. and Halosquirtibacter xylanolyticus sp. nov. are proposed. The type strain of Halosquirtibacter laminarini is DS1-an-13321T (= KCTC 25031T = DSM 115329T) and the type strain of Halosquirtibacter xylanolyticus is DS1-an-2312T (= KCTC 25032T = DSM 115328T).
Similar content being viewed by others


Introduction
Polysaccharides constitute a significant proportion of the carbon inventory in marine organic matter and form a major structural component of both macro- and micro-algae1. Roughly half of this organic matter is released to the marine environment as dissolved and particulate organic matter (DOM, and POM)2. This is further degraded by the activities of marine heterotrophic bacteria, which contribute to carbon cycling in marine ecosystems by producing smaller molecular weight compounds and minerals1,3. Among marine heterotrophic bacteria, Bacteroidota is one of the most abundant groups, after Cyanobacteria and Proteobacteria4. These bacteria engage in different strategies to contribute to regulate the carbon cycle. Indeed, Cyanobacteria are known as a primary producer because of their photoautotrophic ability in marine ecosystems4,5, whereas abundant members of marine Alphaproteobacteria are known to preferentially utilize simple sugars and are found in the water column4,6. In contrast, Bacteroidota are known as polysaccharide degraders7.
Many studies have revealed the ability of Bacteroidota to degrade marine polysaccharides from dissolved or particulate organic matters, and the cell wall component of macro- and micro-algal cells4,7. This highlights the crucial role of this process in sequestering carbon within the marine environment, thereby significantly contributing to the carbon cycle in marine ecosystems4,8. To access this rich source of polysaccharides, Bacteroidota have evolved a unique apparatus for their polysaccharide degradation strategy9. This apparatus was first reported in Bacteroides thetaiotaomicron in the human gut microbiota9,10. It consists of SusD, an outer membrane-bound protein responsible for capturing polysaccharides9, and outer membrane-bound carbohydrate-active enzymes (CAZymes) that partly degrade target polysaccharides into oligosaccharides9,11. Subsequently, these oligosaccharides are imported into the cell periplasm via a membrane-bound SusC transporter9. Other CAZymes then further degrade these oligosaccharides to monosaccharides, which are transported into the cytoplasm through dedicated transporters7,9. This entire process is regulated by a regulator that senses the products of polysaccharide degradation. These SusC, SusD, CAZymes, transporters, and regulators are encoded by genes that are clustered together in a region of the genome and are known as polysaccharide utilization loci (PUL)7.
Bacteroidota was first described as a novel phylum by Krieg et al. in 2010, in the second edition of Bergey’s Manual of Systematic Bacteriology12. The name Bacteroidota was revised (from Bacteroidetes) by Oren and Garrity in 202113. At the time of writing, the phylum Bacteroidota consists of six classes (https://lpsn.dsmz.de/phylum/bacteroidota): Bacteroidia Krieg 2012, Cytophagia Nakagawa 2012, Flavobacteriia Bernardet 2012, Sphingobacteriia Kampfer 2012, Chitinophagia Munoz et al. 2017, and Saprospiria Hahnke et al. 2018. Among these, three classes, Bacteroidia, Cytophagia, and Flavobacteriia, are the largest, and a huge number of studies on the polysaccharide degradation capability of the member bacteria have been reported. To date, members of the class Flavobacteriia are found to be abundant in the macroalgal phycosphere microbiome and in microalgal blooms and are well documented for macro- and microalgal polysaccharide degradation14,15. Several studies have meanwhile demonstrated the ability of members of the class Cytophagia to degrade polysaccharides16,17 and predicted potential polysaccharide degradation18, although there are fewer available literature sources of marine Cytophagia bacteria degrading polysaccharides19. On the other hand, members of the class Bacteroidia are mostly anaerobes and have been well studied for polysaccharide degradation in the mammalian gut microbiota20. Several studies on human gut Bacteroidia bacteria degrading marine polysaccharide under anaerobic conditions also have been reported21. However, few studies on marine Bacteroidia that anaerobically degrade polysaccharides can be found in the literature22.
Xylan is the most abundant form of hemicellulose found in nature and is produced in marine and terrestrial environments23. Xylan from terrestrial plants contains a β-1,4-linkage D-xylopyranosyl backbone and units of acetate, ɑ-L-arabinofuranose, 4-O-methyl-glucuronic acid, or ferulic acid23 whereas xylan from marine sources is homoxylan, which is found only in algae24. The backbone of marine xylan mainly consists of a 1,3-glycoside bond and a mixture of 1,3 and 1,4 glycoside bonds24. The molecular structure of xylan protects the cell walls of plants and algae from abiotic or biotic stresses, and it is very difficult to degrade xylan biologically. Biodegradation of xylan results in the production of xylose and xylooligosaccharides, which have potential roles in biotechnological applications such as biofuel, biomedicine, and food supplements25,26,27,28. Initially, xylan is degraded randomly by endoxylanases GH5, GH10, GH11, or GH3029 to release unbranched xylooligosaccharides, which are then degraded into xylose by β-xylosidase GH39, or GH4330. Several studies have focused on the aerobic degradation of xylan by marine bacteria24,31,32. However, to our knowledge, research on the anaerobic degradation of xylan by marine Bacteroidia bacteria has been limited.
Laminarin serves as the principal energy reserve glycan identified in brown algae33 and some microalgae34, with an estimated annual production of approximately 12–18 gigatons globally35. It is constructed by combining β-(1→3)-linked glucose-based linear chains and a lower ratio of β-(1→6)-linked side chains from glucose moieties35. Laminarin degradation results in the production of glucose and laminarin oligosaccharides, which can potentially be used in biotechnological applications in the biofuel, cosmetics, biomedical, and food industries36. To biologically degrade laminarin, endo-acting β-1,3-glucanases (glycoside hydrolase family 17, GH17) specifically break down the β-(1→3)-linkage in the main chain, while exo-acting β-1,6-glucosidases (GH30) specifically hydrolyze the β-(1→6)-linkage at the side chain of laminarin. Additionally, exo-acting β-1,3-glucosidases (GH3) specifically hydrolyze the β-(1→3)-linkage of oligosaccharides into glucoses35. The ability of laminarin degradation is widely distributed in diverse marine heterotrophic bacteria. However, there has been limited research on the anaerobic degradation of laminarin by marine Bacteroidia bacteria.
The family Prolixibacteraceae of the order Bacteroidales, class Bacteroidia, phylum Bacteroidota was first proposed by Huang et al. 2014. At the time of writing, as described on the List of Prokaryotic Names with Standing in Nomenclature (LPSN, https://www.bacterio.net/), the family accommodates 12 valid genera, including Aquipluma Watanabe et al. 2020, Draconibacterium Du et al. 2014, Gaoshiqia Yu et al. 2023, Mangrovibacterium Huang et al. 2014, Maribellus Zhou et al. 2019, Mariniphaga Iino et al. 2014, Meniscus Irgens 1977, Prolixibacter Holmes et al. 2007, Puteibacter Sun et al. 2020, Roseimarinus Wu et al. 2015, Sunxiuqinia Qu et al. 2011, and Tangfeifania Liu et al. 2014. They have been isolated from various habitats, particularly hypolimnion water37, river sediment38, mangrove sediment39, marine sediment40, seawater22, and crude oil41. Most of the members of the family Prolixibacteraceae are Gram-staining negative, rod-shaped, non-spore-producing bacteria. They have no motility and their oxygen requirement ranges from aerobic to facultative anaerobic conditions. The predominant quinone component is menaquinone 7 (MK-7)42. Several isolation methods have been applied to isolate members of the family Prolixibacteraceae, including enrichment22, a dilution technique38, and nitrogen-free medium cultivation39. In this study, we used another strategy to isolate novel bacteria by using a low nutrient isolation medium, which was prepared from 60% strength seawater (at the collection site) with 1.5% agar (BD) and a piece of filter paper placed on the surface of the agar as the sample carrier19. We isolated two novel bacteria from a sea squirt under anaerobic conditions.
In this study, we identified and characterized two novel anaerobic isolates, DS1-an-13321T (strictly anaerobic) and DS1-an-2312T (facultative anaerobic), through genetic, morphological, biochemical, and chemotaxonomic analyses. These isolates were proposed as novel genus with two novel species within the family Prolixibacteraceae. Genome mining revealed that these isolates contain hundreds of CAZyme genes spanning 65 CAZyme families. Their glycoside hydrolases were the most abundant among CAZymes, and exhibited up to double frequency of GHs per genome compared to the average value in marine Bacteroidota. Additionally, DS1-an-13321T was found to utilize laminarin as a sole carbon source under anaerobic conditions, while DS1-an-2312T could utilize both laminarin and xylan. The final hydrolytic product of laminarin degradation was glucose, whereas xylan degradation primarily yielded xylotriose and xylotetraose. This study is the first to combine genome mining with in vitro experiments to analyze xylan and laminarin degradation in anaerobic marine Bacteroidia, and the results enhance our understanding of the polysaccharide degradation strategies of anaerobic marine Bacteroidia and their role in marine carbon cycling.
Materials and methods
Ecology, isolation, and cultivation of isolates
The source of isolation was a sea squirt collected by a fisherman at a depth of 18 m beneath the surface of seawater in the East Sea, Republic of Korea (38º38’48.5” N, 129º44’29,2” E). The sample was then stored in an ice-pack container and delivered to the laboratory on the same day. For isolation, a low nutrient solid medium prepared by 60% strength seawater (collected from the sampling site) solidified with 1.5% agar (BD) was used. After autoclaving the medium, 50 mg/L cycloheximide (Aldrich Sigma) was added to the agar medium19. All the steps for bacterial isolation were performed in an anaerobic chamber (Coy Lab Products, USA; N2: H2: CO2 = 94.5%:1.5%:5%). The plates were stored in an anaerobic chamber overnight to remove oxygen. A slice taken from the mouth of the squirt was placed on the top of a piece of filter paper (Whatman No.2) on the isolation plates. The plates were placed in an anaerobic jar with a bag of BD GasPak EZ anaerobe container system (BD) and incubated at 23 ℃. After 4–5 days of incubation, the colony surrounding the filter paper was transferred and streaked onto fresh marine agar plates (MA, BD marine agar 2216) under anaerobic conditions. After several transfers, single colonies of the two strains were obtained. The pure cultures of the two strains were preserved in 20% glycerol at -80 ℃, and lyophilized in ampoules stored at 4 ℃.
