
Abstract
Computational HLA typing has surged as a cost-effective strategy to uncover questions regarding the evolution of the HLA system, enabling immunogenic characterization from ancient DNA (aDNA) data. Nevertheless, it remains to be seen whether these methods are suitable for analyzing aDNA generated without target-enrichment. To investigate this, we evaluated the performance of five HLA typing tools using present-day data with simulated profiles typical of aDNA, as well as from high-coverage aDNA genomes downsampled at different read depths. We found that characterization of Class I genes at the first field resolution is feasible at read depths as low as 2x, where it retains an accuracy of ≈ 80%. Next, we used this insight to characterize HLA evolution in Europe from 154 ancient genomes by detecting allele frequency changes throughout distinct prehistoric European populations. We observed important shifts in alleles associated with infectious and autoimmune diseases, most of which are found by contrasting the HLA landscape of Neolithic Farmers to that of present-day. Interestingly, several of these observations are in line with findings that have been previously reported by target-enrichment-based studies. Our results highlight the feasibility of applying HLA typing on shotgun aDNA data to examine the evolution of this loci during important transitions.
Similar content being viewed by others


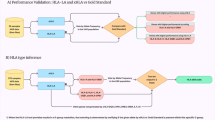
Introduction
The emergence of next generation sequencing has greatly transformed our understanding of immunogenetic variation and its relationship with disease. Notably, these technologies offer a wider scope for studying the genetics of the Human Leukocyte Antigen (HLA) system, allowing researchers to overcome the difficulties of characterizing this region using traditional PCR-based techniques, such as being labor intensive, relatively hard to scale, or presenting a lower typing resolution1. In the last decade, computational strategies to perform HLA typing have been developed by making use of the vast amount of HLA sequencing data available in public repositories2,3. These strategies generally work by aligning high-throughput sequencing reads mapped to the HLA region against a comprehensive panel of known alleles, followed by a prediction performed by a probabilistic model that determines the most likely allele pair for each gene4.
Key applications of HLA typing have already been brought to light by projects that focus on characterizing the HLA region of hundreds of present-day individuals to establish reference panels that comprehend the immunogenic variation of specific populations5,6. Other studies have reported associations between specific HLA alleles and some clinically relevant phenotypes such as susceptibility to infectious diseases7,8,9,10 or autoimmune and inflammatory disorders11,12,13,14,15. Furthermore, advances in DNA extraction techniques and sequencing methods have permitted the comprehensive study of the genetic material from ancient individuals (aDNA). These analyses are challenging because the genetic material of ancient individuals often presents low levels of endogenous DNA content and exhibit distinct patterns of degradation such as fragmentation and cytosine deamination16.
In view of this, recent efforts have harnessed the power of computational HLA typing methods onto paleogenomics data by making use of target-enrichment of the HLA region followed by sequencing. These methods enable the immunogenetic characterization of ancient individuals from different time periods and geographic origins. For instance, a study by Barquera et al.17 analyzed three ancient genomes from African individuals dated to 16th century Colonial Mexico and performed HLA typing, which further supported their Sub-Saharan African origin. Hepatitis B Virus genomes were recovered from these individuals and HLA alleles associated with poor response to this virus infection were characterized, providing key insights on their predisposition to infectious diseases and their overall health status. Similarly, Krause-Kyora et al.18 and Immel et al.19 recovered and studied ancient genetic data from 36 German and 68 Danish individuals retrieved from medieval cemeteries with victims of leprosy and plague, respectively. By characterizing the HLA system from these historical samples, they were then able to detect genetic signatures of susceptibility to their burial-associated disease and to contrast the observed frequencies of relevant HLA alleles to those present in present-day populations.
Another study20 investigated the HLA genetic variation present in 42 individuals dated to the Late Neolithic period in Germany. Alleles associated with an increased predisposition to certain bacterial infections were overrepresented in the ancient population. It was also found that there have been major shifts in the diversity present in the HLA allelic pool between Neolithic farmers in Central Europe and that of present-day Germans. Those shifts are compelling given the context of the so-called “Neolithic Revolution’’, one of the most important events in our recent evolutionary history, where a transition from a hunter-gatherer lifestyle to sedentism occurred among prehistoric West-Eurasian populations. The spread of farming and agriculture shaped the genomic landscape of present-day Europe during this turnover. In particular, domestication processes may have enhanced the risk of infections by zoonotic pathogens, potentially driving selection in immune genes involved in response to such infections21,22,23.
Altogether, these results reveal the feasibility of examining host-pathogen interactions through time using data from specific historical contexts and show that the employment of target-enrichment sequencing strategies enables the precise typing of HLA alleles from tens of ancient individuals. However, we currently do not know if computational methods to perform HLA typing can also be applied using data generated by Whole Genome Sequencing (WGS) without HLA target-enrichment, particularly for aDNA data. Typically, the average read depth for most shotgun-sequenced ancient genomes is exceedingly low (≤ 1 × )24,25; nevertheless, a notable number of samples have been generated to higher depths of coverage19,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45. Being able to employ such datasets would allow the screening of genetic data from individuals that lived through important events that might’ve had an impact on their immunogenetic makeup, such as the peopling of new environments46, epidemics47 or admixture events48.
In this study we carried out an extensive evaluation regarding the performance of multiple HLA typing tools (Supplementary Table 1), focusing on the performance of these strategies on low-coverage WGS aDNA data. We estimated the performance of HLA typing using both a dataset where we introduced damage patterns akin to those present in aDNA, and another one consisting of high coverage aDNA samples downsampled at different levels of read depth. We then used these assessments to develop a framework for the characterization of Class-I HLA alleles from ancient individuals with a read depth ≥ 2x. We highlight the feasibility of performing HLA evolutionary analyses on low-coverage ancient WGS data by studying HLA Class I evolution in Europe, tracing their change in frequency in time and across prehistoric populations with different demographic origin, using data from: European Hunter Gatherers (HG), Neolithic Farmers (NF), Bronze Age (BA) (Fig. 1) and Present Day (PD) individuals from European descent.

