
Abstract
Non-syndromic cleft lip with/without cleft palate (nsCL/P) is one of the most common birth defects and has a multifactorial etiology. To date, over 45 loci harboring common risk variants have been identified. However, the effector genes at these loci, and the cell types that are affected by risk alleles, remain largely unknown. To address this, we combined genetic data from an nsCL/P genome-wide association study (GWAS) with single-cell RNA sequencing data obtained from the heads of unaffected human embryos. Using the recently developed single-cell disease relevance score (scDRS) approach, we identified two major cell types involved in nsCL/P development, namely the epithelium and the HAND2+ pharyngeal arches (PA). Combining scDRS with co-expression networks and differential gene expression analysis, we prioritized nsCL/P candidate genes, some of which were additionally supported by GWAS data (e.g., CTNND1, PRTG, RPL35A, RAB11FIP1, KRT19). Our results suggest that specific epithelial and PA sub-cell types are involved in nsCL/P development, and harbor a substantial fraction of the genetic risk for nsCL/P.
Similar content being viewed by others

Introduction
Non-syndromic cleft lip with or without cleft palate (nsCL/P) is one of the most common birth defects, with a global prevalence of approximately 1 in 1,000 live births1. In addition to complex treatments such as surgery and speech therapy, affected patients are burdened by an increased risk of morbidity2. The etiology of nsCL/P involves both genetic and environmental factors1. To date, genome-wide association studies (GWAS) have identified more than 45 risk loci harboring common variants that are associated with increased nsCL/P risk3. At some loci, candidate genes have been pinpointed by evidence from syndromic forms of facial disorders, the presence of rare variants in affected individuals, or on the basis of results from animal models4. However, how the human risk variants affect the function of nsCL/P candidate genes and the cell types in which they are likely to act, remains largely unclear in most cases, although some initial work has been published5,6,7.
To address these questions, single-cell RNA-sequencing (scRNA-seq) is a promising approach. Instead of analyzing gene expression profiles in bulk from whole tissues, scRNA-seq enables the investigation of gene expression profiles in specific individual cells. When applied to biomaterials of relevance to specific diseases, this allows both the generation of high-resolution transcription maps of cell-types, and the identification of sub-cell types that might contribute to disease pathogenesis.
In the context of craniofacial development, most scRNA-seq studies to date have been performed on murine tissues and have identified cell types that would have been missed in earlier analyses of bulk data. For example, one study identified heterogeneity in gene expression of mesenchymal cells in the anterior palate8, while another found distinct cell populations at the fusion sites of the maxillary, medial-nasal, and lateral-nasal processes9 in mice. To study the role of these cell types in nsCL/P, we and others have utilized these murine single cell expression maps to examine gene expression patterns of candidate genes identified in genetic studies10,11. However, the suitability of murine data for the investigation of nsCL/P is limited. Reasons for this include: (i) differences in morphology and tissue interactions between mice and humans, in particular during the later stages of facial development12; and (ii) the fact that in humans, most genetic nsCL/P associations are located in non-coding (and often non-conserved) regions of the genome, indicating higher-order regulatory mechanisms4. Recently, scRNA-seq data from unaffected human embryos aged four to six weeks were made available13, which partly cover the crucial time period for nsCL/P development between the fourth and tenth week post-conception14.
The joint study of genetic and transcriptomic data has the potential to identify affected cell types and improve the understanding of disease mechanisms during facial development. To date, few computational approaches that combine genetic and single-cell transcriptomic data have been available. However, the recently developed single-cell disease relevance score (scDRS)15 now allows the identification of associations between candidate genes identified via GWAS and individual cells from scRNA-seq data. The aim of the present study was to identify human developmental cell types in which genetically-mediated nsCL/P risk is enriched, which is crucial in terms of unraveling the underlying molecular mechanisms of nsCL/P. For this purpose, the scDRS approach was used to combine scRNA-seq data of unaffected human embryos13 with candidate genes derived from our recent GWAS on nsCL/P3. The identified cell types were then used to determine potential interactions between candidate genes in co-expression networks using high-dimensional weighted correlation network analysis (hdWGCNA)16. We demonstrate how these approaches can facilitate the identification of molecular networks, effector cell-types, and novel candidate genes, thus advancing our understanding of the molecular basis of genetic nsCL/P risk.
Methods
Human embryonic scRNA-seq data
Human embryonic scRNA-seq data13 were downloaded from Gene Expression Omnibus (GSE157329, see data availability section). These data comprised scRNA-seq data from seven unaffected whole human embryos from Carnegie stage (CS) 12 (one embryo), CS 13–14 (three embryos), and CS 15–16 (three embryos), which had been broadly dissected into head, upper and lower trunk, limbs, and viscera. Based on meta-information provided by the authors, the data were reduced to dissection parts ‘head’ and ‘head-upperTrunk’. This led to the exclusion of two embryos without head data, i.e., one embryo respectively from CS 13–14 and CS 15–16. No additional filtering was performed. These data were then re-analyzed using Seurat v4.3.017. Details on analysis parameters are provided in the Supplementary Information. Briefly, to remove potential batch effects, data from different samples were integrated using canonical correlation analysis, as implemented in Seurat. For this purpose, the data were split according to sample (n = 6, including one donor head that was split and analyzed as two samples), and processed as individual Seurat objects prior to integration.