Phylogenetic analysis based on the 16S rRNA gene sequence
For a phylogenetic analysis, the 16S rRNA gene sequences of the two isolates were determined. The 16S rRNA sequences of the two strains were sequenced using four universal primers: 27F (5’-AGAGTTTGATCCTGGCTCAG-3’), 518F (5’-CCAGCAGCCGCGGTAATAC-3’), 805R (5’-GACTACCAGGGTATCTAATC-3’), and 1492R (5’-TACGGYTACCTTGTTACGACTT-3’)19, via Sanger sequencing. The complete sequences were manually assembled using Vector NTI software (Invitrogen). Next, pairwise sequence alignment was performed on EzBioCloud (https://www.ezbiocloud.net/). The related sequences downloaded from EzBioCloud were aligned using ClustalW in BioEdit software (version 7.2.5). The trimmed sequences were then used to make phylogenetic trees in MEGA11 using three algorithms: neighbor-joining (NJ), maximum-likelihood (ML), and maximum-parsimony (MP)19. The estimated matrix on MEGA11 based on the alignment fasta file showed that the optimal model for the MP tree was the general time reversible (GTR) model, and the rates and patterns were Gamma distributed with Invariant sites (G + I). Moreover, the Kimura two-parameter model was used for the NJ algorithm, and tree-bisection-reconnection (TBR) was used for the ML algorithm. The bootstrap resampling method with 1000 replicates was used to evaluate the phylogenetic tree. The sequence of strain Agarivorans albus (accession number AB681878) was obtained from NCBI as the outgroup, and the sequence of Marinilabilia salmonicolor NCIMB 2216T (accession number D12672) was retrieved from NCBI as the type strain of the closest family, Marinilabiliaceae.
Pairwise alignment of 16S rRNA sequences revealed that the strains DS1-an-2312T and DS1-an-13321T had the highest similarity to Puteibacter caeruleilacunae JC036T, with similarity values of 91.57% and 91.48%, respectively. Therefore, the isolated strains were considered to belong to a novel genus in the family Prolixibacteraceae or to a novel family in the order Bacteroidales. To delineate the two strains in terms of phylogeny, the type species of each genus in the family Prolixibacteraceae and the type strains of the second closest family, Marinilabiliaceae, were included in a phylogenetic tree. Based on the phylogenetic position of the two isolates on the phylogenetic tree and their top hit similarity, three strains, Puteibacter caeruleilacunae KCTC 25263T, Prolixibacter bellariivorans KCTC 25261T, and Sunxiuqinia elliptica KCTC 32215T, were selected as the reference strains.
Physiological characterization
The morphology of colonies of the two novel isolates was observed on MA plates after three days of cultivation under anaerobic conditions. Gram staining was performed according to the standard protocol, and the prepared slices were observed under a light microscope (Nikon Eclipse 80i). Scanning electron microscopy (SEM, JEOL JSM-7600F) was used to observe the cell morphology19. To determine their temperature range, the two strains were cultured in marine broth (MB, BD) in test tubes purged with nitrogen gas and then incubated over a temperature range from 10 to 30 ℃ for three days and one week at 4 ℃ and 35 ℃. To determine the pH range, MB in test tubes purged with nitrogen gas was used to determine the growth of the two novel isolates using the following buffer systems at 50 mM: 5.5–6.0 (MES), 6.5–7.0 (PIPES), 7.5–8.0 (HEPES), 8.5 (Bis-Tris), and 9.0–10.0 (CHES)43. In a NaCl tolerance experiment, Zobell broth43 in artificial seawater44 was supplemented with NaCl concentrations of 0, 0.5, and 1–10% (with intervals of 1.0%, w/v). To determine the oxygen requirements, the two isolates were cultivated on MA plates in anaerobic jars under anaerobic conditions provided by a bag of BD GasPak EZ anaerobe container system (BD) and under microaerophilic conditions provided by a bag of BD GasPak EZ CO2 container system. The plates were incubated for seven days at 28 ℃.
The biochemical characteristics of the two novel isolates and their reference strains were determined on MA plates as the basal media at 28 ℃ for three days under anaerobic conditions unless otherwise specified. The hydrolysis of starch was tested by supplying 0.2% starch to MA plates and detecting a clear zone after staining with iodine solution. The hydrolysis of cellulose was assessed on carboxymethyl cellulose agar plates19 using artificial seawater44 with 2% NaCl instead of distilled water, and a clear zone was detected after embedding in Congo Red and washing with 1% (w/v) NaCl solution. The hydrolysis of Tween 20 and Tween 80 was performed by adding 0.1% (v/v) Tweens 20 and 80 to MA plates, respectively. Catalase activity was determined by dropping a 3% H2O2 solution onto the surface of cells19. Oxidase activity was tested by reacting cells with an oxidase reagent (bioMerieux). All five strains were cultured on DNase agar (Difco) using artificial seawater with 2% NaCl instead of distilled water to test DNase activity. Chitin (1%, w/v) was added to MA plates, and chitin hydrolysis activity was detected by the appearance of a clear zone19. Gelatinase activity was evaluated on nutrient gelatin (Remel Gelatin medium) in which distilled water was replaced with artificial seawater44 supplemented with 2% (w/v) NaCl for one week at 25 ℃, and a positive result was recognized by liquidation of the medium45.
To determine anaerobic metabolism, the growth of the two novel isolates on a variety of carbon sources was assessed. An inoculum of the strains was prepared in 5 mL of MB supplemented with 20 mM HEPES (Sigma) and adjusted to pH 7.0 using 2 N NaOH. The strains were cultivated on basal media (BS) supplemented with carbon substrates (0.2%, w/v). The composition of the BS was as follows: 23 g NaCl, 1.3 g KCl, 1 g MgCl2.6H2O, 0.1 g CaCl2.2H2O, 0.5 g NH4Cl, 0.2 g KH2PO4, 3.82 g Na2SO4, 0.08 g FeCl2.4H2O, 1 mL trace elements (KCTC Media No. 918, https://kctc.kribb.re.kr/access/search/viewMedia?sn=918), 1 mL vitamin solution46, 0.1 g yeast extract, 2.5 g NaHCO3, 4.76 g HEPES, and 1 L deionized H2O, adjusted to pH 7.0 using 2 N NaOH. The following carbon substrates were tested: fructose, galactose, glucose, xylose, maltose, lactose, sucrose, and starch. BS without substrates was used as the control. For fermentation conditions, Na2SO4 was omitted from the composition of the basal medium. For sulfate reduction conditions, additional carbon sources (10 mM), which served as an electron donor, were evaluated, including acetate, benzoate, formate, fumarate, hexanoate, octanoate, lactate, propionate, and pyruvate47, and sulfate (27 mM) was tested as an electron acceptor. For an additional anaerobic respiration test, nitrate (10 mM), nitrite (10 mM), sulfite (10 mM), and thiosulfate (10 mM) were added to BS to replace sulfate (27 mM) as electron acceptors.
The growth of the strains was monitored under a microscope (Nikon Eclipse 80i) and compared with that of the control. The concentration of sulfate in the culture broth was measured turbidometrically48. In brief, the supernatant from the centrifuged culture broth was diluted tenfold. The diluted culture broth was mixed with a conditioning reagent in a 1:1 (v/v) ratio. For the preparation of 1 L of conditioning reagent, 150 g of NaCl, 100 mL of glycerol, 60 mL concentrated HCl, 200 mL of absolute ethanol, 60 g of BaCl2, and distilled water were combined to reach a final volume of 1 L, followed by heating and stirring to dissolve the chemicals prior to use. The turbidity was measured at OD 405 nm, and sulfate solutions ranging from 0 to 10 mM were used for calibration. The utilization of mono-, di-, and polysaccharides was quantified by measuring the production of reducing sugars during growth using 3,5-dinitrosalicylic acid (DNS) assay49. Fermentation products were determined after five days of cultivation using high-performance liquid chromatography (HPLC), as described previously50. The chromatograph of the 5-day culture was compared with that of the 0-hour culture. Different and high peaks were identified by aligning the retention times with those of candidate standard compounds. All experiments were performed in duplicate.
Chemotaxonomic characterization
The fatty acid profiles of strains DS1-an-2312T and DS1-an-13321T, as well as their reference strains, were determined after they were grown on MA plates for three days. Cells were harvested for a fatty acid analysis following the procedure outlined in the MIDI protocol (version 6.2). Subsequently, the extracted fatty acid methyl esters were injected into a gas chromatograph system, and their fatty acid components were identified based on the RTSBA 6.0 database.
To determine the quinone type, freeze-dried DS1-an-2312T and DS1-an-13321T cells were mixed with chloroform-methanol (2:1, v/v) and shaken overnight. The extract was collected through paper filtration, concentrated via evaporation, and recovered by adding acetone. The acetone suspension was then applied to a thin-layer chromatography (TLC) plate (Kieselgel 60F254, 20 × 20 cm, Merck) and separated using a combination of petroleum ether-diethyl ether (9:1, v/v). The quinone band on the TLC plate was visualized with UV light, and the samples were harvested and recovered in 100% acetone. The quinone extracts were further analyzed via reversed-phase chromatography using a mobile phase of methanol-isopropanol (7:5, v/v) and a wavelength of 270 nm to detect the quinone components45.
The polar lipids of strains DS1-an-2312T and DS1-an-13321T were extracted from their freeze-dried cells following the detailed methods of Komagata and Suzuki51. The extracted lipids were then applied onto a silica gel TLC plate and developed on the plate in two dimensions. The first dimension involved a combination of chloroform-methanol-water (65:25:4, v/v/v), and the second a combination of chloroform-methanol-acetic acid-water (80:15:12:4, v/v/v/v)45. To identify the polar lipid profiles of the two novel isolates, TLC plates were sprayed with individually appropriate reagents: molybdatophosphoric acid for total lipids, ninhydrin for lipids with amino groups, molybdenum blue for lipids with phosphate groups, and ɑ-naphthol in a sulfuric acid solution for lipids with sugar groups.
