
Abstract
The pyruvate dehydrogenase kinase-3 (PDK3) plays an important role in the regulation of a variety of cancers, including lung, by inhibiting the pyruvate dehydrogenase complex (PDC), shifting energy production towards glycolysis necessary for cancer metabolism. In this study, we aimed to identify potential PDK3 inhibitors using a computer-based drug design approach. Virtual screening of the FDA-approved library of 3839 compounds was carried out, from which Bagrosin and Dehydrocholic acid appeared best due to their strong binding affinity, specific interactions, and potential biological characteristics, and thus were selected for further investigations. Both compounds show strong interactions with functionally important residues of the PDK3 with a binding affinity of − 10.6 and − 10.5 kcal/mol for Bagrosin and Dehydrocholic acid, respectively. MD simulation studies for 100 ns suggest the formation of stable complexes, which is evident from RMSD, RMSF, Rg, and SASA parameters. The PCA and FEL analysis suggested admirable global energy minima for the bagrosin-PDK3 and dehydrocholic acid-PDK3 complexes. Finally, we identified FDA-approved drugs, Bagrosin and Dehydrocholic acid, that offer valuable resources and potential therapeutic molecules for targeting lung cancer. Further clinical investigations are required to validate the clinical utility of selected molecules.
Similar content being viewed by others


Introduction
Lung cancer is one of the most common types in the world1. In the UK alone, there were 46,403 new cases of lung cancer in 2014, which is quite surprising. Even though it’s the third most common cancer overall, it is the top cause of cancer-related deaths, contributing to 18% of deaths globally. It is estimated to account for 127,070 deaths in 20232,3. Unfortunately, in 93 countries, lung cancer is the leading cause of cancer death among men because it has a high mortality rate4. In certain countries, like China, Indonesia, and some African nations, lung cancer rates are still increasing5. India ranks third in the world for both tobacco production and consumption, with approximately 28.6% of its population using tobacco products. Lung cancer accounts for 5.9% of all cancer cases and 8.1% of cancer-related deaths in the country6. Cigarette smoking, responsible for 80% of lung cancer cases and deaths, significantly raises the risk, while additional factors such as secondhand smoke, radon, asbestos, and air pollution7,8. Hence, there is an immediate need to develop therapeutic advances that can overcome the drawbacks of current treatments9.
Energy is necessary for life, and animals obtain their energy through the process of oxidative phosphorylation (OXPHOS) in their mitochondria10. However, most cancer cells have a different way of getting energy, as they can produce lactate from glucose even in the presence of adequate oxygen11,12, a process known as the Warburg effect or Aerobic glycolysis. This change helps cancer cells promote their growth and avoid dying, serving as a target for cancer therapies13. Produced lactate can affect the immune function of cells, making them harder and helping cancer cells move and spread14,15.
The Warburg effect facilitates the transition from oxidative phosphorylation to glycolysis16. This metabolic reprogramming supports rapid proliferation by providing ATP and biosynthetic precursors to grow quickly12. Typically, cells generate around 36 ATP molecules from one glucose molecule through a process known as oxidative phosphorylation, but cancer cells only produce 2 ATP from glycolysis. Despite this inefficiency, glycolysis is much faster, generating ATP up to 100 times more quickly17.
Due to this rapid energy production, cancer cells can efficiently meet their high energy demands. Several signaling pathways, including HIF-1α and PI3K/Akt/mTOR, play critical roles in accelerating glycolysis and enhancing glucose uptake in cancer cells. These pathways not only support the rapid growth and proliferation of cancer cells but also contribute to oncogene activation and tumor suppressor alterations, further promoting tumor development and survival. This metabolic reprogramming is essential for cancer progression and offers potential targets for therapeutic intervention18.
The HIF-1α plays a crucial role in suppressing mitochondrial function under both hypoxic and normoxic conditions by inhibiting pyruvate dehydrogenase (PDH), the enzyme responsible for converting pyruvate into acetyl-CoA for entry into the TCA cycle. This inhibition prevents pyruvate from entering the TCA cycle, thereby reducing oxidative phosphorylation and oxygen consumption. Simultaneously, HIF-1α promotes glycolysis, enabling cancer cells to maintain energy production under low oxygen conditions and supporting their growth and survival in the tumor microenvironment19,20.
The mitochondrial PDC is a group of components that helps convert pyruvate into acetyl-CoA. It plays a crucial role in connecting glycolysis to the tricarboxylic acid cycle. This process also contributes to ATP production, which is vital for energy in our cells21,22. PDC is made up of three parts- pyruvate dehydrogenase (E1), dihydrolipoamide acetyltransferase (E2), and dihydrolipoamide dehydrogenase (E3). The activity of PDC is controlled by a protein called pyruvate dehydrogenase kinase (PDK), which can turn PDC on or off by adding a phosphate group23,24. Inhibition of PDK suppresses aerobic glycolysis by promoting glucose oxidation in the TCA cycle rather than its fermentation into lactate. Genetic inhibition of PDKs has been shown to prevent cancer cell proliferation in both cell cultures and tumor models. The inactivation of the PDC, due to its association with PDK3, has been strongly linked to the development of several cancer types. By inhibiting PDK3, cells can restore PDC activity, enhancing mitochondrial oxidative metabolism and reducing the reliance on glycolysis, which is a hallmark of cancer cells25, and its activity is not affected by high levels of pyruvate. This makes PDK3 a potential target for cancer therapy26.
PDKs, including PDK3, inhibit the conversion of pyruvate to acetyl-CoA, which leads to a shift in cellular energy production from the mitochondria to the cytoplasm. This change in energy production is associated with cancer cell growth and drug resistance25. In hypoxic conditions (low oxygen levels), PDK1 and PDK3 are induced, leading to increased lactic acid production and further inhibition of mitochondrial respiration. This metabolic switch mediated by PDK3 contributes to drug resistance in hypoxic tumors. In cancer cells with high levels of HIF-1α, an oxygen-sensing protein, PDKs inactivate pyruvate dehydrogenase, causing pyruvate to accumulate27. However, PDK3 remains active, ensuring that mitochondrial respiration stays shut down. Blocking PDK3 is essential because it helps make cancer cells more vulnerable to anticancer medications, especially when insufficient oxygen is available in the prostate cancer cell line and gastric cancer cell lines. PDK3 knockdown induced apoptosis and inhibited growth28,29.