The data were normalized and 2,000 highly variable genes were identified before the data were scaled. For the integration of individual Seurat objects, integration anchors between objects were identified and then used to integrate the individual data sets into one data set. The resulting data set was scaled and cell cycle regression was performed as implemented in Seurat. Principal component analysis was performed using the variable features of the data. Clustering was performed by first identifying the shared nearest neighbors of cells and then clustering the cells using the original Louvain algorithm. The resulting data set contained 50,059 cells, which clustered into 25 cell clusters (between 276 and 4,993 cells per cluster, Fig. 1A). The clustering showed no influences attributable to sample batch effects (Fig. S1A). Cluster marker genes were determined (Table S1) and used for cell type annotation of the clusters, as based on the cell type marker genes from the original publication (Supplementary Table S1B from Xu et al. 2023). Identification of differentially expressed genes (DEGs) was performed for epithelial sub-clusters only (Table S2).

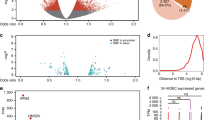
scDRS identifies significant association with nsCL/P candidate genes in epithelium and HAND2+ pharyngeal arches (a) UMAP plot of scRNA-seq data from the heads of five unaffected human embryos from Carnegie stages 12–16. (b) UMAP plot from a colored according to the normalized scDRS for nsCL/P association at the single-cell level in the unweighted setting. (c) Ridgeplot of normalized scDRS according to cell type in the unweighted setting. (d) scDRS disease association at the cell type level in the unweighted setting. Cell types above the dashed line showed significant association with the nsCL/P gene set. Bold cell type labels indicate significant within-cell type heterogeneity in terms of disease association. Anterior presomitic mesoderm (aPSM), frontonasal mesenchyme (FM), log2 fold change (log2FC), pharyngeal arches (PA), posterior presomitic mesoderm (pPSM), sympathetic neurons (SN), single-cell disease relevance score (scDRS).
Identification of nsCL/P candidate cell types using scDRS
Preparation of the scRNA-seq data
To render the scRNA-seq data applicable for scDRS, the scaled data was removed and the Seurat object (containing only count and normalized data) was converted into an .h5ad file. Specific parameters are provided in the Supplementary Information.
Definition of nsCL/P gene set
For the preparation of the genetically-informed gene set, the candidate genes located in topologically associating domains (TADs) of nsCL/P GWAS risk loci were retrieved from Table S9 from Welzenbach et al. 20213. Of the 404 genes located in these TADs, 51 were not detected in the scRNA-seq data. The remaining 353 genes are referred to as ‘TAD genes’. Of these, 87 genes had reached significance in the gene-based test in the original publication, thus providing further genetic support for these genes beyond single-variation association statistics at the risk loci. These genes were used for scDRS analysis, and are referred to as ‘nsCL/P gene set’. For all of these genes, z-scores were retrieved from the MAGMA output file from Welzenbach et al. 20213, and were used as weights in the “weighted setting”.
Application of the scDRS method
Following download (GitHub) and installation of the scDRS package15, scDRSs for all single cells in the scRNA-seq data (n = 50,059) were calculated. For the nsCL/P gene set, this was performed in both settings, i.e., “unweighted” and “weighted”. Downstream analyses were conducted on the scores generated from each gene set. Specific analysis parameters are provided in the Supplementary Information.
Additional quality assessment
To identify potential artifacts, the entire scRNA-seq data set was used. Here, Pearson correlation between the scDRS of each cell and the total number of detected molecules of each cell was calculated.
Co-expression network analysis using hdWGCNA
To generate co-expression networks, the hdWGCNA package version 0.3.1 was used (see section Data availability). First, metacells were created from the single cell matrix, which were then normalized. Next, for both the epithelium and the HAND2+ PA, an expression matrix of the respective metacells was constructed. The soft power thresholds were determined, the co-expression networks were constructed and the matrices were scaled. Then, module eigengenes and eigengene-based connectivity were determined. Specific analysis parameters are provided in the Supplementary Information. For each gene module, the percentage of TAD genes was calculated, and the three top gene modules, i.e., those with the highest percentage of TAD genes, were selected for further analysis. The Circos plots for the co-expression gene modules were created using the R package circlize18.
Gene ontology enrichment analysis
A gene ontology (GO) analysis was performed using the clusterProfiler19 and org.Hs.eg.db20 R packages to identify biological process and molecular function GO terms. For E-9 and PA-14, redundant GO terms were removed (simplify function). Specific analysis parameters are provided in the Supplementary Information.
Identification of new candidate genes using hdWGCNA gene modules and GWAS data
For genes that were identified in the hdWGCNA gene modules, p-values from the gene-based test in our recent GWAS were retrieved from Table S6 from Welzenbach et al. 20213. FDR correction was performed using the Benjamini Hochberg method. For those genes that remained significant (adjusted p-value < 0.05) and were located outside of nsCL/P GWAS TADs, LocusZoom21 was used to create regional association plots from the GWAS summary statistics3. To assess whether the co-expression gene modules were enriched for genes with a genetic association to nsCL/P, a gene set analysis was performed using MAGMA v1.1022 (see section Data availability). The gene sets were created from the six selected co-expression modules (E-9, E-10, E11, PA-12, PA-14, PA-15). The analysis was performed using these gene sets and the MAGMA genes.raw output file from Welzenbach et al. 20213.
Results
Expression patterns of GWAS candidate genes implicate head epithelium and HAND2+ pharyngeal arches in genetically-mediated nsCL/P
Based on scRNA-seq data from the heads of unaffected human embryos (Fig. 1A) and the nsCL/P gene set informed by GWAS results (see Methods), developmental cell types that might underlie nsCL/P etiology were identified using scDRS.