Panel (A). Geographic distribution and population labels based on genetic affinity of 154 prehistoric European samples employed in this study. Panel (B). PCA plot of the 154 ancient individuals from different prehistoric populations. Panel (C). ADMIXTURE plot of the 154 ancient individuals at K = 8.
Results
Assessment of HLA typing performance on present-day WGS data
We evaluated the accuracy achieved by five computational HLA typing strategies: HLA-HD49, HLAScan50, HLA-HD51, OptiType52 and the implementation of a simple baseline for fully-automated allelic calls that we refer to as “Bowtie2 Multiple Alignment (Bowtie2 MA)”, which is partially based on the semi-automated pipeline described in Krause-Kyora et al., (2018)18 (Supplementary Information, Supplementary Table 1). We tested these strategies on a panel of 176 individuals from the One-Thousand Genomes Project (1KGP)53 that had experimental HLA typing available for classical Class I and II genes54,55 (Supplementary Data 1.1, 1.2) by performing allele calls with each method at different read depths(10x-1x) on these individuals (Supplementary Data 1.3). As expected, we noted that all methods displayed diminishing performance as read depth decreased (Supplementary Figure S1, Supplementary Data 1.4). We also found that the highest accuracy was consistently achieved by OptiType for Class I across all depths, with HLAScan scoring last for the typing of both Class I and II genes in every instance (Supplementary Data 1.4). With a mean accuracy of 0.97 both at the first and second fields of resolution, OptiType achieved the highest performance using samples at 10x read depth. The next best result here was obtained by HLA-LA with a mean accuracy of 0.97 at the first field and 0.88 at the second. Despite showing the best performance amongst all evaluated methods for Class I typing, OptiType does not currently support typing of Class II genes; thus, the highest accuracy for Class II typing results at 10x was achieved by HLA-LA with 0.91 at the first field resolution and 0.77 at the second, with HLA-HD coming second with 0.86 and 0.71, respectively.
Given the overall sparse performance for Class II typing (Supplementary Fig. 1, Supplementary Data 1.4), we decided to discard these genes for subsequent analyses. Regarding the influence of the demographic origin of the 1KGP screened samples in the performance of these tools, we noted no significant difference in the accuracy achieved regardless of their overall ancestry (Central European, Eastern Asian, Western African) (Two-Way ANOVA, Supplementary Information, Supplementary Data 2). On the other hand, when analyzing the performance of Class I typing per gene, we detected significantly lower accuracy obtained for HLA-B typing with respect to both HLA-A and HLA-C (Supplementary Information, Supplementary Data 2).
Evaluation of typing accuracy on simulated aDNA
We first assessed the performance of these tools on aDNA sequencing data by simulating DNA fragmentation and deamination patterns similar to those present in ancient samples due to DNA damage and degradation using gargammel56. We incorporated these aDNA profiles onto our samples from 1KGP to generate this 1KGP-aDNA dataset, carried out HLA typing with the computational methods to be tested and measured the accuracy achieved by them. Comparing HLA typing between WGS with and without simulated aDNA profiles, we found an average difference in accuracy of around 0.07 and 0.14 for the first and second field typing resolution using OptiType, respectively (Supplementary Data 3). Moreover, on this evaluation we noted a similar trend to that found when evaluating 1KGP data without aDNA-like damage patterns, where typing accuracy correlated with the read depth of the samples (Fig. 2, Supplementary Figure S3). OptiType continually performed the best out of all the typing methods implemented for the typing of Class I genes (Fig. 2). This method yielded accurate results at the medium (5x) and low (2x) read depths.; particularly for the HLA-C gene at the first field resolution, where this method achieved mean accuracy values of 0.86 and 0.75, respectively. In contrast, three out of the five methods tested were not able to provide a prediction for the data with artificially generated aDNA profiles. The only other method able to handle this type of data was the Bowtie2 MA pipeline, achieving lower accuracy than OptiType for all read depths tested (Fig. 2).

Mean and range of accuracy values achieved on data with artificially introduced aDNA profiles by HLA typing for Class-I genes at first field resolution, for different read depths: 10x-1x (sample size for each read depth were 20, 26, 44, 66, 91, 131, 153, 163, 175 and 176, respectively).
Assessment of HLA typing performance on high-coverage ancient genomes
Next, we evaluated the performance of these HLA typing strategies on a dataset consisting of 56 high-coverage (> 10x) aDNA genomes that were downsampled to 10 different levels of read depth (10-1x) (Supplementary Data 4.1). We compared the typing obtained at each level against the typing produced at high-coverage, which were used as benchmark reference to compare the typing at lower read depths (Supplementary Information) (Supplementary Data 4.2–4.3). In agreement with our evaluation on simulated aDNA profiles, we found a clear association between the concordance and the read depth of the samples, with OptiType consistently attaining the best results for HLA typing on empiric aDNA data (Fig. 3, Supplementary Figure S4). Markedly, this typing tool kept providing results concordant with those obtained at high coverage even when downsampled at read depths as low as 2x, particularly at the first field of resolution, where it achieved mean concordance values of 0.77, 0.68 and 0.89 for HLA-A, HLA-B and HLA-C, respectively (Supplementary Data 3). For this evaluation, the Bowtie2 MA pipeline also displayed poor concordance results relative to OptiType (Fig. 3, Supplementary Figure S4).

Mean and range of concordance values achieved on high-coverage aDNA data by HLA typing for Class I genes at first field resolution, for different read depth levels: 10x-1x (N = 56).
Shifts in HLA allele frequencies traced to neolithic farmers
We decided to leverage the availability of ancient genomes from prehistoric Europe with ≥ 2x average read depth to investigate the evolution of classical HLA Class I allele frequencies across prehistoric populations with a different demographic origin. Using the insight provided by our evaluations, we created a dataset composed of 154 individuals dated to different temporal periods from prehistoric Europe and grouped them based on their genetic affinities to European HG, NF or BA populations as reported in the literature31,35,45 (N = 48, 89 and 17; respectively) (Supplementary Figures S6-S8, Supplementary Data 5.1, 5.2). We conducted HLA typing for Class I genes using OptiType and estimated the allelic frequencies present in each prehistoric population. Then, we estimated present-day HLA allele frequencies following a similar process on a subset of 1KGP individuals of European descent (CEU and GBR). Additionally, we screened HLA allele frequencies of present-day Germany, England, France, and Ireland from the Allele Frequencies Database3 (Supplementary Table S3); all of which were found to work as statistically similar proxies for estimates of present day frequencies (Chi-Squared Test of homogeneity, Supplementary Data 6). To emphasize the validity of using low-coverage data, we also performed this analysis after downsampling the original depth of coverage of all 154 prehistoric European genomes to 2x. HLA typing for Class I genes was again carried out using OptiType, and the allelic frequencies were estimated for each prehistoric and present day population and annotated accordingly.