Dichloroacetate (DCA), M77976, and radicicol are among the several chemotypes of PDK inhibitors that have been found. Inhibitors, such as M77976 and radicicol, bind to the nucleotide-binding pocket, obstructing the ATP hydrolysis and inhibiting the catalytic activity of PDKs30. Furthermore, inhibitors with strong inhibitory potencies that target the lipoamide-binding site include AZD7545, AZ12, and Nov3r. The structural diversity of the inhibitors discovered is a result of the fact that different binding pockets can be targeted to block different PDKs30,31. VER-246608 is an ATP-competitive inhibitor that binds to the ATP pocket of PDKs32.
DCA is a compound similar to pyruvate, which can be taken orally and binds to the N-terminal region of PDK. This binding stops PDK activity, allowing PDC to function again33. As a result, the body shifts from using glycolysis to metabolizing glucose, which leads to apoptosis (cell death), inhibits tumor growth, and could potentially be used as a cancer treatment. A study found that under conditions of low oxygen (hypoxia), PDK3 levels increased, causing resistance to anticancer drugs27. However, when PDK3 was suppressed in cells, this resistance was eliminated. These findings suggest that PDK3 could be a promising target for cancer therapy.
The primary motivation behind drug repurposing is the high cost and lengthy process associated with developing new medications. Establishing the safety, efficacy, and specificity of a novel drug can take several years, if not decades, especially in fields like cancer treatment, where rigorous clinical trials are required before approval. Repurposing can significantly reduce the time and cost associated with bringing effective therapies to patients34.
The use of computational tools in drug discovery has proven successful in identifying several FDA-approved anticancer drugs35. For example, Crizotinib, which treats lung, lymphoma, and esophageal cancers, was identified through structure-based design, while Gefitinib is used to treat non-small cell lung cancer (NSCLC)36. Other anticancer compounds, such as DZD9008 for NSCLC and Resveratrol for breast, skin, and lung cancer37, are currently undergoing clinical trials. The present study focuses on screening FDA-approved compounds to identify new potential inhibitors for PDK3, a protein implicated in cancer metabolism. By leveraging a structure-based drug design approach, we aim to discover novel therapeutic agents for lung cancer. This strategy offers the potential to fast-track drug development by repurposing existing drugs with known pharmacokinetic profiles, ultimately accelerating the path to clinical application.
Materials and methods
The drug database was screened against PDK3 using virtual high-throughput screening. This computational technique identifies potential drug candidates by predicting their likely binding to proteins with high affinity38,39. For docking-based virtual screening and MD simulations, we used InstaDock39, PyMOL40, Discovery Studio Visualizer41, and GROMACS42 tools.
Molecular docking
Molecular docking was applied to determine the favorable receptor-ligand interaction conformation. For this screening procedure, we took the structure of PDK3 from the RCSB Protein Data Bank (PDB ID: 1Y8O)43. The structure was further refined by removing water molecules and heteroatoms with the main structure39. The original structure had missing residues in the amino acid sequences from positions 307–319 and 322–323. To address this, remodelling was carried out using PyMod-3, which employed MODELLER software based on the self-template 6Z45 (version 9.20).
Virtual screening protocol
Structure-based virtual screening was performed on the FDA-approved library to find compounds with greater binding affinity for PDK339. Size parameters were set to X = 71, Y = 65, and Z = 6, and a grid box was defined with X = 153.741, Y = 8.516, and Z = 21.232 as its center. Compounds were selected according to their binding affinity. Multiple docked conformers were generated to analyze interactions. The visualization of close interactions between PDK3 and FDA-approved drugs within a 3.5 Å range. Subsequently, we chose the compounds that interacted with the critical residues of the ATP binding site of PDK3.
PAINS and ADMET properties
The compounds identified through docking analysis were further investigated for their physicochemical properties and ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiles. The drug-likeness of the selected compounds was determined by evaluating their ADMET characteristics and Pan-assay Interference Compounds (PAINS) patterns. After the docking process, the identified hits were analyzed using SwissADME to apply the PAINS filter and assess the ADMET properties of the screened compounds. The SMILES notation of each compound was compatible with both systems. The PAINS filter effectively identified compounds prone to nonspecific binding, ensuring the selection of compounds with more targeted therapeutic potential.
PASS analysis: predicting biological activity
The identified compounds were analyzed using the PASS (Prediction of Activity Spectra for Substances) web tool to predict their potential biological activities44. PASS predicts various pharmacological properties based on structure–activity relationships derived from the chemical features of the molecule. The web server compares the input structure to a large database of predefined biological functions to train its model. Predictions are based on the ratio between the probability of being active (Pa) and the probability of being inactive (Pi). A comprehensive list of potential biological properties is generated for each compound, with a higher Pa value indicating a greater likelihood that the compound will exhibit the predicted biological activity.
Interaction analysis
Interaction analysis was conducted on the selected protein–ligand complexes to investigate the molecular interactions occurring during the binding process. Close contacts within the protein–ligand complex were defined as interactions occurring within a 3.5 Å distance. Critical residues of PDK3 demonstrated particular interactions with the compounds selected for further interaction analysis with active site residues. The docking results correlated these interactions with the binding affinity of PDK3, supporting the selection of these compounds for continued investigation.