First, scDRSs were calculated for each single cell in the scRNA-seq data (n = 50,059) in two settings, i.e., unweighted and weighted (by MAGMA z-scores, see Methods). At the single-cell level and across the two settings, an accumulation of cells with high scDRS was observed in the epithelium (Fig. 1B,C, Fig. S1B). When combining the scDRSs of individual cells over cell-clusters, this accumulation in the epithelium was found to be statistically significant in both the unweighted (p = 0.002, Fig. 1D, Table S3) and the weighted setting (p = 0.01, Fig. S1C, Table S4). In addition, the cell type HAND2+ PA reached statistical significance in the unweighted analysis (p = 0.04; Fig. 1D, Table S3). To identify potential subpopulations of disease-associated cells, all cell types were tested for evidence of within-cell type heterogeneity. In the unweighted setting, significant within-cell type heterogeneity was observed in the epithelium, as well as in sympathetic and GABAergic neurons (Fig. 1C). In the weighted setting, significant within-cell type heterogeneity was observed in the dorsal telencephalon, the sympathetic neurons, the endothelium, and the GABAergic neurons (Fig. S1C).To ensure that the scDRS for each cell and thereby the heterogeneity in association was not caused by technical differences in transcript detection, the correlation between the scDRS of each cell and the total number of molecules detected was tested. No strong support for such a technical bias was found (Pearson correlation coefficient: 0.006).
Given the converging evidence for a role in nsCL/P, and potential heterogeneity within the epithelial cell cluster, this cell cluster was then subdivided into two subclusters, as based on the scDRS p-value of each cell. This resulted in the identification of an associated subcluster (435 cells, p ≤ 0.01) and a non-associated subcluster (717 cells, p ≥ 0.1), from a total of 1,835 cells. DEGs between these subclusters were then identified. A total of 139 DEGs showed higher expression in the subcluster of disease-associated cells (fold change > 1) compared to the subcluster containing non-associated cells (fold change < -1; Fig. 2; Table S2). Of these, 31 genes were among the 353 ‘TAD genes’, and 25 of these 31 genes were among the 87 genes of the ‘nsCL/P gene set’ of previously suggested effector genes (e.g., KRT8, KRT18, TFAP2A, TPM1, ESRP1, and IRF6; Fig. 2; Table S2). Based on these results, we prioritized the remaining six TAD genes as nsCL/P candidate genes (Table 1, ‘DEG’ approach). Notably, for some of them, evidence of an involvement in orofacial clefting phenotypes has already been presented, though not from GWAS data23,24,25.

Marker genes of associated epithelial cells contain known nsCL/P candidate genes. Volcano plot showing differentially expressed genes between nsCL/P associated (scDRS p-value < 0.01) and non-associated (scDRS p-value > 0.1) epithelial cells. Numbers within each group are depicted in the integrated bar plot. Genes with adjusted p-values < 0.05 (dashed horizontal line) and log2FC > 0.1 were considered marker genes for nsCL/P associated cells (red). Genes with adjusted p-values < 0.05 and log2FC < -0.1 were considered marker genes for non-associated cells (blue). Top 10 genes with the lowest p-values are labeled. Non-syndromic cleft lip with/without cleft palate (nsCL/P), log2 fold change (log2 FC), single-cell disease relevance score (scDRS).
scDRS-informed prioritization of candidate genes at GWAS loci
In addition to confirming known nsCL/P risk genes, scDRS within associated cell types can also be used to identify potential novel candidate genes. For this purpose, we used the gene-level downstream application of scDRS, which correlates the scDRS with the expression of genes that are not part of the tested gene set. Specifically, we aimed to identify genes at GWAS loci that were not prioritized as candidate genes due to the presence of another promising gene, or due to the lack of a significant gene-based P-value in the genetic data3. In this analysis, we considered genes with a correlation coefficient of > 0.01. In the unweighted setting, a positive correlation was observed between gene expression and scDRS for 33 genes (Table 1 ‘scDRS TAD gene’ approach, Table S5), with the highest absolute value being observed for RPL35A (Pearson correlation coefficient: 0.15). This gene is located at the 3q29 locus, which harbors the previously proposed candidate genes DLG1 and MELTF. The TAD genes GADD45B (19p13.3), ARHGAP29 (1p22), and MSX1 (4p16.2) showed a positive, albeit less pronounced correlation (Pearson correlation coefficients between 0.048 and 0.055). Our findings provide additional support for prioritizing these as effector genes at their respective loci. Similar results were found in the weighted setting (Table S6).
Co-expression network analysis of epithelium and HAND2+ pharyngeal arches
To identify genes with potential interactions in the previously identified nsCL/P-associated cell types (epithelium and HAND2+ PA), co-expression networks were generated. For each cell type, hdWGCNA identified 18 groups of interconnected, positively correlated genes (so-called ‘gene modules’). Of these, three per cell type were selected for further analysis (see Methods, Tables S7 & S8).