From these analyses, we identified six alleles at the first field resolution (HLA-A: 1, HLA-B: 3, HLA-C: 2), as well as two at the second field (HLA-A: 1, HLA-B: 1) that showed shifts in frequency across the prehistoric and present day European populations, and fell outside the 95 CI of a background distribution comprised of frequency changes from a panel of ≈ 50k genome-wide SNPs used to discern statistically significant shifts. Most of these signals are observed when contrasting the frequencies of NF to that of PD Europe (Fig. 4, Supplementary Data 5.3, 5.4).

Change in frequency observed between different prehistoric populations (N = HG: 48, NF: 89, BA: 17) and present day Europeans for HLA Class I alleles that presented shifts outside the 95 CI of a distribution of frequency changes from a panel of genome-wide SNPs. Panel A describes the trajectory as derived from frequencies obtained from typing at the original ≥ 2x read depth. Panel B describes the trajectory as derived from frequencies obtained from typing downsampled data at 2x read depth. Present-day frequencies are represented by the estimates from 99 CEU individuals, 91 GBR individuals, and the average of four European populations reported in the AlleleFrequencies Database (N = 20,980).
Several of these observations have been reported in studies that investigated HLA evolution using target-enrichment HLA generated data derived from geographically-localized archeological sites20,23. Notably, these HLA alleles have also been associated with susceptibility or resistance to various infectious pathogens, as well as autoinflammatory diseases57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72 (Table 1). Nevertheless, we observe that, after correcting for multiple testing (Benjamini-Hochberg procedure to control for the False Discovery Rate, q = 0.1), only one allele had a significant frequency change: HLA-C*07 (Table 1, Supplementary Data 7).
Immunologically relevant SNPs display different patterns of change in time
Due to the pivotal role that HLA genes play in immunity, we wanted to contrast the changes in frequency observed for these loci with other SNPs involved in immunological processes73,74,75,76,77,78,79 and further characterize changes in the immunological landscape of Europeans across time. Analogous to our analysis on HLA alleles, we employed a distribution of frequency changes from a panel of genome-wide SNPs (N = 36,636) to test for variants that have sustained a significant shift in frequency through time (95% CIs, Table 2, Supplementary Figure S9). We found a total of 37 SNPs that displayed significant changes in frequency (12 increases, 25 decreases) ranging from 19% to up to 52% in allele frequency difference magnitude, most of which also correspond to shifts that seemed to arise when contrasting frequencies present in European Hunter Gatherers and Neolithic Farmers from prehistoric times to those from present-day Europeans (Supplementary Data 8).
Discussion
In this study we gauged the use of five computational strategies for the allelic characterization of HLA genes on WGS aDNA samples. Our evaluations show that OptiType is the most useful tool for computational Class I HLA typing at different levels of read depth and suggest that the allelic characterizations of these genes on aDNA data is attainable. This is especially true at the first field of resolution, which is often enough to find clinically-relevant associations80 and for which we observe mean concordance values of ≥ 0.95 when analyzing data at medium (5x) to high (10x) read-depths, and still achieve mean accuracy results as high as 0.89 for HLA-C at read depths as low as 2x. The typing of the HLA-B was found to be significantly less reliable than other Class I genes, a remark that is in accordance with other observations that have found this gene particularly difficult to type81,82, arguably due to it being the most polymorphic of them as well as having the highest recombination rate2,83.
While our validation highlights meaningful findings at both resolutions, it is clear that typing at the second-field is far less reliable due to the inherent challenges of using low-coverage aDNA data, and more information is needed to provide an accurate result. In line with this, an approximation that standardizes HLA frequency data by collapsing allele calls to the more robust first field resolution seems to help achieve greater accuracy as well as circumvent some of the confounding effects that might arise from variations in sample size or demographic origin, as it’s been reported when comparing the HLA frequencies of hundreds of populations worldwide84. On the other hand, for medium-to-high levels of genomic coverage, the use of OptiType still provides a decent lead for the typing at the second field (Figs. 2 and 3). If needed, manual annotation of variants at this resolution is made easier by first exploring the calls provided by these tools and then using other software to manually validate them (e.g., comparing the typing results and multiple allelic reference sequences using a manual alignment visualizer as described in Pierini et al. (2020)82).
Notably, none of the existing fully-automated typing tools were designed to provide HLA typing of ancient genomes. We believe that there is an area of opportunity in the development of HLA typing computational tools that consider aDNA-specific features into account during the mapping process to improve the accuracy of the reconstructed ancient immunogenetic profiles. Some of the shortcomings arise from the usual intricacies of reference mapping of reads coming from ancient genomes, like an unalterable default behavior that expects reads of a fixed length as input51 or being optimized for present day data available at explicitly high levels of genomic depth of coverage49,50,51. However, we also identified some aspects of certain tools that may hinder their performance, such as the use of outdated HLA allele references for mapping52 or not considering the intronic regions of the HLA region to infer the type50.
In spite of this, we were able to observe promising results for Class I typing using OptiType, showcasing the feasibility of adapting such tools to accommodate some of the difficulties that come with analyzing aDNA. Our study also draws attention to the challenge of typing classical Class II genes on this kind of data, which is not supported by the vast majority of computational typing methods. It is worth noting that studies carried out by a handful of research groups that analyzed these genes on ancient genomes have relied on an unpublished “OptiType 2” version of this tool19,74,85, that has yet to be made publicly available and benchmarked. Although OptiType has proven to be the most reliable tool for the characterization of HLA alleles, we found that Class II genes seem to be notably troublesome to type accurately. It would then be sensible to take caution with any results produced by a tool that hasn’t been openly evaluated on this type of data, and every instance of its use should probably be validated with manual verification of the allelic calls.