Genome analysis
For genomic analysis, genomic DNA was extracted based on the method of Vengadesh et al.52 with several modifications. Briefly, the isolated strains cultured anaerobically for two days in MB were harvested. The cell mass was mixed with proteinase K (20 mg/mL, 20 mM Tris-HCl, pH 8.0) in cetyltrimethylammonium bromide (CTAB) solution. The mixtures were incubated at 37 ℃ for one hour. Next, one volume of phenol-chloroform-isoamyl alcohol (PCI, 25:24:1, v/v/v) was added to the mixture, followed by centrifugation at 11 000 × g. The aqueous phase was collected and transferred to a new tube, and the nucleic acids were precipitated by adding a 0.6 volume of isopropyl alcohol and a one-tenth volume of 3 M sodium acetate. The tubes were inverted to ensure a consistent mixture and then incubated at 4 ℃. After 1–2 h of incubation, the tubes were centrifuged at 16 000 × g for 20 min at 4 ℃. Next, the suspension was removed and replaced twice with 100 µL of 70% ethanol (cooled at -20 ℃), twice. The pellet was dried at room temperature and re-dissolved in DNase-free water. RNA was then removed from the solution by treatment with RNase A at 40 ℃ for one hour. The DNA component subsequently was separated by one volume of PCI and precipitated with alcohol as aforementioned. The DNA was re-dissolved in DNase-free water and used for genome sequencing.
The whole-genome sequences of both novel strains were obtained by a combination of two sequencing methods, short-read Illumina sequencing (Macrogen, Inc., Seoul, Republic of Korea) and Nanopore sequencing. For Illumina sequencing, the short-length DNA of each strain was used to construct a library based on the protocol of the TruSeq DNA PCR-Free Sample Preparation Guide, part #15036187 Rev. D. For nanopore sequencing, high-molecular-weight DNA was used to prepare a library for nanopore sequencing according to the SQK-LSK109 protocol (version GDE_9063_v109_revN_14Aug2019). The genomes of both strains were de novo assembled by Canu (version 2)53 based on a combination of raw data from Nanopore and Illumina sequencing. Medaka (version 1.3.2, https://github.com/nanoporetech/medaka) was used as a polishing tool for assembly by counting the occurrences of each nucleotide at each position on the assembled sequence to predict the true base at that position. The quality of the assembled genome and annotation completeness were assessed by BUSCO (https://busco.ezlab.org/)54. CheckM (version 1.1.3) was used to estimate the contamination and completeness of the genome55. The genome was annotated on Prokka (version 1.12)56. The average nucleotide identity (ANI) tool on EzBioCloud (https://www.ezbiocloud.net/tools/ani)57 and the genome-to-genome distance calculator (version 2.1) on DSMZ (https://ggdc.dsmz.de/ggdc.php#)58 were used for digital DNA-DNA hybridization to distinguish the novel isolates from their closest valid taxa. The whole-genome sequences of the reference strains, including Puteibacter caeruleilacunae JC036T (GCA_005217565), Prolixibacter bellariivorans JCM 13498T (GCF_000621705), Sunxiuqinia elliptica CGMCC 1.9156T (GCF_900113005), Maribellus luteus XSD2T (GCA_003576475), Dracinibacterium orientale FH5T (GCA_000626635), Aquipluma introreducens MeG22T (AP018694), Mangrovibacterium diazotrophicum DSM 27148T (GCF_003610535), Mariniphaga anaerophila DSM 26910T (GCF_900129025), Tangfeifania diversioriginum DSM 27063T (GCF_900141875), and Gaoshiqinia sediminis A06T (GCF_025907915.1), were retrieved from NCBI. The average amino acid identity (AAI) was calculated using the AAI calculator from the Kostas laboratory (http://enve-omics.ce.gatech.edu/aai/)59. Pairwise comparisons for the percentage of conserved proteins (POCP) were calculated based on the method provided by Qin et al.60. The amino acid FASTA sequences of the reference strains were retrieved from NCBI. Functional genes within each genome were also annotated using KEGG and deciphered to pathways using KEGG Decoder61 and KEGG-Expander (https://github.com/bjtully/BioData/tree/masterEGGDecoder). Other databases were used for annotation including the Clusters of Orthologous Genes (COGs)62 and Gene Ontology (GO)63 databases. Carbohydrate-active enzymes were identified through the CAZy database (http://www.cazy.org/)64 and the dbCAN server (https://bcb.unl.edu/dbCAN2/blast.php)65. Biosynthesis gene clusters (BGCs) and metabolic gene clusters (MGCs) were predicted by antiSMASH 7.1.066 and gutSMASH67, respectively. Prophages were predicted in the genomes using PHASTER68. CRISPR-Cas in the genomes was predicted using CRISPRCasFinder69. The subcellular location of proteins in prokaryotes was predicted via PSORTb (version 3.0.3)70. The predicted model of degradation and xylose utilization was drawn by using Affinity Designer (version 1.10.6.1665).
Polysaccharide-degrading ability
To test polysaccharide utilization, two novel strains were anaerobically grown on BS media supplemented with individual polysaccharides (0.1–0.2%, w/v), including alginate, cellulose, chitin, κ-, λ-, and ι-carrageenan, fucoidan, laminarin, starch, and xylan which were obtained from Sigma. Ten microliters of each culture was observed under a light microscope (Nikon Eclipse 80i) every two days to monitor growth. By comparing the cell numbers each day and with those of the control (no carbon source), the growth of both strains was recorded. The production of reducing sugars in the supernatant was detected by DNS assay49.
Growth of DS1-an-13321T and DS1-an-2312T on laminarin and xylan
The ability of strains DS1-an-13321T and DS1-an-2312T to grow on laminarin and the ability of strain DS1-an-2312T to grow on xylan were further studied. During the growth of strains DS1-an-13321T and DS1-an-2312T on laminarin and xylan, black particles and clumped cells were produced in the culture broth, and the density of the cells in culture could not be measured by a spectrophotometer. The growth of DS1-an-13321T and DS1-an-2312T on the corresponding substrates was therefore monitored under a light microscope (Nikon Eclipse 80i). We counted the cells in culture every 6 h under a microscope to track the growth of both strains on laminarin and xylan to make growth curves. Briefly, every 6 h, 100 µL of broth culture was harvested and diluted tenfold. Subsequently, 10 µL aliquots of diluted solution were dropped onto glass slides, stained with safranine (BD), and cells were counted under 400× magnification by a microscope (Nikon Eclipse 80i). Five frames were randomly selected for statistical analysis. Subsequently, the cells in each frame were counted, and the average cell number and standard deviation were calculated considering the dilution factors.
Location of laminarin and xylan-degrading enzymes
For laminarin-degrading enzymes, cells were harvested by centrifugation from the 4-day cultures of strains DS1-an-13321T and DS1-an-2312T on laminarin. The cell pellet was washed and resuspended in phosphate buffer solution (PBS, pH 7.2). Subsequently, the cell suspension was incubated with laminarin (final concentration of 0.1%, w/v) for 6 h at 30 ºC. The supernatant was stirred with 50% (w/v) (NH4)2SO4 (final concentration) to precipitate the extracellular protein. Overnight dialysis was applied to remove the salt. The resulting solution was incubated with laminarin (at a final concentration of 0.1%, w/v) for 6 h at 30 ºC. The enzyme reaction was assessed by detection of reducing sugars by a DNS assay49. The change of reducing sugars from the beginning to the end of the enzyme reaction was determined. The procedure used to determine the location of the active xylan-degrading enzymes was the same as that used for laminarin-degrading enzymes. However, in the enzyme reaction, the final concentration of xylan was 0.2% (w/v).
Degradation of laminarin and xylan by whole cell enzymes
Four-day old strains DS1-an-13321T and DS1-an-2312T cells grown on laminarin (0.1%, w/v) were harvested. The cells were harvested by centrifugation, washed three times, and resuspended in PBS. The cell suspension was then used as a crude enzyme of cell-associated laminarin-degrading enzymes to test for enzyme activity. In the case of DS1-an-2312T grown on xylan (0.2%, w/v), 4-day old DS1-an-2312T cells grown on xylan were harvested by centrifugation. The cell pellet and remaining insoluble xylan were washed three times to remove remaining soluble sugars and resuspended in PBS. The cell suspensions were used as crude enzymes, and enzyme activity was directly tested. The DNS assay was applied to detect the production of reducing sugars during the hydrolysis of laminarin and xylan. The products of the hydrolysis reaction were further analyzed in the next step.
The hydrolysis products of the crude enzymes were assessed by thin layer chromatography (TLC) according to the method of Lee et al.71. In brief, the crude enzymes from strains DS1-an-13321T and DS1-an-2312T were incubated with each substrate (1 mg/mL, final concentration) of glucose (G), laminaribiose (L2), laminaritriose (L3), laminaritetraose (L4), laminaripentaose (L5), laminarihexaose (L6), and laminarin (Ln) at 30 ºC for 24 h. The reaction of the crude enzymes without substrates and the reaction of each substrate without the crude enzymes were included as controls. The enzyme-reaction tubes were centrifuged at 8000 rpm for 2 min at room temperature to harvest the degradation products of the reaction. The supernatant was applied to a TLC plate without concentration. The TLC plate was then developed in a chloroform-acetic acid-water (6:7:1, v/v/v) solvent system71. The TLC plates were visualized by spraying a mixture of ethanol-sulfuric acid (95:5, v/v), followed by drying in an oven at 150 ºC for 5 to 10 min. For hydrolysis products from xylan degradation of DS1-an-2312T, the cell pellet was washed three times with PBS to remove the remaining soluble sugar from the culture. Subsequently, to assess the mode of action of the DS1-an-2312T xylan-degrading enzyme, the reducing sugars present at the initial crude enzyme solution and the reducing sugars that were produced during the enzyme reaction were compared. The limitation of this test is the remaining insoluble xylan in the cell pellet; thus, during enzyme incubation with the whole cell, this remaining xylan that had not yet been consumed was further degraded, and the resulting reducing sugars was released into the suspension. Thus, the presence of hydrolytic products during enzyme reaction of crude enzyme was detected via TLC in the same manner as in the analysis of the laminarin-hydrolytic products.
The oligosaccharides of xylan and laminarin were purchased from Megazyme (Ireland), and laminarin and xylan from oat spelts were purchased from Sigma.