Molecular dynamics simulation
Molecular dynamics (MD) simulation was used to study the structural dynamics and conformational changes in proteins induced by ligand binding45,46. On an HP Z840 computer, all-atom MD simulations were performed at 300 K for 100 ns using the GROMACS 2020-beta simulation package42. MD simulations of PDK3 in combination with FDA-approved drugs were run for one hundred nanoseconds. The three systems were simulated using the GROMOS 54A7 force field and the GROMACS 2020 beta software suite for all atoms. The PRODRG server generated the topological parameters of FDA-approved compounds and then included them in the PDK3 topology to form a protein–ligand complex system.
To establish an equilibrated aqueous environment, the three systems were immersed in a cubic container filled with the TIP3P water model and neutralized by adding Na+ and Cl- ions. To eliminate high-energy disturbances from the initial structures, 1500 energy minimization steps were performed using the steepest descent method until the systems were fully minimized. The energy-minimized systems were then equilibrated in two stages, using the NVT (constant number of particles, volume, and temperature) and NPT (constant number of particles, pressure, and temperature) ensembles. Following equilibration, each system underwent a 100 ns molecular dynamics simulation. The resulting trajectories were analyzed using QtGrace software, with additional checks and validations performed using the built-in capabilities of GROMACS.
Principal component analysis and free energy landscapes
Principal Component Analysis (PCA) is an unsupervised learning approach that identifies principal components (PCs), which represent directions that capture the most variation within the data47. PCA is particularly valuable for detecting significant variations in high-amplitude movements in MD trajectories. In our analysis of the PDK3 MD trajectories, we employed PCA alongside free energy landscape (FEL) analysis to evaluate atomic movements, conformational sampling, and the stability of the compounds bagrosin and dehydrocholic acid before and after their binding with PDK3. This combined approach provides insights into the dynamic behavior and binding characteristics of these compounds within the protein environment.
Results and discussion
Molecular docking
Virtual screening is used to identify potential lead compounds by analyzing a large library of compounds against a specific protein target. During this process, the compounds are docked into the active site of the protein receptor complex, and an energy-scoring function is employed to evaluate their binding affinities38,48,49. An FDA-approved library of 3839 compounds was subjected to docking against PDK3. The virtual screening of compounds against PDK3 was performed using InstaDock, an interactive molecular docking interface that facilitates single-click operations. After the docking process, the top 50 hits were selected based on their predicted ligand efficiency and docking scores. The results indicated that these top 50 compounds demonstrated a remarkable affinity for PDK3, with binding energies ranging from − 11.6 to − 7.7 kcal/mol (see Table S1). These findings suggest that the selected hits possess significant potential for interacting with PDK3, warranting further investigation as potential competitive inhibitors of this target.
PAINS and ADMET properties of compounds
After identifying the top 50 hits through molecular docking, compounds exhibiting PAINS (Pan-Assay Interference Compounds) were excluded from further consideration. These compounds showed a strong affinity for binding to multiple biological targets, which could lead to non-specific off-target effects, making them unsuitable for drug discovery. To address these concerns, PAINS filtration was applied to the selected hits, refining the selection process. The compounds that exhibited the highest affinity towards PDK3 during the molecular docking process—known as the top hits among FDA-approved compounds—were then subjected to further investigation to assess their ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) characteristics.
The pharmacokinetic properties of two promising compounds, bagrosin (− 10.6 kcal/mol) and dehydrocholic acid (− 10.5 kcal/mol) (see Table 1), were evaluated using the SwissADME web server. Bagrosin, in particular, demonstrated key features supporting its potential as an inhibitor and enhancing its druggability. It possesses a balanced lipophilicity, with a consensus Log Po/w of 2.72, which is conducive to good membrane permeability. Additionally, its topological polar surface area (TPSA) of 58.20 Å2 promotes oral bioavailability, and the presence of only one rotatable bond contributes to its rigidity, potentially improving binding strength. Bagrosin meets all major drug-likeness criteria outlined by Lipinski’s rules; however, some modifications may be necessary to mitigate its ability to inhibit certain cytochrome P450 enzymes (CYPs) and to address its slightly elevated lipophilicity (XLOGP3 > 3.5).
Another compound, dehydrocholic acid, exhibits a consensus Log Po/w of 3.19, indicating a favorable balance of lipophilicity that supports both permeability and solubility. It has a topological polar surface area (TPSA) of 88.51 Å2, which promotes optimal oral bioavailability. The presence of four rotatable bonds suggests moderate flexibility, potentially enhancing its binding affinity. In conjunction with the reference molecule, the ADMET features of the compounds under investigation exhibit good gastrointestinal absorption and water solubility characteristics while demonstrating no toxicological patterns. As expected, the chosen compounds exhibited drug-like features.
The PASS analysis: predicting biological activity
The extensive training set of the PASS server encompasses a wide array of bioactive compounds derived from numerous preclinical and clinical studies44. The compounds that underwent ADMET (Table 2) filtration were subsequently examined for their anticipated biological activities. Bagrosin and dehydrocholic acid met the requirements of the PASS criteria for the wanted biological activity prediction. The findings of this study suggest that both compounds demonstrate good anticancer and kinase inhibitory characteristics. When the (Pa) value surpasses 0.7, it indicates a significant probability that the compound will exhibit the expected biological activity. Bagrosin and dehydrocholic acid have shown great potential in various areas, such as inhibiting and antagonizing neurotransmitters, enhancing TP53, antineoplasty inhibiting JAK2, and producing analeptic effects.
Bagrosin has shown potential as a superoxide dismutase inhibitor (Pa = 0.635), an Alzheimer’s disease treatment (Pa = 0.587), a TP53 expression enhancer (Pa = 0.546), an oxygen scavenger (Pa = 0.531) (Table 2). Dehydrocholic acid shows a high probability of acting as a bile-salt sulfotransferase inhibitor (Pa = 0.954) and a protein-disulfide reductase inhibitor (Pa = 0.904). Other potential activities include functioning as an oxidoreductase inhibitor (Pa = 0.771), producing analeptic effects (Pa = 0.719), and having antineoplastic properties (Pa = 0.543). The findings of this study indicate that both compounds exhibit significant biological potential when employed in anticancer interventions that specifically target PDK3.