Regarding the application of this framework to study the evolution of Class I genes in Europe, we found that the largest shifts in HLA allele frequencies happen between Neolithic farmers and present-day populations. These groups present the largest sample sizes, and population genetic analyses suggest that these observations are not caused by population structure, as individuals grouped together in these periods have a similar demographic origin (Fig. 1). The variants with the most pronounced shifts in frequency might reflect the result of selective forces acting on human immunity genes and are mainly associated with susceptibility or resistance to infectious and autoinflammatory diseases in the European population. These results are supported by the fact that we recover half of the signals found when we downsampled the depth of coverage for all individuals to 2x (HLA-B*07, HLA-B*51, HLA-C*07, Supplementary Data 5.4), with the observed trends clearly persisting despite the fact that half of the signals marginally fell outside of the threshold imposed in this study to discriminate statistical significance (Fig. 4; Table 1, Supplementary Data 7).
While our experimental design based on shotgun data from individuals across Europe only detected one significant frequency shift (HLA-C*07; p value = 0.002, Supplementary Data 7) across prehistoric groups and present day (after correcting for multiple tests), we consider that this approach of contrasting changes in HLA allele frequency against the allele frequency of a background distribution of genome-wide SNPs is rather stringent (see methods). Using less strict methods to denote important changes such as a simple test for difference of proportions casts a greater amount of statistically significant changes such as those noted in Immel et al. (2020)20. (Chi-Squared Test for homogeneity of proportions with Bonferroni correction for multiple comparisons, Supplementary Data 9). Nonetheless, the fact that we have replicated such patterns (generated through target-enrichment and sampled at localized regions i.e. Central Europe) using shotgun generated data across Europe, suggests that our observations might be of evolutionary interest. As it implies that these signals might not be restricted to shifts in the HLA frequency to a very localized European region, but instead could be linked to adaptive or demographic events that shaped the genetic landscape across prehistoric Europe. Further studies investigating the HLA repertoire of Hunter-Gatherer, Neolithic Farmer and Bronze Age individuals at larger scales and in regions of Europe will help shed light on this matter.
Although most of the signals observed were deemed as not statistically significant from our approach after correction for multiple comparisons, some interesting observations can be drawn by inquiring into the alleles that displayed the greater shifts in frequency across prehistoric and contemporary Europe. For instance, alleles HLA-B*07 and HLA-B*27, which seemed to be common in European HG populations, appear to fade away with the more recent demographic events that shaped the genetic landscape of present day Europe. In line with previous findings20,23, some of the variants that appear to be less frequent today seem to confer protective effects against viral infections were more frequent in Neolithic farmers, such as HLA-B*27, HLA-B*51 and HLA-C*02, underscoring the clinical relevance of these results. Particularly intriguing are also some of the more distinct signals regarding autoimmune diseases, such as the surge of frequency in allele HLA-C*07 first displayed in Neolithic farmers and its correspondence with the consistent decrease in frequency of HLA-B*27 across time. Both of these alleles are linked to Spondylarthritis, though in the former case it has been described as a protective variant70, while in the latter as a risk factor63. Other alleles related to autoimmune disorders, such as HLA-A*24 or HLA-B*51 (associated to Behçet’s disease susceptibility64,71), show a steady decline in frequency across the different periods; whereas HLA-B*07, which has been linked to a predisposition to Type-1 Diabetes62, seems to be steadily rising in frequency.
Expanding beyond the analysis of classical HLA alleles, we enriched our study by also considering immunologically relevant SNPs to investigate the immunological landscape of ancient Europe in a more comprehensive manner. The patterns of change, as revealed by these SNPs, complement our observations in the frequency of variants linked to autoimmune and autoinflammatory diseases. Taken together, they offer a more comprehensive understanding of the evolving immune repertoire in European populations through different historical periods. There still exists a need for a more comprehensive understanding of how HLA and other immunologically relevant variants have changed over time, and we hope that these findings can help underline the importance of growing the repertoire of paleogenomic data in order to have a more complete picture of the complex demographic events that have sculpted the diverse immunological landscape that we see in the present day. Other complementary analyses, such as using proxy SNPs or HLA genotype imputation could further extend the scope of paleogenomic studies that intend to examine immunogenic information of ancient genomes.
By examining historical transitions and their influence on our immunologic architecture through time, it is possible to obtain key insight on the molecular basis of our immune repertoire, as well as the interplay that is carried out between them and pathogens. We find it encouraging that using WGS low-coverage aDNA we were able to observe some of the results depicted in other studies20,23,74 that employed target-enrichment techniques, which are more sensitive to accurate HLA typing than shotgun-generated data. This observation is of great significance for projects that aim to investigate the evolution of the immunological landscape in human populations, as it provides the potential to leverage a larger sample size to explore the allelic composition of Class I HLA genes throughout time, and takes advantage of the fact that there exist hundreds of partial and complete shotgun-generated ancient genomes publicly available that were not previously considered for HLA typing characterization. As the field of paleogenomics continues to advance, it holds the promise of uncovering how particular past evolutionary events forged the genetic variation of the immunogenic system of human populations. This comprehensive exploration of computational HLA typing strategies on low-coverage aDNA data contributes to the ever-growing body of knowledge in this field, laying some ground for a deeper understanding of the immunogenetic evolution of human populations and its relevance to health and clinical research.
Methods
Assembly of reference panels for HLA-typing analysis
We downloaded Whole Genome Sequencing (WGS) data from the 1KGP53 database for individuals that had available their HLA typing information determined experimentally by previous studies54,55. We obtained this information for a total of 176 individuals for Class I genes and 48 individuals for Class II genes. We then used their PCR-based typing results as our Gold Standard (GS) HLA allele reference panel both at the first and second field of resolution (Supplementary Data 1.1–1.2).