Results and discussion
Isolation, cultivation, and identification
Two strains were isolated from a sea squirt (Fig. 1a) collected at a depth of 18 m of the East Sea, Republic of Korea. A portion of the feeding area of the sea squirt (Fig. 1b) was cut and placed on the surface of filter paper on a low nutrient agar medium that was prepared by 60% seawater collected from the same sampling location. Pure cultures of the two novel strains were obtained by transferring colonies exhibiting gliding traits around the filter paper. The strains were transferred several times onto fresh MA plates until no contaminants were detected. The morphologies of the pure cultures of strains DS1-an-13321T and DS1-an-2312T are presented in Fig. 1c and d, respectively. Strain DS1-an-13321T formed nearly round colonies with a tortilla color in the center and a cream color at the periphery, while strain DS1-an-2312T exhibited irregular colonies with a round tortilla color in the center and broad swarming at the outer edge of the colonies. The cells of both strains were long rods with a length exceeding 20 μm during the log-phase of growth and a width ranging from 0.25 to 0.5 μm (Fig. 1e, f, and Table 1). Interestingly, both strains showed a transition from a long rod to a spherical shape at the end of the stationary phase (Fig. S1), which was also observed from strain Sunxiuqinia faeciviva JAM-BA0302T which changes its morphology from thin, long rods to spherical cells at the late exponential and stationary phases of its growth72. Strains DS1-an-13321T and DS1-an-2312T were maintained on MA plates at 23 ºC and 28 ºC, respectively. For long-term preservation, two novel isolates were preserved by using a liquid-dry method.

Origin, colony, and cell morphology of two novel isolates in the family Prolixibacteraceae. (a) Sea squirt collected at a depth of 18 m under seawater at Sodol Port, East Sea; (b) position at which the collected squirt was cut; (c) and (e): colony morphology and SEM image of DS1-an-13321T cells; (d) and (f): colony morphology and SEM image of DS1-an-2312T cells. Scale bar: 1 cm (a); 1 mm (c, d); 1 μm (e, f).
The 16S rRNA gene sequences of the pure cultures were determined. The analysis of the sequences on the EzBioCloud server (https://www.ezbiocloud.net/) revealed that the highest 16S rRNA gene sequence similarities to Puteibacter caeruleilacunae JC036T for strains DS1-an-13321T and DS1-an-2312T was 91.26% and 91.37%, respectively. Moreover, 16S rRNA similarities of the two novel isolates with type strains of all existing genera in the family Prolixibacteraceae, which is the parent taxon of Puteibacter caeruleilacunae, were calculated (Table S1). This analysis demonstrated that the 16S rRNA similarity between the two isolates and existing members of the family Prolixibacteraceae fell within the range of 86.70 to 91.37%, while a similarity of 98.75% was observed between the two isolates. Based on these results, we hypothesized that the two novel isolates represent a novel genus and two novel species. Therefore, 16S rRNA-based phylogenetic tree, genome-based phylogenetic tree, genomic indices, and polyphasic taxonomic study were performed to evaluate this hypothesis.
16S rRNA-based and genome-based phylogeny
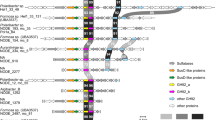
A phylogenetic tree based on 16S rRNA gene sequences by combining three algorithms, ML, NJ, and MP, showed the taxonomic position of the two novel isolates within the families Prolixibacteraceae and Marinilabiliaceae in the phylum Bacteroidota (Fig. 2). Interestingly, strain DS1-an-13321T formed a single cluster with strain DS1-an-2312T, but the two strains were separated from all other representatives in the family Prolixibacteraceae, even Puteibacter caeruleilacunae JC036T (approximately 91% of 16S rRNA similarity).

16S rRNA-based maximum-likelihood phylogenetic tree constructed with MEGA11 software (version 11.0.13) indicating the positions of two novel strains, DS1-an-13321T and DS1-an-2312T, with their closest representatives belonging to the family Prolixibacteraceae and the closest family Marinilabiliaceae. The strain Agarivorans albus NBRC 102603T (GenBank accession number AB076561) was used as the outgroup. GenBank accession numbers are presented in parentheses. The 16S rRNA gene sequences were aligned by ClustalW, and the resulting file was trimmed in BioEdit software (version 7.2.5). Bootstrap values > 50% based on 1000 replicates are presented at nodes. The closed circles indicate the consensus of the nodes recovered by using three algorithms, ML, NJ, and MP, respectively. The open circles indicate the consensus of recovered nodes found from two out of three algorithms. Bar, 0.05 substitutions per nucleotide position.
Based on 92 core genes from their genomes, a genome-based phylogenetic tree was constructed under the UBCG pipeline using the ML algorithm to clarify the taxonomic position of the two isolates within the families Prolixibacteraceae and Marinilabiliaceae (Fig. 3). The Gene Support Index (GSI), which quantifies the number of single-gene trees supporting a branch in a UBCG tree, was used to evaluate the robustness of the branching. A higher GSI indicates stronger branch support73. In contrast to the 16S rRNA-based phylogenetic tree, the genome-based phylogenetic tree revealed that the two novel isolates clustered with their closest relative, Puteibacter caerulelacunae JC036T, at a branch node supported by a high GSI value of 92, indicating robust support from all UBCGs. This cluster formed a clade with other representatives of the family Prolixibacteraceae, and was distinct from the clades of the families Marinilabiliaceae and Marinifilaceae. Despite this difference, the 16S rRNA-based phylogenetic tree showed that the cluster of the two novel isolates was closer to the cluster of known representatives in the family Prolixibacteraceae and was separated from the clusters of the families Marinilabiliaceae and Marinifilaceae. The differences between the topologies of the 16S rRNA-based phylogenetic tree and that of the genome-based phylogenetic tree may be attributed not only to the distinct evolutionary information they provide but also to the number of genes evaluated in the genome-based tree (92 core genes). This larger number of genes offers a more comprehensive view of the taxonomic position of the two novel isolates and their relatives. Thus, the integration of the results from both the 16S rRNA-based and genome-based phylogenetic trees indicates that the two novel strains can be classified as a new genus within the family Prolixibacteraceae.

Maximum-likelihood phylogenetic tree showing the relationships among DS1-an-2312T, DS1-an-13321T and their closely related species based on 92 core genes identified via the UBCG pipeline. GenBank accession numbers of the whole genome sequences are given in parentheses. Agarivorans albus JCM 21469T (GCA_019670105.1) was used as the outgroup. Gene Support Indices (GSIs) are given at the branch nodes. Bar, 0.1 substitutions per site.
To evaluate the taxonomic proposal of the two novel isolates, the genomic indices of the two isolates against the type species in all genera of the family Prolixibacteraceae, including the ANI, dDDH, AAI, and POCP values, were calculated (Table 2). The ANI and dDDH values between strain DS1-an-13321T and strain DS1-an-2312T were 73.99% and 20.9%, respectively. These values were under the cutoff values of the ANI (95%)57 and dDDH (70%)58 for species demarcation. In addition, the ANI and dDDH values between the two isolates and other representatives in the family Prolixibacteraceae ranged from 66.17 to 67.76% and 24.0–37.4%, respectively. All these values were much lower than the cutoff values of the ANI (95%) and dDDH (70%). Hence, the two novel isolates were deemed candidates for two novel species classified in the family Prolixibacteraceae. For a comparison of the amino acid sequences, the AAI and POCP values between the two novel isolates and representatives in the family Prolixibacteraceae ranged from 48.33 to 52.35% and 29.01–37.37%, respectively. Both of these ranges fell under the cutoff values for genus demarcation of AAI (60%)74 and POCP (50%)60 but strain DS1-an-13321T shared 71.34% and 73.55% for the AAI and POCP values, respectively, with strain DS1-an-2312T. Therefore, the genomic indices provide strong evidence for the proposal of strains DS1-an-13321T and DS1-an-2312T as candidates for two novel species in a novel genus in the family Prolixibacteraceae.
Physiological characterization
The two novel strains showed differences with other representatives in the family Prolixibacteraceae. Both strains DS1-an-13321T and DS1-an-2312T could hydrolyze starch and DNA, while the closest strain Puteibacter caeruleilacunae JC036T43 cannot hydrolyze starch or DNA, Prolixibacter bellariivorans JCM 13498T75, Maribellus luteus XSD2T22, and Gaoshiqia sediminis A06T76 cannot hydrolyze DNA, and Sunxiuqinia elliptica CGMCC 1.9156T77, Draconibacterium orientale FH5T40, and Mangrovibacterium diazotrophicum DSM 27148T39 cannot hydrolyze starch (Table 1). Additionally, both novel isolates could not utilize fructose, while strains Puteibacter caeruleilacunae JC036T43, Sunxiuqinia elliptica CGMCC 1.9156T77, Draconibacterium orientale FH5T40, Aquipluma nitroreducens MeG22T37, and Gaoshiqia sediminis A06T76 can utilize fructose. Only strain DS1-an-13321T was strictly anaerobic, while most others are facultative anaerobic, and only strain Sunxiuqinia elliptica CGMCC 1.9156T is strictly aerobic77. Interestingly, even though DS1-an-13321T and DS1-an-2312T belong to one genus, only strain DS1-an-2312T could utilize xylose (Table 1). The detailed characteristics of the two novel isolates and representatives in the family Prolixibacteraceae are presented in Table 1.
Chemotaxonomic characterization
The fatty acid profiles of the two novel isolates and their reference strains are shown in Table 3. The two novel isolates were distinguished from each other and from their closest valid species by the different major fatty acid components and their proportions. The major fatty acid components (> 10% of the total fatty acids) of strain DS1-an-13321T were iso-C15:0 (28.08%), anteiso-C15:0 (21.27%), and iso-C15:0 3OH (13.58%). In addition, the predominant fatty acid components (> 10% of the total fatty acids) of strain DS1-an-2312T were anteiso-C15:0 (20.08%), iso-C15:0 (16.51%), iso-C17:0 3OH (12.83%), and iso-C15:0 3OH (10.4%). The primary components (> 10% of the total fatty acids) of their three reference strains included iso-C15:0, anteiso-C15:0, and iso-C17:0 3OH. The primary isoprenoid quinone of both novel strains was menaquinone-7 (MK-7), which is common to other species in the family Prolixibacteraceae. The polar lipids of strain DS1-an-13321T included phosphatidylethanolamine (PE), phosphatidylserine (PS), two amino-lipids (AL1–2), an aminophospholipid (APL), a phospholipid (PL), and an unidentified lipid (L2) (Fig. S2a). The polar lipid components of strain DS1-an-2312T consisted of phosphatidylethanolamine (PE), and three aminophospholipids (APL1–3) (Fig. S2b).