Of the epithelial gene modules, the following were selected: E-9 (348 genes / including 12 nsCL/P TAD genes); E-10 (73/3); and E-11 (201/8) (Fig. 3A, Table S7). The eigengene values within the epithelial cluster were then plotted in their UMAP space of scRNA-seq data (Fig. 3C). While for E-9 and E-11, these appeared to be restricted to the upper part and lower part of the UMAP plot respectively, the highest values for E-10 did not appear specific. For E-9, the hub genes (i.e., genes with the largest number of connections within the module’s network) included the nsCL/P TAD genes TFAP2A, TPM1, and ARHGAP29, thus providing further support for the hypothesis that they play a causal role in nsCL/P at their respective loci (Table S9)24,26. Enrichment analyses for E-9 using GO terms identified odontogenesis, wound healing, actin filament organization, the canonical Wnt signaling pathway, and Cadherin binding (Fig. 3D, Table S10). In contrast to E-9, the hub genes of E-10 and E-11 contained no nsCL/P TAD genes, and the GO term analysis results were non-specific (pituitary gland development and central nervous system neuron differentiation for E-10; regulation of neuron differentiation, ribosome binding, and unfolded protein binding for E-11; Table S10). Together, this suggests that the most relevant epithelial gene module in terms of nsCL/P risk may be E-9.

Co-expression gene modules of epithelium and HAND2+ PA. (a) Circos plot of nsCL/P genes in epithelial co-expression gene modules. The outer track shows the chromosomal cytoband, the inner track shows the positions of TADs described in Welzenbach et al. 20213. The colors of the connecting lines correspond to the respective gene module. The strength of the connecting lines reflects the pairwise correlation coefficient between two genes multiplied by a factor of 30 for illustration purposes. (b) Circos plot of nsCL/P genes (black) and potential novel candidate genes (gray) in HAND2+ PA co-expression gene modules. Panel layout as described in a. (c) UMAP plots of epithelium (E-9, E-10, E-11) and HAND2+ PA (PA-12, PA-14, PA-15) colored according to the module eigengene values for each co-expression gene module. (d) Bar plots of selected GO terms for biological process (E-9 & PA-14) and molecular function (E-9) for the epithelial co-expression gene module E-9 and the HAND2+ PA co-expression gene module PA-14. The vertical dashed line is set at p-value -log10 of 0.05. (e) Venn diagram of gene overlap between epithelial gene module E-9 and HAND2+ PA gene modules PA-12, PA-14, and PA-15. Epithelium (E), gene ontology (GO), non-syndromic cleft lip with/without cleft palate (nsCL/P), pharyngeal arches (PA), topologically associating domains (TADs).
For HAND2+ PA, the selected gene modules were: PA-12 (111 genes / including 5 nsCL/P TAD genes), PA-14 (145/7), and PA-15 (83/4; Fig. 3B, Table S8). The highest module eigengene values for PA-12 and PA-15 were distributed evenly over the cluster, while the highest values for PA-14 were more concentrated at the bottom of the UMAP space (Fig. 3C). For PA-12, the identified hub genes included MRC2, which is located at the nsCL/P risk locus 17q23.2 but showed no significant gene-based association in Welzenbach et al. 20213. Notably, while a set of seven genes is located at this locus, none has garnered sufficient research evidence to date to be considered the effector gene. Therefore, our results now prioritize MRC2 as a candidate gene for functional studies. The hub genes of PA-14 contained the nsCL/P candidate genes TPM1 and ZFP36L2, while those in PA-15 included the nsCL/P candidate gene KRT18 (Table S11). The GO term analysis generated no significantly enriched terms for PA-12 or PA-15 (Table S10). However, for PA-14, the GO terms included T-cell differentiation and the development of the roof of the mouth, tongue, muscle tissue, arteries, and mesenchyme (Fig. 3D, Table S10).
When comparing the three epithelial gene modules and the three HAND2+ PA gene modules, a limited overlap of between 1 and 11 genes was observed (Fig. 3E, Fig. S2A/B, Table S12 e.g. ZFP36L2 and TPM1). This suggests that most genes located at GWAS loci act or interact in only one of the two cell types.
MAGMA gene set analysis identifies association between epithelial co-expression gene module and nsCL/P
To examine the joint association of genes within hdWGCNA-identified co-expression gene modules and nsCL/P, a MAGMA gene set analysis was performed using the six selected gene modules as individual gene sets (see above) and the nsCL/P GWAS summary statistics from Welzenbach et al. 20213. This analysis revealed a significant association with nsCL/P for gene module E-9 (p = 5.98 × 10− 5), and provides further evidence that this gene module is enriched with genes that are associated with nsCL/P (Table S13).
Identification of novel nsCL/P candidate genes
To identify potential novel nsCL/P candidate genes, an analysis was performed of genes that are located outside the GWAS-TADs and which represent plausible novel candidate genes given similarities in scDRS and co-expression networks. First, genes whose expression patterns are positively correlated (correlation coefficient > 0.01) with the scDRS of individual cells (see above), but which are located outside of any known GWAS locus, were identified. Here, KRT19, EPCAM, and the Claudin family members CLDN6, CLDN4, and CLDN7 were moderately correlated with the scDRS in the unweighted setting (Pearson correlation coefficients 0.17 to 0.29; Table 1 ‘scDRS gene’ approach, Table S5). None of these genes showed significance in the MAGMA gene-based test in Welzenbach et al. 20213. However, KRT19 and EPCAM were among the epithelial co-expression modules (E-9 and E-11, respectively). Similar results were obtained for the weighted setting (Table S6).
Second, genes that were listed among the selected hdWGCNA gene modules (E-9, E-10, E-11, PA-12, PA-14, and PA-15), and which were significant in the gene-based test, were examined. No new candidate genes were identified in any of the three epithelial gene modules. However, evidence was generated to suggest that HYAL2 and BFAR (both in PA-12), CTNND1 (PA-14), and PRTG (PA-15) represent novel nsCL/P candidate genes from the HAND2+ PA gene modules (Table 1 “hdWGCNA + GWAS’ approach, Table S8). An examination of the association structure around these genes in the GWAS summary statistics yielded nominally significant genetic support for the loci harboring CTNND1 and PRTG (Fig S2 C/D). This suggests that these loci might reach conservative thresholds for genetic associations of common variation in future studies involving increased power. Interestingly, previous studies already linked rare variants in CTNND127 and low-frequency coding variants in PRTG28 to nsCL/P.