Obtention and processing of 1KGP and high coverage aDNA samples
For all individuals from the GS reference panel, their corresponding WGS FASTQ files were downloaded from the 1KGP database. After sample pre-processing (trimming and quality control), reads from all individuals were aligned to the human reference genome GRCh37/hg19 with the Burrows-Wheeler Aligner (BWA) (v0.7.10)86 using bwa-aln with parameters -l 16,500 -n 0.01 and -o 2. BAM files were generated, and all reads that aligned to the HLA system and had a mapping quality greater than 30 were extracted using SAMTools (v1.19)88 using parameters -F 4, -q 30 and -L to bed coordinates corresponding to the HLA region (6 28477700 33448400). For each individual, we generated a subdataset at different levels of read depth according to the original read depth present (10x-1x), using samtools view -s.
BAM files from 56 high coverage aDNA genomes were obtained from their corresponding publication repositories (Supplementary Data 4.1) and reads mapped to the HLA region were extracted. For typing methods that required FASTQ files as input, we converted the BAM file from the HLA region to the FASTQ format using samtools fastq. A subdataset consisting of differential read depth instances (10x-1x) was generated for each individual in a similar manner to the processing of the 1KGP dataset.
Incorporation of artificial aDNA profiles onto a 1KGP dataset
We generated a dataset composed of HLA reads from samples obtained from the 1KGP53 database that exhibit damage patterns akin to those present in aDNA samples (1KGP-aDNA) using gargammel56, a suite of programs aimed at simulating ancient DNA fragments and deamination patterns from modern data. From these tools, we adapted and made use of fragSim to simulate aDNA fragmentation in our samples, following a log-normal distribution with location 4.2 and scale 0.16 by using parameters: --norev --loc 4.2 --scale 0.16.
Subsequently, we employed deamSim to specify the deamination patterns to be present in our artificially damaged samples providing a misincorporation matrix that represents the empirical rates of misincorporation present in aDNA due to cytosine deamination; this misincorporation matrix is made available with the rest of the gargammel suite and is based on the profile of an individual found in La Braña who lived approximately 7,000 years ago87. Finally, for each individual we generated a subdataset of instances at different read depths (10x-1x) using samtools view -s.
Computational HLA typing
We implemented publicly available computational methods for HLA typing that have been reported in the literature to perform well on WGS data18,49,50,51,52 (Supplementary Table 1). All methods were run on each subdataset of the 1KGP, 1KGP introduced-aDNA profiles and the high coverage aDNA samples using default parameters or those recommended in their respective software documentation (Supplementary Table 1, Supplementary Information). After each successful run, the typing results for the supported HLA genes were annotated at both the first and second-field resolution. Computational typing methods evaluated were HLA-HD49, HLAScan50, HLA-LA51, OptiType52 and a pipeline based on the semi-automated method first described in a study of aDNA signatures of leprosy susceptibility18, that in this study we refer as “Bowtie2 Multiple Alignment (Bowtie2 MA)” (Supplementary Information). For each of the 176 1KGP individuals, the allelic predictions produced by these methods were compared against the GS reference at the first and second field resolution for HLA Class I (HLA-A, HLA-B, HLA-C) to assess their typing accuracy. Class II (HLA-DRB1, HLA-DQB1) typing accuracy was measured in a similar manner with the aforementioned computational tools for the 48 individuals with typing results present in the GS reference.
Assessment of computational HLA typing performance
For both the 1KGP and 1KGP-aDNA datasets, HLA typing results from all methods were directly compared to the PCR-based HLA genotyping reported in the GS reference panel at the first and second-field resolution. For each sample, typing accuracy was calculated as the ratio between the number of correctly typed alleles and the total number of alleles. This approximation was undertaken at the first and second field resolution across classical Class I (HLA-A, HLA- B, HLA-C) and Class II (HLA-DRB1, HLA-DQB1) genes. Assessment of the accuracy achieved by each method was carried out on every subdataset. The accuracy values of all methods were annotated for each group of genes and averaged to the total number of samples typed (Supplementary Data 1.3).
For the evaluation of typing of ancient genomes that have been sequenced at high coverage (> 10x) and had their WGS data publicly available18,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43 (Supplementary Data 4.1), these samples were typed using the evaluated strategies (OptiType and Bowtie2 MA) at their original read depth, and these allelic calls were used as the reference to measure the concordance at different read depths(10x-1x), calculating the proportion of allelic calls at that particular read depth that matched the typing at high coverage (> 10x) both at the first and second resolution fields. These results were annotated and summarized as described above (Supplementary Data 4.2–4.3).
Generation of aDNA dataset to assess HLA evolution in Prehistoric Europe
We employed 154 aDNA samples derived from people that lived in Europe at three different time periods: European hunter-gatherers (HG, N = 48), farmers from the Neolithic (NF, N = 89), and Bronze Age (BA, n = 17) individuals. Ancient genomes were downloaded from their respective publication repositories (Supplementary Data 5.1) as BAM files. Reads that aligned to the HLA region were extracted using SAMTools. Additionally, for the subsequent analysis at low coverage, these BAM files were downsampled to a standardized read depth of 2x, which corresponds to the read depth that entailed a high enough number of individuals from each period while still comprising enough read depth to achieve ≈ 80% of accuracy from computational HLA typing.
Principal component analysis
PCA was performed using smartpca from the EIGENSOFT package using the “numoutlieriter: 0” and “r2thresh: 0.2” parameters. For each ancient individual (N = 154), a PCA was conducted together with 991 present-day individuals from 67 European (EU), Near Eastern (NE) and Caucasian (Cau) populations extracted from the Human Origins panel v289,90 and each ancient sample (Supplementary Data 5.1). Individual .evec files were plotted using Procrustes transformation21 using all SNPs as described in Sanchez-Quinto et al. (2019)28. The result was displayed using an in-house R script from the vegan library. A preliminary PCA (Supplementary Figure S8) incorporating a larger panel (N = 184) of ancient individuals from prehistoric Europe was carried out to refine our dataset so that it was constituted by genetic affinities to prehistoric European populations: HG, NF and BA (Supplementary Information).