Interaction analysis
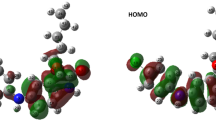
The structure of PDK3 highlights important residues involved in substrate binding (Fig. 1). Our analysis revealed that both bagrosin and dehydrocholic acid fit well to the active site pocket of PDK3 (Fig. 2) and exhibit notable binding affinity towards critical residues within the ATP-binding site, specifically Gly327 and Phe324, respectively (Fig. 2B and E). The interaction distance between PDK3 residue Gly327 and bagrosin is measured at 3.3 Å, while the distance between Phe324 and dehydrocholic acid is 2.4 Å (Table 3). Ideally, key interactions, such as hydrogen bonds, should fall within a distance range of approximately 2.5–3.5 Å, with van der Waals contacts typically within 3–4 Å. Our findings demonstrate a strong association between both compounds and the ATP-binding pocket, which is recognized as a primary interaction site for various established PDK3 inhibitors. Both bagrosin and dehydrocholic acid show strong binding affinity to the active site of PDK3, effectively to the ATP-binding pocket (Fig. 2C and F). The results suggest that PDK3 can be pharmacologically inhibited through competitive binding to PDK3.

Structural overview of PDK3, highlighting the key binding sites critical for its function.

Molecular docking of Bagrosin and Dehydrocholic acid with PDK3. (A) A cartoon representation showing Bagrosin and Dehydrocholic acid docked at the active site of PDK3, along with residues involved in polar interactions. (B) Localization of Bagrosin in the PDK3 binding pocket. (C) Surface view of PDK3 with Bagrosin bound. (D) 2D interaction diagram illustrating the interactions between Bagrosin and PDK3. (E) Localization of Dehydrocholic acid in the PDK3 binding pocket. (F) Surface view of PDK3 with Dehydrocholic acid bound. (G) 2D interaction diagram illustrating the interactions between Dehydrocholic acid and PDK3.
A comprehensive study was conducted to identify the types and characteristics of non-covalent interactions within the protein–ligand complex. A 2D depiction of the docked complex is presented in Fig. 2D and G. These diagrams illustrate the interactions between the ATP-binding residues Gly327 and Phe324 of PDK3 with both compounds, bagrosin and dehydrocholic acid, through hydrogen bonding. Additionally, these compounds interact with several other key residues surrounding the ATP-binding site in PDK3, including Ile330, Glu247, Tyr326, Lys250, Pro124, Ala127, Gln128, Ile131, His243, and Lys250. Notably, Gly327 plays a critical role in the ATP-binding process with PDK3 (Table 3). These findings indicate that bagrosin and dehydrocholic acid occupy a region analogous to where ATP is co-crystallized, suggesting that these compounds can inhibit ATP competitively. The interaction analysis highlights the potential of bagrosin and dehydrocholic acid as effective inhibitors of PDK3 by obstructing the ATP-binding site.
MD simulations
Structural deviations in PDK3
The root mean square deviation (RMSD) provides useful information on conformational dynamics and structural variations in protein50,51,52. The RMSD analysis was used to evaluate the variations in the backbone conformation of PDK3 before and after a ligand’s interaction. The RMSD plots of the PDK3-bagrosin and PDK3-dehydrocholic acid complexes displayed time-dependent graphs illustrating the changes and deviations in the protein backbone during the simulations. The simulation trajectory results are shown in (Fig. 3A) and were then used to examine the stability of the complex. The RMSD figure indicates a small reduction in fluctuations in ligand-bound systems compared to the free PDK352,53.

Structural dynamics of PDK3 in the presence of Bagrosin (Red) and Dehydrocholic acid (Green). (A) RMSD plot comparing the structural deviations of PDK3 with and without the compounds. (B) RMSF plot showing residue-level fluctuations of PDK3 in the presence and absence of Bagrosin and Dehydrocholic acid. (C) Probability Density Function (PDF) plot of RMSD before and after Bagrosin and Dehydrocholic acid binding to PDK3. (D) PDF plot of RMSF before and after Bagrosin and Dehydrocholic acid binding to PDK3.
All three systems achieved equilibrium and maintained stability throughout the 100-ns simulation period. The fluctuations in RMSD were not significant enough to cause substantial structural deviations, though they indicate some adaptations within the systems. Notably, the PDK3 complex with Bagrosin exhibited a slightly lower RMSD value of 0.28 nm compared to native PDK3 at 0.32 nm and the PDK3-Dehydrocholic acid complex at 0.29 nm, suggesting a more stable interaction with Bagrosin (Table 4). Additionally, the Probability Density Function (PDF) plot shown in the lower panel (Fig. 3C) further illustrates a marginal reduction in the RMSD values, reinforcing the stability of the PDK3-Bagrosin complex.
An analysis of root-mean-square fluctuation (RMSF) is useful for identifying the residual vibrations present in a protein molecule during MD simulation54,55. RMSF analysis of the PDK3 backbone and its interactions with bagrosin and dehydrocholic acid (Fig. 3B,D) characterize the local variations occurring throughout the simulation. The high variations in the RMSF values indicate the presence of loops and unstructured areas in the protein. The average variation in local structural flexibility of each residue was analyzed in PDK3 before and after interaction with the drug. The figure shows that all systems exhibit a comparable RMSF pattern with minor randomized variations. The plot shows residual fluctuations reflect a consistent and slightly elevated pattern when bagrosin and dehydrocholic acid bind, suggesting that the protein–ligand complexes are distressed yet remain stable. PDK3-Bagrosin and PDK3-Dehydrocholic acid have a somewhat higher RMSF (0.13 nm) than the native PDK3 (0.12 nm), indicating some increased flexibility upon ligand binding (Table 4). The residues inside the PDK3 binding pocket that engage with bagrosin and dehydrocholic acid were rather stable throughout the simulation, as shown by the RMSF study.