Anaerobic metabolism
The type of anaerobic metabolism of strains DS1-an-13321T and DS1-an-2312T was assessed. Under fermentation conditions, both strains were able to utilize galactose, glucose, lactose, maltose, sucrose, and starch. Strain DS1-an-2312T was also able to utilize xylose, whereas DS1-an-13321T was not able to utilize xylose. The presence of sulfate did not change the carbon utilization pattern of either strain, as also observed under fermentation conditions, and a decrease of sulfate was not observed (data not shown). Additionally, strain DS1-an-13321T could weakly grow on pyruvate, but grew actively on glucose. Strain DS1-an-2312T could grow on lactate or pyruvate. The growth of strains DS1-an-13321T and DS1-an-2312T on glucose was also observed in the presence of nitrate and thiosulfate, but no reduction of nitrate or thiosulfate was observed. Interestingly, only strain DS1-an-2312T could grow in the presence of sulfite. However, according to the API 20NE test, neither DS1-an-13321T nor DS1-an-2312T could perform nitrate/nitrite reduction under anaerobic conditions. Moreover, in the presence of sulfite, the growth of DS1-an-13321T was suppressed, and in the presence of nitrite, the growth of both strains was suppressed. These results indicated that strains DS1-an-13321T and DS1-an-2312T have fermentation metabolism even in the presence of a high concentration of sulfate (27 mM). This high concentration of sulfate in the basal medium mimicked the sulfate concentration in natural sea water78.
The fermentation products of strains DS1-an-13321T and DS1-an-2312T from the above experiments were analyzed through HPLC50. By comparing the retention times of standard SCFAs under the same analysis conditions, the major fermentation products of strains DS1-an-13321T and DS1-an-2312T on galactose, glucose, maltose, lactose, sucrose, and starch and those of strain DS1-an-2312T on xylose were acetic acid and propionic acid.
Polysaccharide utilization
For polysaccharide utilization, strains DS1-an-13321T and DS1-an-2312T were tested for growth on alginate, cellulose, chitin, κ-, λ-, and ι-carrageenan, fucoidan, laminarin, starch, and xylan in basal media. Their growth was observed under a microscope. Strain DS1-an-13321T was found to utilize laminarin and starch as the sole carbon source, while strain DS1-an-2312T could utilize xylan in addition to laminarin and starch (Table 4). However, despite observing growth in the cultures, the DNS assay could not detect any reducing sugars in the culture broth. This finding suggests that the strains effectively utilized all the reducing sugars generated during polysaccharide degradation. This observation might be related to the “selfish” lifestyle of polysaccharide degraders in the phylum Bacteroidota7,79.
Several studies have reported that members of the family Prolixibacteraceae can utilize polysaccharides. For instance, Mangrovibacterium lignilyticum is enriched in media containing lignin80, Tangfeifania diversioriginum is capable of hydrolyzing starch81, and Sunxiuqinia indica82 and Gaoshiqia sediminis76 contain 176 genes and 297 genes related to CAZymes, respectively. However, there has not yet been a study based on a combination of in silico analysis and in vitro experiments to assess polysaccharide utilization among members of the family Prolixibacteraceae. Interestingly, both novel isolates could utilize laminarin, which was not previously reported for members of the family Prolixibacteraceae, and strain DS1-an-2312T could degrade xylan. Therefore, in this study, we further investigated laminarin and xylan utilization by the isolates through in vitro examination and in silico genomic mining.
Growth of DS1-an-13321T and DS1-an-2312T on laminarin and xylan
Based on the cell counting method, with counts taken every six hours for growth on laminarin and every 24 h for growth on xylan, the growth curves of DS1-an-13321T and DS1-an-2312T on laminarin and xylan are shown in Fig. 4. The growth of strain DS1-an-13321T on laminarin reached the late log phase on day 3, while the late log phase of strain DS1-an-2312T occurred on day 4. The growth of strain DS1-an-2312T on xylan reached the late log phase on day 3. Based on these results, cultures at their late log phase were harvested for further enzyme activity studies.

Growth curves of strains DS1-an-13321T and DS1-an-2312T on laminarin (a) and xylan (b) under anaerobic conditions.
The production of reducing sugars was measured using the DNS assay during the enzyme reactions of cell-associated proteins from strains DS1-an-13321T and DS1-an-2312T with laminarin on day 2 and 6, respectively. However, no detectable production of reducing sugar was observed in the enzyme reactions involving concentrated proteins from the dialysis solution of their broth cultures with laminarin. This indicated that their laminarin-degrading enzymes were cell-associated proteins. Similarly, the xylan-degrading enzymes of strain DS1-an-2312T were also identified as cell-associated proteins, as evidenced by the production of reducing sugars in enzyme reactions of the strain’s cell-associated proteins with xylan.
To trace the mode of action of the laminarin- and xylan-degrading enzymes of DS1-an-13321T and DS1-an-2312T, cell-associated enzymes were harvested from each strain and the degradation products were determined by a TLC analysis. Based on the Rf values of the enzyme reactant and standard compounds, the major hydrolytic product on the laminarin or laminarin oligosaccharides of both strains was identified as glucose (Fig. 5, and Fig. S3). These results indicated that the laminarin-degrading enzymes of both strains exhibited exo-hydrolytic activities. For the xylan-degrading enzyme, the hydrolytic products by the crude enzyme of strain DS1-an-2312T on xylan were xylosetriose and xylotetraose (Fig. 6), indicating that the cell-associated xylan-degrading enzyme of DS1-an-2312T exhibited endo-hydrolytic enzyme activities.

TLC plate analysis of hydrolysis products released from the reaction of cell-associated laminarin-degrading enzymes DS1-an-13321T on laminarin oligosaccharides and laminarin for 24 h at 30 ºC. Laminarin oligosaccharides: G, glucose; L2, laminaribiose; L3, laminaritriose; L4, laminaritetraose; L5, laminaripentaose; L6, laminarihexaose; Ln, laminarin. The solvent system: chloroform-acetic acid-water (6:7:1, v/v/v). e: enzyme reaction product. c: control reaction product. M: mixture of laminarin oligosaccharides.

TLC plate analysis of hydrolysis products released from the reaction of xylan-degrading enzyme DS1-an-2312T on xylan after 24 h at 30 ºC. The solvent system: chloroform-acetic acid-water (6:7:1, v/v/v). e.Xn: crude enzyme reacting with xylan; ce: control of crude enzyme reacting without the presence of substrate. M: mixture of xylan oligosaccharides, including: X1, xylose; X2, xylobiose; X3, xylotriose; X4, xylotetraose; X5, xylopentaose; X6, xylohexaose.
Genome analysis
The whole genomes of DS1-an-13321T and DS1-an-2312T, determined by a combination of Nanopore and Illumina platforms, were obtained with high completeness (BUSCO values: 94.3% and 93.5%, respectively). Both the DS1-an-13321T and DS1-an-2312T genomes comprised a single circular chromosome with sizes of 4,465,088 bp and 5,187,288 bp, respectively, and had G + C content 35.9% and 36.5%, respectively (Table S2).
The whole-genome sequence of strain DS1-an-13321T contained 3,545 predicted genes including 3,341 coding genes and eight pseudogenes. Among these, there were 158 tRNAs, five noncoding RNAs, and 41 rRNA genes (15 5S rRNAs, 13 16S rRNAs, and 13 23S rRNAs) (Table S2). On the other hand, genome analysis of strain DS1-an-2312T revealed that the strain harbored 3,807 predicted genes, consisting of 3,634 coding genes and six pseudogenes. Among these, there were 128 tRNAs, five noncoding RNAs, and 40 rRNA genes (14 5S rRNAs, 13 16S rRNAs, and 13 23S rRNAs) (Table S2).
AntiSMASH and gutSMASH revealed that the genome of strain DS1-an-13321T encodes one BGC belonging to the nonribosomal peptide synthetase family and nine MGCs, while the genome of strain DS1-an-2312T encodes one BGC belonging to the linear azoline-containing peptides family and nine MGCs. In strain DS1-an-13321T, the three COGs with the greatest number of genes were related to cell wall/membrane/envelope biogenesis, the mobilome (prophases, transposons), translation, ribosomal structure, and biogenesis. Moreover, in strain DS1-an-2312T, the three COGs with the greatest number of genes were related to cell wall/membrane/envelope biogenesis, inorganic ion transport and metabolism, and carbohydrate transport and metabolism (Fig. S4).
Genome mining revealed that the genomes of strains DS1-an-13321T and DS1-an-2312T contain a high number of genes encoding carbohydrate-active enzymes (CAZymes). The CAZy database (http://www.cazy.org/), which contains information about carbohydrate-active enzymes, including glycoside hydrolase (GH, cleavage of glycosidic bonds), glycosyl transferase (GT, construction of glycosidic bonds), polysaccharide lyase (PL, nonhydrolytic hydrolysis of glycosidic bonds), carbohydrate esterase (CE, cleavage of carbohydrate esters), auxiliary activity (AA, a redox enzyme that works in conjunction with other CAZymes), and carbohydrate-binding modules (CBM, adhesion to carbohydrates), was used to assess the detailed composition of CAZymes in the genomes of the two isolates, and the results are presented in Table S3. Strain DS1-an-13321T encoded a total of 155 CAZymes consisting of 84 GHs, 32 GTs, 13 PLs, seven CEs and 19 CBMs, while strain DS1-an-2312T encoded a total of 249 CAZymes, an amount 1.6 times greater than that of DS1-an-13321T, consisting of 128 GHs, 37 GTs, 27 PLs, 35 CEs, and 22 CBMs. The number of GH genes per genome in strain DS1-an-13321T was 18.81 (GHs/Mb), while in strain DS1-an-2312T it was 24.46 (GHs/Mb); both of these values are significantly greater than the average value of 12 GHs/Mb in the genomes of other members of marine bacteria of the class Bacteroidia83. The genomes of DS1-an-13321T and DS1-an-2312T contained 27 and 34 PULs, respectively, which are approximately one-third lower than the average number of PULs in human gut Bacteroides and similar to the number of PULs found in the genus Prevotella (average 23 PULs/genome)20,84.