Unsupervised ADMIXTURE
Ancestry components were inferred using ADMIXTURE v1.391 based on: 1,758 present-day individuals from 179 world-wide populations from the Human Origins panel v2, 99 CEU individuals from the 1KGP53 and the 154 ancient individuals incorporated in this study (Supplementary Data 5.1). SNPs were ascertained using transversions as in Sánchez-Quinto et al. (2019)28 from the Human Origins panel v2. Common modes among the different runs were identified, and clusters were aligned across different values of K using pong (Supplementary Figure S6). Pong results suggested that K = 8 was the last greatest K at which more than 80% of runs produced the same model solution. A preliminary ADMIXTURE analysis (Supplementary Figure S7) incorporating a larger panel (N = 184) of ancient individuals from prehistoric Europe was carried out to refine our dataset so that it was constituted by genetic affinities to prehistoric European populations: HG, NF and BA (Supplementary Information, Supplementary Data 5.2).
Estimation of HLA allele frequencies throughout time in Europe
HLA typing was conducted using OptiType on our European aDNA dataset. Then, we simply computed the estimated allele frequencies at both the first and second field of resolution for each period. Manual validation of a subset of the calls produced by OptiType for six European high-coverage genomes was done using TARGT82, downsampling HLA reads present to high (10x), medium (5x) and low (2x) coverage. This validation involved manually assessing 108 alleles (six individuals screened at three different depth of coverage thresholds, assessing whether the corresponding alleles made by OptiType at each coverage level was concordant with the typing made at high-coverage at the first and second field resolution (Supplementary Table 5).
Present-day allelic frequencies for Class I HLA alleles were calculated using a panel of 99 CEU and 91 GBR individuals from 1KGP. Additionally, the frequency of HLA alleles that displayed significant changes throughout time was contrasted to those reported in the AlleleFrequencies Database3 for populations from Central/Northwestern Europe (Supplementary Table 6). We found no significant difference between the HLA allelic frequencies from any of these present-day different sources for HLA allele frequencies (Supplementary Data 6).
Generation of a background distribution of shifts in HLA allele frequency
To discern which changes in frequency from the HLA alleles investigated we would deem as significant, a set of background distributions was constructed for each pairwise comparison of scores of changes in allelic frequencies between European hunter-gatherers, Neolithic farmers, and Bronze Age and present-day individuals, respectively. Starting from a panel of 2,412,321 genome-wide SNPs transversions with a MAF of 5% in 1KGP populations53, we used plink v. 1.9392 to prune variants that were in approximate linkage disequilibrium with each other using --indep-pairwise 200 25 0.4. After this, we filtered out instances that had a minor allele frequency of less than 0.01 and a call rate greater than 80% across the different periods evaluated, using --maf 0.02 and --geno 0.2 as parameters in plink v. 1.9392, respectively. Lastly, we kept only SNPs that exhibited values of background selection and recombination rate similar (located in the same percentile ± 10) to those present in classical Class I HLA genes, as described in McVicker G. et al. (2009)93 and The International HapMap Consortium (2005)83. After all these filters, the frequency of 36,616, 60,031 and 56,588 variants (corresponding to HLA-A, HLA-B and HLA-C counts, respectively) were used to calculate differences in frequency between each of the periods evaluated. We used these values of difference in frequency between periods to generate a distribution of scores for each of the six following pairs of time periods per gene: HG-NF, HG-BA, HG-PD, NF-BA, NF-PD and BA-PD. We compared the observed changes in frequencies for HLA alleles with those distributions to discern which HLA alleles suffered a significant change in frequency than what would be expected by chance (95% Confidence Intervals (CIs), Supplementary Information).
Changes in frequency of immunologically relevant SNPs
We screened SNP alleles using our dataset of ancient individuals and compiled a total of 209 variants that have been previously described to play a role in distinct immunological processes73,74,75,76,77,78,79 and described in the literature before May 2023 (Supplementary Data 7.1).
In a similar way to the analysis of HLA allele frequencies, a distribution of scores of changes in frequency was constructed for changes between periods (HG-NF, HG-BA, HG-PD, NF-BA, NF-PD and BA-PD). Starting from a panel of 2,412,321 genome-wide SNPs, we used plink v. 1.9392 to prune variants that were in approximate linkage disequilibrium with each other using --indep-pairwise 200 25 0.4. After this, we filtered out instances that had a minor allele frequency of less than 0.01 and a call rate greater than 80% across the different periods evaluated, using --maf 0.02 and --geno 0.2 as parameters, respectively.
Statistical analyses
All statistical tests presented in this work were performed using the software R, v3.6.2.
Data availability
Sequence data for all ancient genomes employed in this work were obtained from their respective publications, which are referenced in Supplementary Data 4.1 and Supplementary Data 5.1. Sequence data for 1KGP individuals were obtained from the official database: The International Genome Sample Resource (IGSR).
References
Gabriel, C. et al. HLA typing by nextgeneration sequencing -- getting closer to reality. Tissue Antigens 83, 65–75 (2014).
Barker, D. J. et al. The IPD-IMGT/HLA database. Nucleic Acids Res. 51, D1053–D1060 (2022).
Gonzalez-Galarza, F. F. et al. Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 48, (2019).
Bauer, D. C., Zadoorian, A., Wilson, L. O. W. & Thorne, N. P. Evaluation of computational programs to predict HLA genotypes from genomic sequencing data. Briefings Bioinformatics 2, bbw097 (2016).
Nordin, J., Ameur, A., Lindblad-Toh, K., Gyllensten, U. & Meadows, J. R. S. SweHLA: the high confidence HLA typing bio-resource drawn from 1000 Swedish genomes. Eur. J. Hum. Genet. 28, 627–635 (2019).
Trachtenberg, E. et al. HLA class I (A, B, C) and class II (DRB1, DQA1, DQB1, DPB1) alleles and haplotypes in the Han from southern China. Tissue Antigens 70, 455–463 (2007).
Hernandez et al. Prevalent HLA Class II Alleles in Mexico City Appear to Confer Resistance to the Development of Amebic Liver Abscess. PLOS ONE10, e0126195 (2015).
Dubaniewicz, A., Lewko, B., Moszkowska, G., Zamorska, B. & Stepinski, J. Molecular subtypes of the HLA-DR antigens in pulmonary tuberculosis. Int. J. Infect. Dis. 4, 129–133 (2000).