Structural compactness
The radius of gyration (Rg) is a frequently used metric to determine the density of proteins and protein–ligand complexes56. We calculated the time evolution of the Rg using the simulated trajectory of all three systems52. Based on the Rg plot, when bagrosin and dehydrocholic acid are present, PDK3 remains consistent at a value between 2.15 and 2.19 nm during the trajectory (Fig. 4A). The findings suggest that the binding to bagrosin and dehydocholic acid and the structural dynamics and folding of PDK3 remain consistently stable with a little increase. The Rg values for PDK3 exhibit a consistent distribution both before and after binding with bagrosin and dehydocholic acid, as shown in the PDF plot (Fig. 4C).

Analysis of the structural evolution of PDK3 over a 100 ns MD simulation. (A) Rg plot showing the compactness of free PDK3 and its complexes with Bagrosin and Dehydrocholic acid. (B) SASA plot showing the exposure of PDK3 to the solvent over time. Black, red, and green lines represent free PDK3, the Bagrosin-PDK3 complex, and the Dehydrocholic acid-PDK3 complex, respectively. (C) PDF plot of Rg before and after Bagrosin and Dehydrocholic acid binding. (D) PDF plot of SASA before and after compound binding.
The portion of a protein molecule exposed to solvent is referred to as the solvent-accessible surface area (SASA)57. The changes in SASA of PDK3 over time were studied regarding its binding with bagrosin and dehydrocholic acid. PDK3-Dehydrocholic acid has the highest SASA (186.55 nm2) compared to PDK3-Bagrosin (181.35 nm2) and native PDK3 (181.31 nm2), showing a more exposed structure (Table 4). Based on the SASA investigation, the PDK3 structure remains stable throughout the simulation, even when exposed to bagrosin and dehydrocholic acid (Fig. 4B). The SASA distribution exhibits a similar equilibration pattern to the Rg values, with a slight increase in ligand-bound systems but not significantly impacting the overall compactness (Fig. 4B and D).
Interaction dynamics in the PDK3 complexes: H-bond analysis
Hydrogen bonds play an essential part in folding protein structures and assembling their conformations58. The intramolecular H-bond count in PDK3 has been measured over time using MD trajectories (Fig. 5A). The outcomes allow us to analyze the intramolecular bonding consistency of PDK3 both before and after its binding to bagrosin and dehydrocholic acid. The figure demonstrates a change in the number of hydrogen bonds established inside PDK3 before and after bagrosin and dehydrocholic acid-binding. PDK3-Bagrosin forms the most hydrogen bonds (282), implying stronger interactions. The graph indicates that the hydrogen bonds produced inside PDK3 were stable and played an essential part in determining the shape of the protein structure. The dynamic analysis shows that a little rise in intramolecular hydrogen bonds within PDK3-bagrosin complexes resulted in a somewhat more condensed structure than the free PDK3 form. On the other hand, PDK3-dehydrocholic acid exhibited a comparable amount of intramolecular hydrogen bonds. The PDF illustrating intramolecular hydrogen bonding in all systems was also shown (Fig. 5B).

Hydrogen bond analysis of PDK3 in complex with Bagrosin and Dehydrocholic acid. (A) Intramolecular hydrogen bonds within PDK3. (B) PDF plot showing the probability of hydrogen bond formation between Bagrosin, Dehydrocholic acid, and PDK3. (C) Hydrogen bonds formed between PDK3 and Bagrosin. (D) Hydrogen bonds formed between PDK3 and Dehydrocholic acid. All bond pairs within a 0.35 nm distance were considered.
The interaction between bagrosin and dehydrocholic acid with PDK3 was investigated in terms of the stability of hydrogen bonding. This was done by analyzing the changes in intermolecular hydrogen bonds over time. The bagrosin-PDK3 complex established 2 stable hydrogen bonds, whereas the dehydrocholic acid complex formed 3 (Fig. 5C and D). The PDF analysis demonstrated that the intramolecular hydrogen bonds in both systems exhibited a consistent level, with a greater PDF value corresponding to an increased number of hydrogen bonding interactions (Fig. 5B). The bagrosin and dehydocholicacid remained stationary at their original docking site on the PDK3 protein owing to stable intermolecular hydrogen bonding, which helped stabilize the complexes between the protein and ligands.
Principal component and free energy landscape analyses
We studied the first two principal components (PCs) derived from the PCA analysis of PDK3 and its complexes with bagrosin and dehydrocholic acid. Figure 6 illustrates the exploration of several conformations inside the fundamental subspace of PDK3, PDK3-bagrosin, and PDK3-dehydrocholic acid. The Cα atom of PDK3 determines the conformational states along the EV1 and EV2. The graph demonstrates that the PDK3-bagrosin and PDK3-dehydrocholic acid complexes have nearly the same essential region as PDK3 in its unbound form. The PDK3-bagrosin complex and free-PDK3 are seen to occupy the same subspace in both EVs. The flexibility of the PDK3-bagrosin complex is greater on EV1 compared to EV2. Simultaneously, the PDK3-dehydrocholic acid complex exhibits more flexibility on EV1 in the direction of positive projection (Fig. 7). PCA reveals that the binding of Bagrosin and Dehydrocholic acid affects the conformational movements of PDK3. Both compounds reduce the protein’s conformational space, indicating a stabilizing effect on PDK3. This stabilization might be key for the protein to perform its biological functions more efficiently when interacting with these compounds.

Principal Component Analysis of PDK3 dynamics. The red and green trajectories show the dynamic changes after Bagrosin and Dehydrocholic acid binding, respectively.

Free energy landscape analysis of PDK3. The Gibbs free energy landscapes were generated for (A) free PDK3, (B) the PDK3-Bagrosin complex, and (C) the PDK3-Dehydrocholic acid complex during a 100 ns MD simulation.