Discussion
In recent years, multiple genetic studies on nsCL/P have identified genomic risk loci, and suggested local candidate genes in the associated regions3,25,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43. However, since most of the associated regions map to non-coding parts of the genome and can thus be presumed to have context-specific effects, biological interpretation of these discoveries requires the identification of the affected cell types. This knowledge would in turn inform the context in which functional studies should be performed, which is essential for understanding the molecular mechanisms of nsCL/P development. For some established candidate genes, expression patterns have already been reported, e.g., IRF6 expression in neural crest and epithelial cells6,44,45,46,47,48, and TFAP2A expression in facial mesenchyme, nervous system, epithelial, and neural crest cells49,50,51. In addition, in a previous study involving single-cell transcriptome analyses in mice10, our group showed that the murine homologs of certain nsCL/P candidate genes are expressed predominantly in either epithelial cell types (e.g., candidate genes IRF6, TFAP2A, ESRP1) or mesenchymal-like cell types (e.g., candidate genes ALX1, ALX3, GREM1). The present study complemented previous research by performing a systematic examination of the joint gene expression of nsCL/P candidate genes from GWAS, with the aim of detecting the human developmental cell types that mediate genetic nsCL/P risk.
Based on expression data from unaffected human embryonic heads, our scDRS analysis implicated the epithelium and HAND2+ PA as primary cell types with an involvement in genetic nsCL/P risk. This confirms, and further refines, observations from our previous study in mice, which showed that individual nsCL/P genes were expressed in epithelial and mesenchymal cell types10. Epithelial cells are involved in manifold processes during lip and palate formation. These processes include: (i) epithelial seam formation, which is required for the fusion processes of the upper lip and the palate, as well as those between the medial-nasal, the lateral nasal, and the maxillary prominences52,53,54,55; (ii) epithelial-to-mesenchymal transitions, which allow for movement of cells as facial prominences grow, as well as the removal of epithelial seams56,57,58; (iii) the formation of the periderm, which covers the developing epithelium59; and (iv) cell adhesion and migration60,61. We speculate that the heterogeneity we observed in the overall epithelial cell cluster might recapitulate different expression patterns associated with these different functions. Indeed, one of the top markers of the associated epithelial cells is IRF6, which is a particularly relevant gene in periderm formation, and has been shown to be crucial in the development of the palate62,63. However, we note that the data include the whole head and, therefore, the possibility remains that the within-cell type heterogeneity might also be caused by epithelial cells originating from other regions of the head, rather than the facial processes. The second major cell-type we identified were HAND2+ PA, one of three clusters that were annotated as PA, which give rise to the bones and connective tissue of the head64. The transcription factor HAND2, which characterized the specific PA cluster associated with nsCL/P, was previously found to be expressed in the neural-crest derived mesenchyme of the PA of mice65 and involved with patterning in the PA of zebrafish66. Together, the findings of specific cell types within PA and epithelium suggest that there may be specific sub-cell types that are involved in nsCL/P, which should be assessed with spatial and functional data in future studies.
Having identified relevant cell-clusters, the respective expression data can be explored using co-expression network analysis in order to identify genes that are potentially subject to the same gene regulation or which interact on a molecular level. This can help to prioritize effector genes at established genomic risk loci or identify new candidate genes. Importantly, our co-expression modules identified known interactions, such as IRF6 and TFAP2A, which have been shown to act jointly in a genetic pathway5,6, as well as IRF6 and TP63, the latter of which has been reported to activate IRF6 expression67. This suggests that some of the newly identified genes within the same gene modules are promising genes for further functional studies, for example, TPM1 and ZFP36L2, which occurred in the same gene module in the epithelial cells and in the HAND2+ PA. Interestingly, recent studies found that ZFP36L2 was significantly associated with nsCL/P and one of its subtypes, i.e., non-syndromic cleft lip only, in GWAS data from a Chinese Han population68,69. We also found evidence for a role of RPL35A, which is located in a larger deletion region in patients with craniofacial abnormalities70. Our data provide further support for the suspected genes GADD45B, ARHGAP29, and MSX1, though these had not been prioritized in Welzenbach et al. 20213. The present analyses also identified new candidate genes located outside of GWAS loci, such as CTNND1 and PRTG, which were implicated through their expression patterns in the HAND2+ PA gene modules. For both genes, genetic support is provided by our in-house GWAS data, but also through rare variants identified by exome sequencing of multiplex families for CTNND127 and low-frequency coding variants in PRTG28.
To obtain information on the biological relevance of the gene modules, we performed GO term analyses. The results reflect the before discussed processes the epithelium and PA are involved in, e.g. epithelial morphogenesis, mesenchyme development, and migration. Additionally, they support functional hypotheses such as cadherin-binding via CTNND1, involvement of several members of the Wnt-family, and muscle tissue development, all of which have been previously implicated in nsCL/P27,71,72,73,74,75,76,77,78. The association of one gene module with T-cell differentiation could provide an exciting link to immunological factors, which requires further examination. The differences in biological functions between the gene modules of epithelium and PA together with a very small gene overlap in genes between the gene modules, suggest that the genetic risk for nsCL/P is split on different biological, and maybe complementary, functions across those two cell types.