The genomes of strains DS1-an-13321T and DS1-an-2312T contained genes predicted to be involved in laminarin degradation. The genome of strain DS1-an-13321T harbored genes encoding for four GH3, one GH16, and one GH30, while strain DS1-an-2312T harbored genes encoding for seven GH3, two GH16, and one GH30 (Table S4), which has been reported to contribute to the degradation of laminarin in other marine bacteria14,85,86. Unlike other laminarin degraders of Gramella spp.85, Formosa spp.86 (class Flavobacteriia) or Bacteroides spp.87,88 (class Bacteroidia), neither strain DS1-an-13321T nor DS1-an-2312T harbored genes encoding GH3 and GH16, which in general collocate with each other and collocate with SusC/SusD (a signature for the PUL structure). Instead, GH3 and GH16 of strains DS1-an-13321T and DS1-an-2312T were located separately, and each gene was collocated with SusC/SusD in the genomes. For strain DS1-an-13321T, we found that PUL8 contained a tandem of SusD/TBDR, an unknown protein, and two copies of GH3. Additionally, these two GH3s were predicted to be located in the periplasmic space (PSORTb scores of 9.44 and 9.76). Furthermore, strain DS1-an-13321T harbored an unidentified PUL (21_un_PUL), and the gene cluster contained SusD (K4L44_09375), SusC (K4L44_09380), TonB-dependent receptor (TBDR) (K4L44_09385), two unknown proteins (K4L44_09390, K4L44_09405), superoxide dismutase, Ni (K4L44_09395), GH3 (K4L44_09400), IS4 family transposase (K4L44_09410), and GH30 (K4L44_09415) (Fig. S5a). Within this gene cluster, we detected GH3 (cleavage β-1,3-glucan) and GH30 (cleavage β-1,6-glucan), which PSORTb could predict at multiple locations on the cell. In contrast, strain DS1-an-2312T consisted of three PULs (PUL9, PUL29, and an unidentified PUL (12_un_PUL)), which contained GH3 without the presence of GH16 and GH30. We also found that the GH10 and GH5 genes in PUL9 and PUL29, respectively, were predicted to degrade the xylan main chain, indicating that these two PULs may play a role in xylan degradation rather than laminarin degradation. Additionally, in the 12_un_PUL (Fig. S5b), we detected several genes of SusC/SusD and GH3. The GH3 was predicted to locate in the periplasmic space with PSORTb score 9.44. To identify the active gene cluster responsible for laminarin degradation, further transcriptomic analysis is required.
In this study, the novel strain DS1-an-2312T was identified as an anaerobic bacterium capable of utilizing xylan as a sole carbon source. Whole-genome analysis of strain DS1-an-2312T revealed the presence of CAZymes involved in the effective degradation of the natural polymer xylan (Tables S4, and S5). We found that strain DS1-an-2312T harbored genes encoding two GH5, one GH10, one GH30, and three GH141 enzymes (Table S4). Notably, using PULDB, all four potential xylan utilization loci were identified, PUL8, PUL9, PUL25, and PUL29 (Fig. S5c). A detailed analysis of these PUL indicated that PUL9 and PUL29 would have greater potential for xylan degradation in strain DS1-an-2312T (Table S5). Specifically, PUL9 contained SusC and SusD, which are responsible for capturing polysaccharides and delivering oligosaccharides into the cytoplasm9,11. It also contained a GH10 enzyme, which exhibited the highest amino acid similarity of 26% (covering 80% of the sequence) to endo-1,4-β-xylanase (UniProt accession code G4MLU0) and 24.4% (covering 81% of the sequence) to a reported GH10 module glycoside hydrolase of Caldicellulosiruptor danielii (PDB accession code 6D5C_A). Additionally, the PUL9 of strain DS1-an-2312T contained a GH3 enzyme, which exhibited the highest similarity (43.5%, covering 83% of the sequence) to β-xylosidase of Formosa agariphila (UniProt accession code T2KMH0). Similarly, PUL29 contained the SusC, SusD, and a multidomain protein consisting of one GH5 subfamily 46 domain and two CBM6 modules. The multidomain protein exhibited 32.8% similarity to endoglucanase C of Acetivibrio thermocellus (UniProt accession code A3DJ77). Moreover, these two enzymes, GH5 and GH10, were not found in the genome of strain DS1-an-13321T, which cannot utilize xylose and xylan as a sole carbon source under anaerobic conditions (Table 4). Additionally, through genome analysis, physiological characterization, and TLC analysis, it was inferred that strain DS1-an-2312T exhibited strong xylan degradation capabilities (Tables 1 and 4, Fig. S5 and 6). In the xylan degradation process, endo-1,4-β-xylanase and β-xylosidase enzymes degrade xylan to xylooligosaccharides (Fig. 7). The genome of strain DS1-an-2312T encoded both endo-1,4-β-xylanase (GH10) and β-xylosidase (GH3) enzymes (Table S5). Additionally, arabinofuranosidase GH30, a multisubstrate-specific family enzyme, acts as an endo-1,4-β-xylanase and degrades xylooligosaccharides. Subsequently, xylooligosaccharides are transported into the cell membrane. Bacterial strains typically use active transport mechanisms, with some routes utilizing high and low affinity transporters. Only the genome of strain DS1-an-2312T, not DS1-an-13321T, encoded the xylose transporter (XylE), which is a low-affinity transporter associated with xylooligosaccharide transportation via a proton motive force89,90, and xylose isomerase (XylA), which facilitates the reversible conversion of D-xylose into D-xylulose91.

Predicted schematic model of the xylan degradation and xylose utilization pathway in DS1-an-2312T under anaerobic conditions. The enzymes in the pathway are endo-1,4-β-xylanase (GH10, GH5 + CBM6 + CBM6), xylose transporter XylE, xylose isomerase XylA, xylulose kinase (XK), phosphoketolase (PK).
The whole-genome sequence of DS1-an-2312T was analyzed and the xylose metabolic pathway of the strains was modelled (Fig. 7). It was hypothesized that the metabolic pathway of the novel species involves xylose isomerase, as indicated by the presence of genes such as xylose isomerase (K5X82_00205) in its genome. In the isomerase pathway, the xylose transporter XylE (K5X82_00210) is responsible for the uptake of xylooligosaccharides. Xylooligosaccharides are degraded into D-xylose at the periplasm under the function of GH3 (K5X82_03105) (Fig. 7). This model hypothesis was supported by a TLC experiment (Fig. 6), where no detectable D-xylose was detected in the enzyme-reaction supernatant after removing the whole-cell-associated proteins of strain DS1-an-2312T. The xylose isomerase xylA (K5X82_00205) enzyme converts D-xylose to D-xylulose, which is phosphorylated to D-xylulose-5-phosphate by the xylulokinase enzyme90,92,93. The phosphoketolase enzyme further degrades D-xylulose-5-phosphate (a 5-carbon compound) into acetyl phosphate (a 2-carbon compound) and glyceraldehyde-3-phosphate (a 3-carbon compound). Some anaerobic bacteria, such as Clostridium sp., and lactic acid bacteria, can cleave xylulose-5-P by phosphoketolase into these compounds89,93,94,95,96.
Taken together, the results of this study not only support two novel strains that represent a novel genus with two novel species in the family Prolixibacteraceae, class Bacteroidia, phylum Bacteroidota but also expand our understanding of the strategies employed by marine Bacteroidia bacteria to access and degrade polysaccharides anaerobically. By mimicking natural nutrient conditions for isolation, pure cultures of the type strains of the two novel species were obtained. Both strains were capable of fermenting glucose, galactose, maltose, lactose, sucrose, and starch, with only DS1-an-2312T exhibiting the ability to utilize xylose. The major fermentation products of strains DS1-an-13321T and DS1-an-2312T were acetic acid and propionic acid. Genome mining revealed that both novel species contained rich sources of CAZymes. In vitro experiments demonstrated that both novel species could degrade laminarin and starch, with only DS1-an-2312T capable of utilizing xylan under anaerobic conditions. Both strains possessed cell-associated laminarin-degrading enzymes, exhibiting exo-hydrolytic enzyme activity and producing glucose as the major final product. In addition, strain DS1-an-2312T possessed a cell-associated xylan-degrading enzyme with endo-hydrolytic enzyme activity, producing xylotriose and xylotetraose as the major final products. These results highlight the potential biotechnological applications of the two novel species and their strategies for adaptation under anoxic conditions in marine ecosystems through fermentation and polysaccharide degradation. For further study of the molecular mechanism of laminarin and xylan degradation in both novel species, future work will involve transcriptomic and proteomic analyses.
Description of Halosquirtibacter gen. nov.
Halosquirtibacter gen. nov. (Ha.lo.squirt.i.bac’ter. N.L. fem. n. Halosquirt, an animal genus, L. n. squirt, acolloquinal term for the sea squirt; N.L. masc. n. bacter, a rod; N.L. masc. n. Halosquirtibacter, a rod from the sea squirt Halocynthia).
Cells are Gram-stain-negative, anaerobic, rod-shaped, and oxidase- and catalase-negative. Prominent fatty acid components are iso-C15:0, anteiso-C15:0, iso-C15:0 3-OH, and iso-C17:0 3-OH. The major respiratory quinone type is menaquinone-7 (MK-7). Glucose, galactose, maltose, lactose, sucrose, and starch are fermented to produce a mixture of acid as major products. The genus Halosquirtibacter belongs to the family Prolixibacteraceae, phylum Bacteroidota. The type species is Halosquirtibacter laminarini.
Description of Halosquirtibacter laminarini sp. nov.
Halosquirtibacter laminarini sp. nov. (la.mi.na.ri’ni. N.L. gen. n. laminarini, of laminarin, referring to its ability to hydrolyze laminarin).
Cells are Gram-strain-negative, mesophilic, neutrophilic, strictly anaerobic, long rod-shaped at the log phase, and spherical at the end of the stationary phase of growth. They are oxidase- and catalase-negative. Round and bright brown colonies appeared on the surface of MB agar plates. Growth occurs at 15–30 ℃ (optimum, 20–30 ℃), at pH 6.0–8.5 (optimum, 6.5–8.5), and with 2–4% NaCl (optimum, 2–3%). H2S is produced. Positive for hydrolysis of gelatin, DNA, laminarin, and starch. Galactose, glucose, maltose, lactose, sucrose, and starch are fermented to produce acetic acid and propionic acid as the major products. The major fatty acid components are iso-C15:0, anteiso-C15:0, iso-C15:0 3-OH, and iso-C17:0 3-OH. Menaquinone 7 (MK-7) is the major quinone. The polar lipid profile comprises phosphatidylethanolamine (PE), two unidentified amino-lipids (AL1–2), one unidentified aminophospholipid (APL), one unidentified phospholipid (PL), one identified lipid (L), and one phosphatidylserine (PS).
The type strain DS1-an-13321T (= KCTC 25031T = DSM 115329T) was isolated from a sea squirt at a depth of 18 m under the surface of seawater. The genome contains one circular chromosome that is 4.47 Mb long. The G + C content is 35.9%, as calculated from whole-genome sequencing.
Description of Halosquirtibacter xylanolyticus sp. nov.
Halosquirtibacter xylanolyticus sp. nov. (xy.la.ni.ly’ti.cus. N.L. neut. n. xylanum, xylan; Gr. masc. adj. lytikos, dissolving; N.L. masc. adj. xylanilyticus, xylan-dissolving).