The conformational behavior and folding states of free energy landscapes (FELs) were analyzed by constructing them from simulated trajectories59. The stability of the protein and protein–ligand complexes in the presence of a solvent was evaluated using MD simulation trajectories. The lowest energy states and the arrangement of molecular structures for PDK3, PDK3-Bagrosin, and PDK3-Dehydrocholic acid complexes were derived from the analysis of two principal components, PC1 and PC2. The contoured maps depicting the FELs of these complexes are shown in Fig. 7. The FEL plots indicate that the binding of Bagrosin and dehydrocholic acid to PDK3 has a minimal impact on the size and location of the energy landscapes, which are confined to 1–2 stable global minima. A deeper shade of blue in the FELs suggests the presence of conformations with lower energy levels that are close to the native states, as illustrated in Fig. 7.
The graph illustrates that PDK3 is constrained to a single, expansive global minimum with 1–2 basins. In contrast, the PDK3-Bagrosin and PDK3-Dehydrocholic acid complexes exhibit nearly identical configurations, characterized by a prominent global minimum accompanied by 3–4 smaller local basins that vary in population (Fig. 7A–C). Both compounds stabilize the PDK3 protein, leading to a lower energy state compared to free PDK3, suggesting a favorable binding process. The presence of a more concentrated energy basin in the bound complexes (Fig. 7B and C) indicates that the PDK3 protein adopts a specific, stable conformation upon binding, further supporting the potential role of these compounds as stabilizing agents. In summary, the MD simulation and PCA of PDK3 in the presence of Bagrosin and dehydrocholic acid demonstrate that both compounds remain stable over the 100-ns simulations, exhibiting minor conformational changes. Based on these findings, it can be inferred that Bagrosin and dehydrocholic acid exhibit significant affinity and stability when binding to PDK3, resulting in potent inhibition of the PDK3 protein.
Discussion
Lung cancer continues to be a leading cause of cancer-related deaths worldwide, highlighting the urgent need for novel therapeutic strategies. Targeting metabolic pathways, particularly PDK3, has emerged as a promising approach for treating various cancers, including lung cancer26. PDK3 plays a pivotal role in metabolic reprogramming by facilitating the Warburg effect, which shifts energy production from mitochondria to the cytoplasm. By inhibiting the PDC, PDK3 prevents the conversion of pyruvate to acetyl-CoA, thereby promoting glycolysis and supporting cancer metabolism.
This study explores a strategic approach to targeting PDK3 through the repurposing of FDA-approved compounds, presenting a promising avenue for modulating pathways with anticancer properties. By employing virtual screening methods, we identified potential inhibitors of PDK3 through a computational approach involving molecular docking and MD simulations. Two compounds, bagrosin and dehydrocholic acid, emerged as strong candidates for PDK3 inhibition. These compounds demonstrated significant binding affinities for the ATP-binding site of PDK3, specifically interacting with key residues such as Gly327 and Phe324.
MD simulations further confirmed the stability of both compounds within the PDK3 binding pocket, exhibiting minimal structural deviations over the 100 ns simulation period. Structural analysis revealed that both compounds formed stable hydrogen bonds, contributing to the inhibition of PDK3 activity. Additionally, PAINS and ADMET filtering stages indicated that these compounds possess favorable pharmacokinetic and safety profiles, suggesting a low likelihood of off-target effects. Furthermore, bagrosin and dehydrocholic acid exhibited good absorption and bioavailability, making them suitable candidates for further preclinical development. PASS analysis predicted a diverse range of potential biological activities, with both compounds displaying anticancer and kinase inhibitory properties.
Bagrosin is classified as a hydantoin derivative and is recognized for its antiepileptic properties, specifically its anticonvulsant effects. It is reported to bind within a hydrophobic pocket located in domain-3 of the protease, which could potentially disrupt dimer formation. Bagrosin functions by blocking voltage-gated sodium channels in neurons and decreasing calcium movement through neuronal membranes, thereby stabilizing neuronal functions. Clinical studies suggest that bagrosin may be safer than other similar medications, making it a promising option in the management of epilepsy and potentially other neurological conditions60.
Dehydrocholic acid is recognized for its ability to induce choleresis, which is the increased secretion of bile. Studies have demonstrated that dehydrocholic acid enhances the permeability of tight junctions in the canalicular membranes, facilitating the direct interchange of bile and plasma. It is commonly used in medical settings to treat cholestatic hepatitis, where it promotes bile production from hepatocytes, thereby improving bile flow and maintaining cleanliness in the biliary ducts. This action contributes to reducing the occurrence of hepatitis, making dehydrocholic acid an important therapeutic agent in managing liver-related conditions61.
The combination of virtual screening, docking, and MD simulations has proven to be an effective strategy for identifying novel PDK3 inhibitors. This approach accelerates the discovery of therapeutics by utilizing compounds with established pharmacokinetic profiles, significantly shortening the drug development timeline. Bagrosin and dehydrocholic acid emerged as potential inhibitors of PDK3, a critical target in cancer metabolism. These FDA-approved compounds demonstrated strong binding affinities for the ATP-binding site of PDK3, with MD simulations confirming the stability of their interactions. Additionally, the pharmacokinetic and safety profiles of these compounds suggest that they hold substantial promise for repurposing as anticancer agents, particularly in the treatment of lung cancer.
Conclusions
In conclusion, targeting metabolic reprogramming pathways, particularly through the inhibition of PDK3, presents a viable strategy for cancer treatment. This study utilized computational approaches, including virtual screening, molecular docking, and molecular dynamics simulations, to identify two FDA-approved compounds, bagrosin and dehydrocholic acid, as promising PDK3 inhibitors. Both compounds demonstrated strong binding affinities to the ATP-binding site of PDK3 and interacted with key residues such as Gly327 and Phe324, contributing to their inhibitory potential. MD simulations further confirmed the stability of these compounds within the PDK3 binding pocket, reinforcing their viability as inhibitors. Additionally, the pharmacokinetic and safety profiles, evaluated through PAINS and ADMET filtering, suggest that these compounds have favorable drug-like properties and are unlikely to cause off-target effects. The compounds also exhibited good absorption and bioavailability and a predicted range of biological activities, including anticancer properties. By repurposing these FDA-approved drugs, the study offers a streamlined pathway toward developing novel therapeutics for lung cancer. Bagrosin and dehydrocholic acid, with their established pharmacokinetic profiles and promising anticancer potential, warrant further preclinical investigations to provide a cost-effective solution for developing new cancer therapies.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Dwyer, L. L. et al. Disparities in lung cancer: A targeted literature review examining lung cancer screening, diagnosis, treatment, and survival outcomes in the United States. J. Racial Ethn. Health Disparities 11, 1489–1500 (2024).