In summary, we combined human scRNA-seq data with genetic information on nsCL/P risk and identified nsCL/P-associated cell types and potential sub-cell types, which might harbor a considerable part of the genetic risk. Co-expression networks in these cell types allowed us to identify established and potential new gene-gene interactions. We also demonstrated how to identify new candidate genes based on these networks by revisiting the initial GWAS data.
Data availability
The original scRNA-seq data from Xu et al. are available via Gene Expression Omnibus accession number GSE157329 or via https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157329. Our re-analyzed scRNA-seq data have been deposited at Zenodo in Seurat object format (DOI: 10.5281/zenodo.12742819). hdWGCNA documentation: https://smorabit.github.io/hdWGCNA/index.html. MAGMA: https://cncr.nl/research/magma/.
References
Mangold, E., Ludwig, K. U. & Nöthen, M. M. Breakthroughs in the genetics of orofacial clefting. Trends Mol. Med. 17 (12), 725–733 (2011).
Christensen, K., Juel, K., Herskind, A. M. & Murray, J. C. Long term follow up study of survival associated with cleft lip and palate at birth. Br. Med. J. 328 (7453), 1405–1406 (2004).
Welzenbach, J. et al. Integrative approaches generate insights into the architecture of non-syndromic cleft lip with or without cleft palate. Hum. Genet. Genomics Adv. 2 (3), 1–14 (2021).
Thieme, F. & Ludwig, K. U. The role of noncoding genetic variation in isolated Orofacial Clefts. J. Dent. Res. 96 (11), 1238–1247 (2017).
Carroll, S. H. et al. An Irf6-Esrp1/2 regulatory axis controls midface morphogenesis in vertebrates. Dev, 147 (24) (2020).
Kousa, Y. A. et al. The TFAP2A-IRF6-GRHL3 genetic pathway is conserved in neurulation. Hum. Mol. Genet. 28 (10), 1726–1737 (2019).
Kousa, Y. A., Fuller, E. & Schutte, B. C. IRF6 and AP2A Interaction regulates epidermal development. J. Invest. Dermatol. 138 (12), 2578–2588 (2018).
Ozekin, Y. H., O’Rourke, R. & Bates, E. A. Single cell sequencing of the mouse anterior palate reveals mesenchymal heterogeneity. Dev. Dyn. 252 (6), 713–727 (2023).
Li, H., Jones, K. L., Hooper, J. E. & Williams, T. The molecular anatomy of mammalian upper lip and primary palate fusion at single cell resolution. Dev. 146 (12) (2019).
Siewert, A. et al. Analysis of candidate genes for cleft lip ± cleft palate using murine single-cell expression data. Front. Cell Dev. Biol., 11 (April), 1–11 (2023).
Cui, X. et al. Genetic variants in BCL-2 family genes influence the risk of non-syndromic cleft lip with or without cleft palate. Birth Defects Res. 116 (1), 1–12 (2024).
Yu, K., Deng, M., Naluai-Cecchini, T., Glass, I. A. & Cox, T. C. Differences in oral structure and tissue interactions during mouse vs. human palatogenesis: Implications for the translation of findings from mice. Front. Physiol. 8 (Mar), 1–12 (2017).
Xu, Y. et al. A single-cell transcriptome atlas profiles early organogenesis in human embryos. Nat. Cell. Biol. 1–12 (2023).
Dixon, M. J., Marazita, M. L., Beaty, T. H. & Murray, J. C. Cleft lip and palate: Understanding genetic and environmental influences. Nat. Rev. Genet. 12 (3), 167–178 (2011).
Zhang, M. J. et al. Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNA-seq data. Nat. Genet. (2022).
Morabito, S., Reese, F., Rahimzadeh, N. & Miyoshi, E. hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell. Rep. Methods. 3 (6), 100498 (2023).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell. 184 (13), 3573–3587.e29, (2021).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics. 30 (19), 2811–2812 (2014).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2 (3), 100141 (2021).
Carlson, M. org.Hs.eg.db: Genome wide annotation for Human. R package version 3.8.2. (2019).
Boughton, A. P. et al. LocusZoom.js: Interactive and embeddable visualization of genetic association study results. Bioinformatics. 37 (18), 3017–3018 (2021).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11 (4), 1–19 (2015).
Leslie, E. J. et al. Expression and mutation analyses implicate ARHGAP29 as the etiologic gene for the cleft lip with or without cleft palate locus identified by genome-wide association on chromosome 1p22. Birth Defects Res. Part. - Clin. Mol. Teratol. 94 (11), 934–942 (2012).
Savastano, C. P. et al. Impact of rare variants in ARHGAP29 to the etiology of oral clefts: Role of loss-of-function vs missense variants. Clin. Genet. 91 (5), 683–689 (2017).
Yu, Y. et al. Genome-wide analyses of non-syndromic cleft lip with palate identify 14 novel loci and genetic heterogeneity. Nat. Commun. 8 (2), 1–11 (2017).
Milunsky, J. M. et al. TFAP2A mutations result in branchio-oculo-facial syndrome. Am. J. Hum. Genet. 82 (5), 1171–1177 (2008).
Cox, L. L. et al. Mutations in the epithelial Cadherin-p120-Catenin complex cause mendelian non-syndromic cleft lip with or without cleft palate. Am. J. Hum. Genet. 102 (6), 1143–1157 (2018).
Leslie, E. J. et al. Association studies of low-frequency coding variants in nonsyndromic cleft lip with or without cleft palate. Am. J. Med. Genet. Part. A. 173 (6), 1531–1538 (2017).