Nishida, N. et al. Genome-Wide Association Study Confirming Association of HLA-DP with Protection against Chronic Hepatitis B and viral clearance in Japanese and Korean. PLoS ONE 7, e39175 (2012).
Cruz-Robles, D., Reyes, P. A., on-Padilla, M. & niz, O. M. On, V.-A. MHC class I and class II genes in Mexican patients with Chagas disease. Hum. Immunol. 65, 60–65 (2004).
Dallmann-Sauer, M. et al. The complex pattern of genetic associations of leprosy with HLA class I and class II alleles can be reduced to four amino acid positions. PLoS Pathog. 16, e1008818 (2020).
Hirata, J. et al. Genetic and phenotypic landscape of the major histocompatibilty complex region in the Japanese population. Nat. Genet. 51, 470–480 (2019).
Yatskiu, H. A., Zlotnikova, M. V., Sukalo, A. V. & Goncharova, R. I. Characteristic spectra of class I and II HLA-alleles in patients with different clinical forms of juvenile idiopathic arthritis in the Republic of Belarus. Doklady Natl. Acad. Sci. Belarus. 64, 209–216 (2020).
Erlich, H. A. et al. Next generation sequencing reveals the Association of DRB3*02:02 with type 1 diabetes. Diabetes 62, 2618–2622 (2013).
Enczmann, J. et al. Next Generation Sequencing Identifies the HLA-DQA1*03:03 Allele in the Type 1 Diabetes Risk-Associated HLA-DQ8 Serotype. Genes 12, 1879 (2021).
Orlando, L. et al. Ancient DNA analysis. Nat. Reviews Methods Primers 1, (2021).
Barquera, R. et al. Origin and Health Status of First-Generation Africans from Early Colonial Mexico. Current Biology 30, 2078–2091.e11 (2020).
Krause-Kyora, B. et al. Ancient DNA study reveals HLA susceptibility locus for leprosy in medieval europeans. Nature allentCommunications 9, (2018).
Immel, A. et al. Analysis of genomic DNA from medieval Plague victims suggests long-term effect of Yersinia pestis on Human immunity genes. Mol. Biol. Evol. 38, 4059–4076 (2021).
Immel, A. et al. Genome-wide study of a neolithic Wartberg grave community reveals distinct HLA variation and hunter-gatherer ancestry. Commun. Biology 4, (2021).
Skoglund, P. Origins and Genetic Legacy of Neolithic Farmers and Hunter-Gatherers in Europe. Science 336, (2012).
Armelagos, G. J., Goodman, A. H. & Jacobs, K. H. The origins of agriculture: Population growth during a period of declining health. Popul. Environ. 13, 9–22 (1991).
Barquera, R. & Krause, J. An ancient view on host pathogen interaction across time and space. Curr. Opin. Immunol. 65, 65–69 (2020).
Allentoft, M. E. et al. Population genomics of post-glacial western Eurasia. Nature 625, 301–311 (2024).
Allentoft, M. E. et al. 100 ancient genomes show repeated population turnovers in Neolithic Denmark. Nature 625, 329–337 (2024).
Moreno-Mayar, J. V. ictor. Early human dispersals within the Americas. Science 362, (2018).
Marchi, N. et al. The genomic origins of the world’s first farmers. Cell 185, 1842–1859.e18 (2022).
Sanchez-Quinto, F. et al. Megalithic tombs in western and northern Neolithic Europe were linked to a kindred society. Proc. Natl. Acad. Sci. 116, 9469–9474 (2019).
Rasmussen, M. et al. The genome of a late pleistocene human from a Clovis burial site in western Montana. Nature 506, 225–229 (2014).
Schlebusch, C. M. Southern African ancient genomes estimate modern human divergence to 350,000 to 260,000 years ago. Science 358, (2017).
De Damgaard, B. et al. P. The first horse herders and the impact of early Bronze Age steppe expansions into Asia. Science 360, (2018).
Gamba, C. et al. Genome flux and stasis in a five millennium transect of European prehistory. Nat. Communications 5, (2014).
Cassidy, L. M. et al. A dynastic elite in monumental neolithic society. Nature 582, 384–388 (2020).
Sikora, M. et al. The population history of northeastern Siberia since the Pleistocene. Nature 570, 182–188 (2019).
Jones, E. R. et al. Upper palaeolithic genomes reveal deep roots of modern eurasians. Nature Communications 6, (2015).
Lazaridis, I. et al. Ancient human genomes suggest three ancestral populations for present-day europeans. Nature 513, 409–413 (2014).
Llorente, M. G. Ancient Ethiopian genome reveals extensive eurasian admixture in Eastern Africa. Science 250 (2015).
Svensson, E. et al. Genome of Pecstera Muierii skull shows high diversity and low mutational load in pre-glacial Europe. Curr. Biol. 31, 2973–2983e9 (2021).
Gunther et al. Population genomics of Mesolithic Scandinavia: investigating early postglacial migration routes and high-latitude adaptation. PLoS Biol. 16, e2003703 (2018).
Sikora, M. Ancient genomes show social and reproductive behavior of early Upper Paleolithic foragers. Science 358, (2017).
Ebenesersdóttir, S. S. Ancient genomes from Iceland reveal the making of a human population. Science 360, (2018).
Amorim, C. E. G. et al. Understanding 6th-century barbarian social organization and migration through paleogenomics. Nat. Commun. 9, (2018).
Margaryan, A. et al. Population genomics of the viking world. Nature 585, 390–396 (2020).
Fu, Q. et al. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 514, 445–449 (2014).
Broushaki, F. Early Neolithic genomes from the eastern Fertile Crescent. Science 353, (2016).
Main, P., Attenborough, R. D., Chelvanayagam, G. & Gao, X. Project MUSE - The Peopling of New Guinea: Evidence from Class I Human Leukocyte Antigen. Human Biology 73, 365–383.
Klunk, J. et al. Evolution of immune genes is associated with the black death. Nature 611, 312–319 (2022).
Deng, Z. et al. Adaptive admixture of HLA Class I allotypes enhanced genetically determined strength of natural killer cells in East asians. Mol. Biol. Evol. 38, 2582–2596 (2021).