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 73, 17–48 (2023).
Arnold, M. et al. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995–2014 (ICBP SURVMARK-2): A population-based study. Lancet Oncol. 20, 1493–1505 (2019).
Jha, P. Avoidable global cancer deaths and total deaths from smoking. Nat. Rev. Cancer 9, 655–664 (2009).
Gupta, D., Gupta, N., Singh, N. & Prinja, S. Economic evaluation of targeted therapies for anaplastic lymphoma kinase–and ROS1 fusion-positive non–small cell lung cancer in India. JCO Global Oncol. 10, e2300260 (2024).
Vieira, A. R. et al. Fruits, vegetables and lung cancer risk: A systematic review and meta-analysis. Ann. Oncol. 27, 81–96 (2016).
Thun, M. J. et al. 50-year trends in smoking-related mortality in the United States. N. Engl. J. Med. 368, 351–364 (2013).
Parkin, D. M., Bray, F., Ferlay, J. & Jemal, A. Cancer in africa 2012. Cancer Epidemiol. Biomark. Prev. 23, 953–966 (2014).
Stacpoole, P. W. Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. J. Natl. Cancer Inst. 109, 071 (2017).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: The next generation. cell 144, 646–674 (2011).
Liu, J. et al. RRAD inhibits the Warburg effect through negative regulation of the NF-κB signaling. Oncotarget 6, 14982 (2015).
Iansante, V. et al. PARP14 promotes the Warburg effect in hepatocellular carcinoma by inhibiting JNK1-dependent PKM2 phosphorylation and activation. Nat. Commun. 6, 7882 (2015).
Gottfried, E. et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107, 2013–2021 (2006).
Goetze, K., Walenta, S., Ksiazkiewicz, M., Kunz-Schughart, L. A. & Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 39, 453–463 (2011).
Xie, J. et al. Beyond Warburg effect–dual metabolic nature of cancer cells. Sci. Rep. 4, 4927 (2014).
Shestov, A. A. et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. elife 3, e03342 (2014).
Semenza, G. L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20, 51–56 (2010).
Denko, N. C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705–713 (2008).
Vaupel, P., Schmidberger, H. & Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 95, 912–919 (2019).
Sugden, M. & Holness, M. Therapeutic potential of the mammalian pyruvate dehydrogenase kinases in the prevention of hyperglycaemia. Curr. Drug Targ. Immune Endocr. Metab. Disord. 2, 151–165 (2002).
Harris, R. A., Huang, B. & Wu, P. Control of pyruvate dehydrogenase kinase gene expression. Adv. Enzyme Regul. 41, 269–288 (2001).
Patel, M. S. & Korotchkina, L. G. Regulation of mammalian pyruvate dehydrogenase complex by phosphorylation: Complexity of multiple phosphorylation sites and kinases. Exp. Mol. Med. 33, 191–197 (2001).
Roche, T. A. & Hiromasa, Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell. Mol. Life Sci. 64, 830–849 (2007).
Lu, C.-W. et al. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am. J. Pathol. 179, 1405–1414 (2011).
Anwar, S., Shamsi, A., Mohammad, T., Islam, A. & Hassan, M. I. Targeting pyruvate dehydrogenase kinase signaling in the development of effective cancer therapy. Biochim. Biophys. Acta. Rev. Cancer 1876, 188568. https://doi.org/10.1016/j.bbcan.2021.188568 (2021).
Lu, C.-W., Lin, S.-C., Chen, K.-F., Lai, Y.-Y. & Tsai, S.-J. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J. Biol. Chem. 283, 28106–28114 (2008).
Feng, L., Cheng, K., Zang, R., Wang, Q. & Wang, J. miR-497-5p inhibits gastric cancer cell proliferation and growth through targeting PDK3. Biosci. Rep. 39, BSR20190654 (2019).
Wang, L.-Y. et al. KDM4A coactivates E2F1 to regulate the PDK-dependent metabolic switch between mitochondrial oxidation and glycolysis. Cell Rep. 16, 3016–3027 (2016).
Pecoraro, C. et al. 1, 2, 4-Amino-triazine derivatives as pyruvate dehydrogenase kinase inhibitors: Synthesis and pharmacological evaluation. Euro. J. Med. Chem. 249, 115134 (2023).
Kato, M., Li, J., Chuang, J. L. & Chuang, D. T. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure 15, 992–1004 (2007).
Gan, L. et al. Targeting the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis to discover potent PDK inhibitors through structure-based virtual screening and pharmacological evaluation. Euro. J. Med. Chem. 264, 116008 (2024).
Tso, S.-C. et al. Structure-guided development of specific pyruvate dehydrogenase kinase inhibitors targeting the ATP-binding pocket. J. Biol. Chem. 289, 4432–4443 (2014).
Nowak-Sliwinska, P., Scapozza, L. & i Altaba, A. R. Drug repurposing in oncology: Compounds, pathways, phenotypes and computational approaches for colorectal cancer. Biochim. Biophys. Acta (BBA) Rev. Cancer 1871, 434–454 (2019).
Cui, J. J. et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal–epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 54, 6342–6363 (2011).
Ohbayashi, N. et al. Structural basis for the inhibition of cyclin G-associated kinase by Gefitinib. ChemistryOpen 7, 713–719 (2018).
Wang, M. et al. Sunvozertinib, a selective EGFR inhibitor for previously treated non–small cell lung cancer with EGFR exon 20 insertion mutations. Cancer Discov. 12, 1676–1689 (2022).