Rahimov, F. et al. Disruption of an AP-2α binding site in an IRF6 enhancer is strongly associated with cleft lip. Nat. Genet. 40 (11), 1341–1347 (2008).
Ludwig, K. U. et al. Meta-analysis reveals genome-wide significance at 15q13 for nonsyndromic clefting of both the lip and the Palate, and functional analyses implicate GREM1 as a plausible causative gene. PLoS Genet. 12 (3), 1–21 (2016).
Leslie, E. J. et al. Genome-wide meta-analyses of nonsyndromic orofacial clefts identify novel associations between FOXE1 and all orofacial clefts, and TP63 and cleft lip with or without cleft palate. Hum. Genet. 136 (3), 275–286 (2017).
Ludwig, K. U. et al. Imputation of orofacial clefting data identifies novel risk loci and sheds light on the genetic background of cleft lip ± cleft palate and cleft palate only. Hum. Mol. Genet. 26 (4), 829–842 (2017).
Mostowska, A. et al. Common variants in DLG1 locus are associated with non-syndromic cleft lip with or without cleft palate. Clin. Genet. 93 (4), 784–793 (2018).
Mukhopadhyay, N. et al. Whole genome sequencing of orofacial cleft trios from the Gabriella Miller Kids First Pediatric Research Consortium identifies a new locus on chromosome 21. Hum. Genet. 139 (2), 215–226 (2020).
Mukhopadhyay, N. et al. Genome-wide association study of multiethnic nonsyndromic orofacial cleft families identifies novel loci specific to family and phenotypic subtypes. Genet. Epidemiol. 46, 3–4 (2022).
Moreno, L. M. et al. FOXE1 association with both isolated cleft lip with or without cleft palate, and isolated cleft palate. Hum. Mol. Genet. 18 (24), 4879–4896 (2009).
Birnbaum, S. et al. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat. Genet. 41 (4), 473–477 (2009).
Beaty, T. H. et al. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat. Genet. 42 (6), 525–529 (2010).
Mangold, E. et al. Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nat. Genet. 42 (1), 24–26 (2010).
Ludwig, K. U. et al. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci, 44 (9), 968–971 (2012).
Beaty, T. H. et al. Confirming genes influencing risk to cleft lip with/without cleft palate in a case-parent trio study. Hum. Genet. 132 (7), 771–781 (2013).
Sun, Y. et al. Genome-wide association study identifies a new susceptibility locus for cleft lip with or without a cleft palate. Nat. Commun., 6 (2015).
Leslie, E. J. et al. A multi-ethnic genome-wide association study identifies novel loci for non-syndromic cleft lip with or without cleft palate on 2p 24.2, 17q23 and 19q13. Hum. Mol. Genet. 25 (13), 2862–2872 (2016).
Kondo, S. et al. Mutations in IRF6 cause Van Der Woude and popliteal pterygium syndromes. Nat. Genet. 32 (2), 285–289 (2002).
Knight, A. S., Schutte, B. C., Jiang, R. & Dixon, M. J. Developmental expression analysis of the mouse and chick orthologues of IRF6: The gene mutated in Van Der Woude syndrome. Dev. Dyn. 235 (5), 1441–1447 (2006).
Richardson, R. J., Dixon, J., Jiang, R. & Dixon, M. J. Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Hum. Mol. Genet. 18 (14), 2632–2642 (2009).
Goudy, S. et al. Cell-Autonomous and non-cell-autonomous roles for Irf6 during development of the Tongue. PLoS One, 8 2 (2013).
Kousa, Y. A. & Schutte, B. C. Toward an orofacial gene regulatory network. Dev. Dyn. 245 (3), 220–232 (2016).
Schorle, H., Meiert, P., Buchertt, M., Jaenisch, R. & Mitchellt, P. J. Transcription factor AP-2 essential for cranial closure and craniofacial development. 381 (May), 235–238 (1996).
Nguyen, T. T. et al. TFAP2 paralogs regulate midfacial development in part through a conserved ALX genetic pathway. Dev, 151 (1) (2024).
Woodruff, E. D., Gutierrez, G. C., Van Otterloo, E., Williams, T. & Cohn, M. J. Anomalous incisor morphology indicates tissue-specific roles for Tfap2a and Tfap2b in tooth development. Dev. Biol., 472 (January) 67–74 (2021).
Gaare, J. D. & Langman, J. Fusion of nasal swellings in the mouse embryo: Regression of the nasal fin. Am. J. of Anatomy. 150, 477–499 (1977).
Gaare, J. D. & Langman, J. Fusion of nasal swellings in the mouse embryo: Surface coat and initial contact. Am. J. of Anatomy. 1503 (3), 461–475 (1977).
Abramyan, J. & Richman, J. M. Recent insights into the morphological diversity in the amniote primary and secondary palates. Dev. Dyn. 244 (12), 1457–1468 (2015).
Kim, S. et al. Convergence and extrusion are required for Normal Fusion of the mammalian secondary palate. PLoS Biol. 13 (4), 1–24 (2015).
Losa, M. et al. Face morphogenesis is promoted by pbx-dependent EMT via regulation of snail1 during frontonasal prominence fusion. Dev. 145 (5) (2018).
Fitchett, J. E. & Hay, E. D. Medial edge epithelium transforms to mesenchyme after embryonic palatal shelves fuse. Dev. Biol. 131 (2), 455–474 (1989).