Cells are gram-stain-negative, mesophilic, neutrophilic, long rod-shaped at the log phase and spherical at the end of the stationary phase of growth. They are oxidase- and catalase- negative. Irregularly shaped and bright brown colonies appeared on the surface of MB agar plates. Growth occurs at 10–32 ℃ (optimum, 20–30 ℃), at pH 6.0–8.0 (optimum, 7.0–7.5), and with 1–4% NaCl (optimum, 2–3%). H2S is produced. Positive for hydrolysis of gelatin, DNA, laminarin, starch, and xylan. Galactose, glucose, xylose, maltose, lactose, sucrose, and starch are fermented to produce acetic acid and propionic acid as the major products. The major fatty acid components are iso-C15:0, anteiso-C15:0, iso-C15:0 3-OH, and iso-C17:0 3-OH. Menaquinone 7 (MK-7) is the major quinone. The polar lipid profile comprises phosphatidylethanolamine (PE) and three unidentified aminophospholipids (APL1–3).
The type strain DS1-an-2312T (= KCTC 25032T = DSM 115328T) was isolated from a sea squirt at a depth 18 m under the surface of sea water. The genome contains one circular chromosome that is 5.19 Mb long. The G + C content is 36.52%, as calculated from whole-genome sequencing.
Data availability
The DNA sequences generated from the study are available at the National Center for Biotechnology Information (NCBI). GenBank/EMBL/DDBJ accession numbers of 16S rRNA gene sequences of the strains DS1-an-13321T and DS1-an-2312T are MZ851973 and MZ851974, respectively, and the genome sequences are CP081303 and CP082230, respectively.
References
Arnosti, C. et al. The biogeochemistry of marine polysaccharides: sources, inventories, and bacterial drivers of the carbohydrate cycle. Ann. Rev. Mar. Sci. 13, 81–108 (2021).
Krause-Jensen, D. & Duarte, C. M. Substantial role of macroalgae in marine carbon sequestration. Nat. Geosci. 9, 737–742 (2016).
Bäumgen, M. et al. A new carbohydrate-active oligosaccharide dehydratase is involved in the degradation of ulvan. J. Biol. Chem. 279, 101210 (2021).
Fernández-Gómez, B. et al. Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7, 1026–1037 (2013).
Lau, N. S., Matsui, M. & Abdullah, A. A. A. Cyanobacteria: photoautotrophic microbial factories for the sustainable synthesis of industrial products. BioMed Res. Int. 2015, 754934 (2015).
Schada von Borzyskowski, L. et al. Marine Proteobacteria metabolize glycolate via the β-hydroxyaspartate cycle. Nature. 575, 500–504 (2019).
Krüger, K. et al. In marine Bacteroidetes the bulk of glycan degradation during algae blooms is mediated by few clades using a restricted set of genes. ISME J. 13, 2800–2816 (2019).
Sidhu, C. et al. Dissolved storage glycans shaped the community composition of abundant bacterioplankton clades during a North Sea spring phytoplankton bloom. Microbiome. 11, 77 (2023).
Foley, M. H., Cockburn, D. W. & Koropatkin, N. M. The Sus operon: a model system for starch uptake by the human gut Bacteroidetes. Cell. Mol. Life Sci. 73, 2603–2617 (2016).
Martens, E. C., Koropatkin, N. M., Smith, T. J. & Gordon, J. I. Complex glycan catabolism by the human gut microbiota: the Bacteroidetes sus-like paradigm. J. Biol. Chem. 284, 24673–24677 (2009).
McKee, L. S. et al. Polysaccharide degradation by the Bacteroidetes: mechanisms and nomenclature. Environ. Microbiol. Rep. 13, 559–581 (2021).
Noel, R. & Whitman, W. B. Phylum XIV. Bacteroidetes phyl. nov. In Bergey’s Manual of Systematic Bacteriology: Volume 4: The Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes (ed. Krieg, N. R.) (Springer Science & Business Media, 2011).
Oren, A. & Garrity, G. M. Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 71, 005056 (2021).
Kappelmann, L. et al. Polysaccharide utilization loci of North Sea Flavobacteriia as basis for using SusC/D-protein expression for predicting major phytoplankton glycans. ISME J. 13, 76–91 (2019).
Lu, D. C., Wang, F. Q., Amann, R. I., Teeling, H. & Du, Z. J. Epiphytic common core bacteria in the microbiomes of co-located green (Ulva), brown (Saccharina) and red (Grateloupia, Gelidium) macroalgae. Microbiome 11, 126 (2023).
Xie, S. et al. Glycosyltransferase-related protein GtrA is essential for localization of type IX secretion system cargo protein cellulase Cel9A and affects cellulose degradation in Cytophaga hutchinsonii. Appl. Environ. Microbiol. 88, e0107622 (2022).
Zhao, D. et al. Identification of the type IX secretion system component, PorV (CHU_3238), involved in secretion and localization of proteins in Cytophaga hutchinsonii. Front. Microbiol. 12, 742673 (2021).
Gómez-Pereira, P. R. et al. Genomic content of uncultured Bacteroidetes from contrasting oceanic provinces in the North Atlantic Ocean. Environ. Microbiol. 14, 52–66 (2012).
Nguyen, T. T. H. et al. Three marine species of the genus Fulvivirga, rich sources of carbohydrate-active enzymes degrading alginate, chitin, laminarin, starch, and xylan. Sci. Rep. 13, 6301 (2023).
Wexler, A. G. & Goodman, A. L. An insider’s perspective: Bacteroides as a window into the microbiome. Nat. Microbiol. 2, 17026 (2017).
Shang, Q. et al. Gut microbiota fermentation of marine polysaccharides and its effects on intestinal ecology: an overview. Carbohydr. Polym. 179, 173–185 (2018).
Zhou, L. Y., Yu, Z. L., Xu, W., Mu, D. S. & Du, Z. J. Maribellus luteus gen. nov., sp. nov., a marine bacterium in the family Prolixibacteraceae isolated from coastal seawater. Int. J. Syst. Evol. Microbiol. 69, 2388–2394 (2019).
Naidu, D. S., Hlangothi, S. P. & John, M. J. Bio-based products from xylan: a review. Carbohydr. Polym. 179, 28–41 (2018).
Zhao, F. et al. A novel class of xylanases specifically degrade marine red algal β1,3/1,4-mixed-linkage xylan. J. Biol. Chem. 299, 105116 (2023).
Dong, C. D. et al. Bioprocess development for the production of xylooligosaccharide prebiotics from agro-industrial lignocellulosic waste. Heliyon. 9, e18316 (2023).
Mhetras, N., Mapre, V. & Gokhale, D. Xylooligosaccharides (XOS) as emerging prebiotics: its production from lignocellulosic material. Adv. Microbiol. 9, 14–20 (2019).
Quiñones, T. S. et al. Production of xylooligosaccharides from renewable agricultural lignocellulose biomass. Biofuels. 6, 147–155 (2015).
Yan, F. et al. Preparation and nutritional properties of xylooligosaccharide from agricultural and forestry byproducts: a comprehensive review. Front. Nutr. 9, 977548 (2022).
Nordberg Karlsson, E., Schmitz, E., Linares-Pastén, J. A. & Adlercreutz, P. Endo-xylanases as tools for production of substituted xylooligosaccharides with prebiotic properties. Appl. Microbiol. Biotechnol. 102, 9081–9088 (2018).
Lagaert, S., Pollet, A., Courtin, C. M. & Volckaert, G. β-xylosidases and α-L-arabinofuranosidases: accessory enzymes for arabinoxylan degradation. Biotechnol. Adv. 32, 316–332 (2014).
Dutschei, T. et al. Marine Bacteroidetes enzymatically digest xylans from terrestrial plants. Environ. Microbiol. 25, 1713–1727 (2023).
Sun, H. N. et al. Diversity of marine 1,3-xylan-utilizing bacteria and characters of their extracellular 1,3-xylanases. Front. Microbiol. 12, 721422 (2021).
Sterner, M. & Gröndahl, F. Extraction of laminarin from Saccharina latissima seaweed using cross-flow filtration. J. Appl. Phycol. 33, 1825–1844 (2021).
Chen, J. et al. Laminarin, a major polysaccharide in stramenopiles. Mar. Drugs. 19, 576 (2021).
Becker, S. et al. Laminarin is a major molecule in the marine carbon cycle. Proc. Natl. Acad. Sci. U S A. 117, 6599–6607 (2020).
Huang, Y., Jiang, H., Mao, X. & Ci, F. Laminarin and laminarin oligosaccharides originating from brown algae: preparation, biological activities, and potential applications. J. Ocean. U China. 20, 641–653 (2021).
Watanabe, M., Kojima, H. & Fukui, M. Aquipluma nitroreducens gen. nov., sp. nov., a novel facultatively anaerobic bacterium isolated from a freshwater lake. Int. J. Syst. Evol. Microbiol. 70, 6408–6413 (2020).
Du, J. et al. Draconibacterium sediminis sp. nov., isolated from river sediment. Int. J. Syst. Evol. Microbiol. 65, 2310–2314 (2015).
Huang, X. F. et al. Mangrovibact diazotrophicumphicum gen. nov., sp. nov., a nitrogen-fixing bacterium isolated from a mangrove sediment, and proposal of Prolixibacteraceae fam. Nov Int. J. Syst. Evol. Microbiol. 64, 875–881 (2014).
Du, Z. J., Wang, Y., Dunlap, C., Rooney, A. P. & Chen, G. J. DraconibactOrientaleentale gen. nov., sp. nov., isolated from two distinct marine environments, and proposal of Draconibacteriaceae fam. Nov Int. J. Syst. Evol. Microbiol. 64, 1690–1696 (2014).
Iino, T., Sakamoto, M. & Ohkuma, M. Prolixibacter denitrificans sp. nov., an iron-corroding, facultatively aerobic, nitrate-reducing bacterium isolated from crude oil, and emended descriptions of the genus Prolixibacter and Prolixibacter bellariivorans. Int. J. Syst. Evol. Microbiol. 65, 2865–2869 (2015).
Iino, T. et al. Description of Mariniphaga anaerophila gen. nov., sp. nov., a facultatively aerobic marine bacterium isolated from tidal flat sediment, reclassification of the Draconibacteriaceae as a later heterotypic synonym of the Prolixibacteraceae and description of the family Marinifilaceae fam. Nov Int. J. Syst. Evol. Microbiol. 64, 3660–3667 (2014).