Ali, S. et al. Identification and evaluation of inhibitors of lipase from Malassezia restricta using virtual high-throughput screening and molecular dynamics studies. Int. J. Mol. Sci. 20, 884 (2019).
Mohammad, T., Mathur, Y. & Hassan, M. I. InstaDock: A single-click graphical user interface for molecular docking-based virtual high-throughput screening. Brief. Bioinform. 22, bbaa279 (2021).
DeLano, W. L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 40, 82–92 (2002).
Studio, D. Discovery studio. Accelrys [2.1] 420 (2008).
Van Der Spoel, D. et al. GROMACS: Fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005).
Kato, M., Chuang, J. L., Tso, S. C., Wynn, R. M. & Chuang, D. T. Crystal structure of pyruvate dehydrogenase kinase 3 bound to lipoyl domain 2 of human pyruvate dehydrogenase complex. EMBO J. 24, 1763–1774 (2005).
Lagunin, A., Stepanchikova, A., Filimonov, D. & Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 16, 747–748 (2000).
Deng, Y. & Roux, B. Computations of standard binding free energies with molecular dynamics simulations. J. Phys. Chem. B 113, 2234–2246 (2009).
Naqvi, A. A., Mohammad, T., Hasan, G. M. & Hassan, M. I. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr. Top. Med. Chem. 18, 1755–1768 (2018).
Adnan, M. et al. Targeting inhibition of microtubule affinity regulating kinase 4 by Harmaline: Strategy to combat Alzheimer’s disease. Int. J. Biol. Macromol. 224, 188–195. https://doi.org/10.1016/j.ijbiomac.2022.10.115 (2023).
da Silva Rocha, S. F., Olanda, C. G., Fokoue, H. H. & Sant’Anna, C. M. Virtual screening techniques in drug discovery: Review and recent applications. Curr. Top. Med. Chem. 19, 1751–1767 (2019).
Shamsi, A. et al. MARK4 inhibited by AChE inhibitors, donepezil and rivastigmine tartrate: Insights into Alzheimer’s disease therapy. Biomolecules 10, 789. https://doi.org/10.3390/biom10050789 (2020).
Anwar, S. et al. Structure-based investigation of MARK4 inhibitory potential of Naringenin for therapeutic management of cancer and neurodegenerative diseases. J. Cell. Biochem. 122, 1445–1459 (2021).
Yousuf, M. et al. Structure-guided design and development of cyclin-dependent kinase 4/6 inhibitors: A review on therapeutic implications. Int. J. Biol. Macromol. 218, 394–408 (2022).
Rathi, A. et al. Exploring the potential of baicalin and resveratrol as PIM-1 kinase inhibitors: Therapeutic targeting of prostate and breast cancers. J. Mol. Liquids 396, 124026 (2024).
Taiyab, A. et al. Exploring MTH1 inhibitory potential of Thymoquinone and Baicalin for therapeutic targeting of breast cancer. Biomed. Pharmacother. 173, 116332 (2024).
Anwar, S. et al. Rosmarinic acid exhibits anticancer effects via MARK4 inhibition. Sci. Rep. 10, 10300 (2020).
Liu, X. et al. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 13, 23–37 (2018).
Anjum, F. et al. Impact of single amino acid substitutions in parkinsonism-associated deglycase-PARK7 and their association with Parkinson’s disease. J. Pers. Med. 12, 220. https://doi.org/10.3390/jpm12020220 (2022).
Ali, S. A., Hassan, M. I., Islam, A. & Ahmad, F. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr. Protein Pept. Sci. 15, 456–476. https://doi.org/10.2174/1389203715666140327114232 (2014).
Amir, M. et al. Investigation of conformational dynamics of Tyr89Cys mutation in protection of telomeres 1 gene associated with familial melanoma. J. Biomol. Struct. Dyn. 39, 35–44. https://doi.org/10.1080/07391102.2019.1705186 (2021).
Altis, A., Otten, M., Nguyen, P. H., Hegger, R. & Stock, G. Construction of the free energy landscape of biomolecules via dihedral angle principal component analysis. J. Chem. Phys. 128 (2008).
Iftikhar, H., Ali, H. N., Farooq, S., Naveed, H. & Shahzad-ul-Hussan, S. Identification of potential inhibitors of three key enzymes of SARS-CoV2 using computational approach. Comput. Biol. Med. 122, 103848 (2020).
Zhang, X. et al. Dehydrocholic acid ameliorates sodium taurocholate-induced acute biliary pancreatitis in mice. Biol. Pharm. Bull. 43, 985–993 (2020).
Acknowledgements
MIH acknowledges the Council of Scientific and Industrial Research for financial support [Project No. 27(0368)/20/EMR-II]. The authors extend their appreciation to Taif University, Saudi Arabia for supporting this work through project number (TU-DSPP-2024-140). AS thanks to Ajman University of the payment of APC.
Funding
This work is supported by the Indian Council of Medical Research (Grant No. ISRM/12(22)/2020) and Taif University, Saudi Arabia, Project Number (TU-DSPP-2024–140).
Author information
Authors and Affiliations
Contributions
Z.F.K.: Conceptualization, Investigation, Data curation, Validation, Writing—original draft, A.R.: Investigation, Validation, Formal analysis, A.K.: Validation, Formal analysis, F.A.: Methodology, Investigation, Validation, A.C.: Investigation, Validation, Formal analysis, A.T.: Validation, Formal analysis, Writing—review and editing, A.S.: Data curation, Validation, Formal analysis, Writing—review and editing, Md. I.H.: Conceptualization, Funding acquisition, Supervision, Project administration, Writing—review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khan, Z.F., Rathi, A., Khan, A. et al. Exploring PDK3 inhibition in lung cancer through drug repurposing for potential therapeutic interventions. Sci Rep 14, 29672 (2024). https://doi.org/10.1038/s41598-024-78022-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-78022-0