Jin, J. Z. & Ding, J. Analysis of cell migration, transdifferentiation and apoptosis during mouse secondary palate fusion. Development. 133 (17), 3341–3347 (2006).
Merker, H. & V. M’Boneko and Development and morphology of the periderm of mouse embryos (days 9–12 of gestation). Acta Anat. 133 (4), 325–336 (1988).
Ji, Y., Hao, H., Reynolds, K., McMahon, M. & Zhou, C. J. Wnt signaling in neural crest ontogenesis and oncogenesis. Cells, 8, 10, (2019).
Lough, K. J., Byrd, K. M., Spitzer, D. C. & Williams, S. E. Closing the gap: Mouse models to study adhesion in secondary palatogenesis. J. Dent. Res. 96 (11), 1210–1220 (2017).
Kousa, Y. A. et al. IRF6 and SPRY4 signaling interact in Periderm Development. J. Dent. Res. 96 (11), 1306–1313 (2017).
G. De La Garza et al., Interferon regulatory factor 6 promotes differentiation of the periderm by activating expression of grainyhead-like 3. J. Invest. Dermatol., 133 (1), 68–77 (2013).
Graham, A. Development of the pharyngeal arches. Am. J. Med. Genet. 119 A (3), 251–256 (2003).
Liu, N. et al. DNA binding-dependent and -independent functions of the Hand2 transcription factor during mouse embryogenesis. Development. 136 (6), 933–942 (2009).
Coffin Talbot, J., Johnson, S. L. & Kimmel, C. B. hand2 and dlx genes specify dorsal, intermediate and ventral domains within zebrafish pharyngeal arches. Development. 137 (15), 2507–2517 (2010).
Thomason, H. A. et al. Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. J. Clin. Invest. 120 (5), 1561–1569 (2010).
Lin-Shiao, E. et al. P63 establishes epithelial enhancers at critical craniofacial development genes. Sci. Adv. 5 (5), 1–15 (2019).
Sun, J. et al. Genetic association and functional validation of ZFP36L2 in non-syndromic orofacial cleft subtypes. J. Hum. Genet. 69, 3–4 (2024).
Gianferante, D. M. et al. Genotype-phenotype association and variant characterization in Diamond Blackfan anemia caused by pathogenic variants in RPL35A. Haematologica. 106 (5), 1303–1310 (2021).
Chiquet, B. T. et al. Variation in WNT genes is associated with non-syndromic cleft lip with or without cleft palate. Hum. Mol. Genet. 17 (14), 2212–2218 (2008).
Nikopensius, T. et al. Genetic variants in COL2A1, COL11A2, and IRF6 contribute risk to nonsyndromic cleft palate. Birth Defects Res. Part. - Clin. Mol. Teratol. 88 (9), 748–756 (2010).
Nikopensius, T. et al. Variation in FGF1, FOXE1, and TIMP2genes is associated with nonsyndromic cleft lip with or without cleft palate. Birth Defects Res. Part. - Clin. Mol. Teratol. 91 (4), 218–225 (2011).
Mostowska, A. et al. Genotype and haplotype analysis of WNT genes in non-syndromic cleft lip with or without cleft palate. Eur. J. Oral Sci. 120 (1), 1–8 (2012).
Feng, C. et al. C392T polymorphism of the < em > Wnt10a gene in non-syndromic oral cleft in a northeastern Chinese population, Br. J. Oral Maxillofac. Surg., vol. 52, no. 8, pp. 751–755, Oct. (2014).
Lu, Y. et al. Variations in WNT3 gene are associated with incidence of non-syndromic cleft lip with or without cleft palate in a northeast Chinese population. Genet. Mol. Res. 14 (4), 12646–12653 (2015).
Pengelly, R. J. et al. Deleterious coding variants in multi-case families with non-syndromic cleft lip and/or palate phenotypes. Sci. Rep. 6 (November 2015), 1–8 (2016).
Zieger, H. K. et al. Prioritization of non-coding elements involved in non-syndromic cleft lip with/without cleft palate through genome-wide analysis of de novo mutations. Hum. Genet. Genomics Adv. 4 (1), 100166 (2023).
Funding
K.U.L is member of the Cluster of Excellence ImmunoSensation - EXC2151–390873048, funded by Deutsche Forschungsgemeinschaft (DFG), and has received support from the DFG (LU 1944/3 − 1).
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.S., K.U.L.; Analysis and curation of data: A.S., S.H.; Investigation: A.S., S.H., K.U.L.; Resources: E.M., K.U.L; Writing - original draft preparation: A.S., K.U.L.; Writing - review and editing: A.S., S.H., E.M., K.U.L.; All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Acknowledgements.
The authors thank Friederike David, Carina Mathey, and Sabrina Henne for technical support during data analysis. The authors gratefully acknowledge the granting of access to the Bonna cluster, which is hosted by the University of Bonn.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Siewert, A., Hoeland, S., Mangold, E. et al. Combining genetic and single-cell expression data reveals cell types and novel candidate genes for orofacial clefting. Sci Rep 14, 26492 (2024). https://doi.org/10.1038/s41598-024-77724-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-77724-9
Keywords
This article is cited by
-
5’tiRNA-35-GlyTCC-3 and 5’tiRNA-33-CysGCA-11 target BMP6, CUL1 and SPR of non-syndromic cleft palate
BMC Oral Health (2025)
-
Single cell spatial transcriptomics links Wnt signaling disruption to extracellular matrix development in a cleft palate model
Scientific Reports (2025)
-
Identification of functional non-coding variants associated with orofacial cleft
Nature Communications (2025)