Sun, W. et al. Puteibacter caeruleilacunae gen. nov., sp. nov., a facultatively anaerobic bacterium isolated from Yongle Blue hole in the South Seana sea. Int. J. Syst. Evol. Microbiol. 70, 1623–1629 (2020).
Iizuka, T., Jojima, Y., Fudou, R. & Yamanaka, S. Isolation of myxobacteria from the marine environment. FEMS Microbiol. Lett. 169, 317–322 (1998).
Lee, H. et al. Flavisolibacter carri sp. nov., isolated from an automotive air-conditioning system. Antonie Van Leeuwenhoek. 111, 1969–1976 (2018).
Wolin, E., Wolin, M. J. & Wolfe, R. Formation of methane by bacterial extracts. J. Biol. Chem. 238, 2882–2886 (1963).
Spring, S. et al. Sulfate-reducing bacteria that produce exopolymers thrive in the calcifying zone of a hypersaline cyanobacterial mat. Front. Microbiol. 10, 441572 (2019).
Nelson, P. SW-846 test method 9038: sulfate (turbidimetric). In Test Methods for Evaluating Solid Waste, Physical/chemical Methods, 1–6 (USEPA, 1986).
Deshavath, N. N., Mukherjee, G., Goud, V. V., Veeranki, V. D. & Sastri, C. V. Pitfalls in the 3, 5-dinitrosalicylic acid (DNS) assay for the reducing sugars: interference of furfural and 5-hydroxymethylfurfural. Int. J. Biol. Macromol. 156, 180–185 (2020).
Ryu, S. W. et al. Gut microbiota Eubacterium callanderi exerts anti-colorectal cancer activity. Microbiol. Spectr. 10, e0253122 (2022).
Komagata, K. & Suzuki, K. I. Lipid and cell-wall analysis in bacterial systematics. Method Microbiol. 19, 161–207 (1988).
Natarajan, V. P., Zhang, X., Morono, Y., Inagaki, F. & Wang, F. A modified SDS-based DNA extraction method for high quality environmental DNA from seafloor environments. Front. Microbiol. 7, 195544 (2016).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S. & Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek. 110, 1281–1286 (2017).
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P. & Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14, 1–14 (2013).
Rodriguez-R, L. M. & Konstantinidis, K. T. Bypassing cultivation to identify bacterial species. Microbe Mag. 9, 111–118 (2014).
Qin, Q. L. et al. A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 196, 2210–2215 (2014).
Graham, E., Heidelberg, J. & Tully, B. Potential for primary productivity in a globally-distributed bacterial phototroph. ISME J. 12, 1861–1866 (2018).
Galperin, M. Y. et al. COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 49, D274–D281 (2021).
Seth Carbon et al. The gene ontology resource: enriching a gold mine. Nucleic Acids Res. 49, D325–D334 (2021).
Cantarel, B. L. et al. The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238 (2009).
Zheng, J. et al. dbCAN3: automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 51, W115–W121 (2023).
Blin, K. et al. antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 51, W46–W50 (2023).
Pascal Andreu, V., Roel-Touris, J., Dodd, D., Fischbach, M. A. & Medema, M. H. The gutSMASH web server: automated identification of primary metabolic gene clusters from the gut microbiota. Nucleic Acids Res. 49, W263–W270 (2021).
Arndt, D. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21 (2016).
Couvin, D. et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251 (2018).
Yu, N. Y. et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 26, 1608–1615 (2010).
Lee, K. C. et al. Purification and characterization of a thermostable laminarinase from Penicillium Rolfsii c3-2 (1) IBRL. BioResources. 9, 1072–1084 (2014).
Takai, K. et al. Sunxiuqinia faeciviva sp. nov., a facultatively anaerobic organoheterotroph of the Bacteroidetes isolated from deep subseafloor sediment. Int. J. Syst. Evol. Microbiol. 63, 1602–1609 (2013).
Na, S. I. et al. Up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 56, 281–285 (2018).
Luo, C., Rodriguez-r, L. M. & Konstantinidis, K. T. MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res. 42, e73–e73 (2014).
Holmes, D. E., Nevin, K. P., Woodard, T. L., Peacock, A. D. & Lovley, D. R. Prolixibacter bellariivorans gen. nov., sp. nov., a sugar-fermenting, psychrotolerant anaerobe of the phylum Bacteroidetes, isolated from a marine-sediment fuel cell. Int. J. Syst. Evol. Microbiol. 57, 701–707 (2007).
Yu, W. X., Liang, Q. Y., Xuan, X. Q., Du, Z. J. & Mu, D. S. Gaoshiqia sediminis gen. nov., sp. nov., isolated from coastal sediment. Int. J. Syst. Evol. Microbiol. 73, 005855 (2023).
Qu, L. et al. Sunxiuqinia elliptica gen. nov., sp. nov., a member of the phylum Bacteroidetes isolated from sediment in a sea cucumber farm. Int. J. Syst. Evol. Microbiol. 61, 2885–2889 (2011).
Zhu, G. et al. Quantifying the seawater sulfate concentration in the Cambrian Ocean. Front. Earth Sci. 9, 767857 (2021).
Cuskin, F. et al. Human gut Bacteroidetes can utilize yeast mannan through a selfish mechanism. Nature. 517, 165–169 (2015).
Sun, C., Zeng, X., Lai, Q., Wang, Z. & Shao, Z. Mangrovibacterium lignilyticum sp. nov., a facultatively anaerobic lignin-degrading bacterium isolated from mangrove sediment. Int. J. Syst. Evol. Microbiol. 70, 4502–4507 (2020).
Liu, Q. Q., Li, X. L., Rooney, A. P., Du, Z. J. & Chen, G. J. Tangfeifania diversioriginum gen. nov., sp. nov., a representative of the family Draconibacteriaceae. Int. J. Syst. Evol. MicroBiol. 64, 3473–3477 (2014).
Li, J., Qi, M., Lai, Q., Wang, G. & Shao, Z. Sunxiuqinia indica sp. nov., isolated from deep sea. Int. J. Syst. Evol. Microbiol. 70, 4186–4192 (2020).
Tang, K., Lin, Y., Han, Y. & Jiao, N. Characterization of potential polysaccharide utilization systems in the marine Bacteroidetes Gramella flava JLT2011 using a multi-omics approach. Front. Microbiol. 8, 220 (2017).
Gálvez, E. J. et al. Distinct polysaccharide utilization determines interspecies competition between intestinal Prevotella spp. Cell. Host Microbe. 28, 838–852 (2020).
Kabisch, A. et al. Functional characterization of polysaccharide utilization loci in the marine Bacteroidetes ‘Gramella forsetii’ KT0803. ISME J. 8, 1492–1502 (2014).
Unfried, F. et al. Adaptive mechanisms that provide competitive advantages to marine Bacteroidetes during microalgal blooms. ISME J. 12, 2894–2906 (2018).
Singh, R. P., Rajarammohan, S., Thakur, R. & Hassan, M. Linear and branched β-glucans degrading enzymes from versatile Bacteroides uniformis JCM 13288T and their roles in cooperation with gut bacteria. Gut Microbes. 12, 1826761 (2020).
Déjean, G. et al. Synergy between cell surface glycosidases and glycan-binding proteins dictates the utilization of specific beta (1, 3)-glucans by human gut Bacteroides. mBio. 11, e00095–e00020 (2020).
Domingues, R. et al. Xylose metabolism in bacteria—opportunities and challenges towards efficient lignocellulosic biomass-based biorefineries. Appl. Sci. 11, 8112 (2021).
Khankal, R., Chin, J. W. & Cirino, P. C. Role of xylose transporters in xylitol production from engineered Escherichia coli. J. Biotechnol. 134, 246–252 (2008).
Van Maris, A. J. A. et al. Development of efficient xylose fermentation in Saccharomyces cerevisiae: Xylose isomerase as a key component. Adv. Biochem. Engin /Biotechnol. 108, 179–204 (2007).
Li, J. et al. Molecular mechanism of environmental D-xylose perception by a XylFII-LytS complex in bacteria. Proc. Natl. Acad. Sci. U S A. 114, 8235–8240 (2017).
Desai, T. A. & Rao, C. V. Regulation of arabinose and xylose metabolism in Escherichia coli. Appl. Environ. Microbiol. 76, 1524–1532 (2010).
Gu, Y., Jiang, Y., Yang, S. & Jiang, W. Utilization of economical substrate-derived carbohydrates by solventogenic clostridia: pathway dissection, regulation and engineering. Curr. Opin. Biotechnol. 29, 124–131 (2014).
Liu, L. et al. Phosphoketolase pathway for xylose catabolism in Clostridium acetobutylicum revealed by 13 C metabolic flux analysis. J. Bacteriol. 194, 5413–5422 (2012).
Tanaka, K. et al. Two different pathways for D-xylose metabolism and the effect of xylose concentration on the yield coefficient of L-lactate in mixed-acid fermentation by the lactic acid bacterium Lactococcus lactis IO-1. Appl. Microbiol. Biotechnol. 60, 160–167 (2002).
Acknowledgements
The authors thank Prof. Dr. Bernhard Schink from the University of Konstanz (Germany) and Prof. Dr. Aharon Oren from Edmond J. Safra Campus, The Hebrew University of Jerusalem (Israel), for their help with the nomenclature of the new genus and two novel species names. The authors also thank Mrs. Mi-Kyung Eom and Dr. Li Zhun at KCTC for maintaining the two novel isolates and for providing the SEM images, respectively. This research was supported by The Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program (KGM5232423) and a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. NRF-2021M3H9A1030164).
Author information
Authors and Affiliations
Contributions
T.T.H.N. performed experiments of isolation, identification, and characterization of the bacterial strains including anaerobic metabolism, polysaccharide degradation, and genome analysis. T.T.H.N also wrote the manuscript. T.Q.V. analyzed genomes, constructed a UBGC genome tree, determined POCP indices, and drew the xylan degradation pathway. H.L.H carried out the genome analysis for xylose utilization and proposed the schematic model of xylose utilization, and wrote the paragraphs regarding xylose utilization. S-G.K. supervised the experiments and finalized the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nguyen, T.T.H., Vuong, T.Q., Han, H.L. et al. Halosquirtibacter laminarini gen. nov., sp. nov. and Halosquirtibacter xylanolyticus sp. nov., marine anaerobic laminarin and xylan degraders in the phylum Bacteroidota. Sci Rep 14, 24329 (2024). https://doi.org/10.1038/s41598-024-74787-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-74787-6