Kawaguchi, S., Higasa, K., Shimizu, M., Yamada, R. & Matsuda, F. HLAHD: an accurate HLA typing algorithm for nextgeneration sequencing data. Hum. Mutat. 38, 788–797 (2017).
Ka, S. et al. HLAscan: genotyping of the HLA region using next-generation sequencing data. BMC Bioinform. 18, (2017).
Dilthey, A. T. et al. HLA*LA—HLA typing from linearly projected graph alignments. Bioinformatics 35, 4394–4396 (2019).
Szolek, A. et al. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics 30, 3310–3316 (2014).
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Erlich, R. L. et al. Next-generation sequencing for HLA typing of class I loci. BMC genomics 12, 42 (2011).
Huang, Y. et al. HLAreporter: a tool for HLA typing from next generation sequencing data. Genome medicine 7, 25 (2015).
Renaud, G., Hanghøj, K., Willerslev, E. & Orlando, L. Gargammel: a sequence simulator for ancient DNA. Bioinformatics 33, 577–579 (2016).
Machens, A. et al. Correlation of Thymic Pathology with HLA in Myasthenia Gravis. Clin. Immunol. 91, 296–301 (1999).
Adamashvili, I. et al. Soluble HLA-I (s-HLA-I) synthesis in systemic lupus erythematosus. Rheumatol. Int. 23, 294–300 (2003).
Qiu, X., Zhang, F., Chen, D. & Azad, K. & Tao, N. HLA-B*07 is a high risk allele for familial cervical cancer. Asian Pac. J. Cancer Prevention 12.
Mendoza-Ramirez et al. Class I MHC Polymorphisms Associated with Type 2 Diabetes in the Mexican Population. Genes 13, 772 (2022).
Tziotzios, C. et al. Genome-wide association study in frontal fibrosing alopecia identifies four susceptibility loci including HLA-B*07:02. Nat. Commun. 10, 1–9 (2019).
Shen, J. et al. HLA-B*07, HLA-DRB1*07, HLA-DRB1*12, and HLA-C*03:02 strongly associate with BMI: data from 1.3 million healthy Chinese adults. Diabetes 67, 861–871 (2018).
Colbert, R. A., Navid, F. & Gill, T. The role of HLA-B*27 in spondyloarthritis. Best Pract. Res. Clin. Rheumatol. 31, 797–815 (2017).
Takeno, M. The association of Behcet’s syndrome with HLA-B51 as understood in 2021. Curr. Opin. Rheumatol. 34, 4–9 (2022).
Shao, L. N., Wang, N., Zhou, S. H. & Wang, Z. Associations between human leukocyte antigen C locus polymorphism and psoriatic arthritis in populations of European and middle eastern descent: a meta-analysis. Ann. Saudi Med. 40, 338–346 (2020).
Dubaniewicz, Szczerkowska, Z. & HLA-A, B. C antigens in pulmonary sarcoidosis in Polish population. Archivum Immunologiae et Therapiae Experimentalis 55.
Akilov, O. E. & Mumcuoglu, K. Y. Association between human demodicosis and HLA class I. Clin. Exp. Dermatol. 28, 70–73 (2003).
Simmonds, M. J. et al. A novel and major association of HLA-C in Graves’ disease that eclipses the classicalHLA-DRB1effect. Hum. Mol. Genet. 16, 2149–2153 (2007).
Schweiger, S. et al. Genetic risk for cooccurrence of type 1 diabetes and celiac disease is modified by HLA C and killer immunoglobulinlike receptors. Tissue Antigens 84, 471–478 (2014).
de Winter, J. J. et al. HLA-C*07 in axial spondyloarthritis: data from the German Spondyloarthritis Inception Cohort and the Spondyloarthritis Caught Early cohort. Genes Immun. 20, 671–677 (2019).
Montes-Cano, M. A. et al. HLA and non-HLA genes in Behçet’s disease: a multicentric study in the Spanish population. Arthritis Res. Therapy 15, R145 (2013).
Manca, M. A. et al. Association of HLA-A*11:01, -A*24:02, and -B*18:01 with prostate Cancer risk: a case-control study. Int. J. Mol. Sci. 24, 15398 (2023).
Mathieson, I. et al. Genome-wide patterns of selection in 230 ancient eurasians. Nature 528, 499–503 (2015).
Childebayeva, A. et al. Population Genetics and Signatures of Selection in Early Neolithic European Farmers. Molecular Biology and Evolution 39, (2022).
Kerner, G. et al. Genetic adaptation to pathogens and increased risk of inflammatory disorders in post-Neolithic Europe. Cell genomics 3, 100248 (2023).
Hristova, M. et al. The role of IL-17 rs2275913, IL-17RC rs708567 and TGFB1 rs1800469 SNPs and IL-17A serum levels in patients with lupus nephritis. Rheumatol. Int. 41, 2205–2213 (2021).
Zheng, R. et al. Genome-wide association study identifies two risk loci for tuberculosis in Han Chinese. Nat. Commun. 9, 1–9 (2018).
Sanchez-Mazas, A. HLA studies in the context of coronavirus outbreaks. Swiss Med. Wkly. 150, w20248–w20248 (2020).
Le, M. K. et al. 1,000 ancient genomes uncover 10,000 years of natural selection in Europe. bioRxiv: the preprint server for biology 2022.08.24.505188 (2022).
Madden, K. & Chabot-Richards, D. HLA testing in the molecular diagnostic laboratory. Virchows Arch. 474, 139–147 (2018).
Eberle, M. et al. HLA-B typing by allele separation followed by direct sequencing. Tissue Antigens 49, 365–375 (1997).
Pierini, F. et al. Targeted analysis of polymorphic loci from low-coverage shotgun sequence data allows accurate genotyping of HLA genes in historical human populations. Sci. Rep. 10, 1–14 (2020).
International HapMap Consortium. A haplotype map of the human genome. Nature 437, 1299–1320 (2005).
McVicker, G., Gordon, D., Davis, C. & Green, P. Widespread Genomic Signatures of Natural Selection in Hominid Evolution. PLOS Genetics5, (2009).Arrieta-Bolaños, E., Hernández-Zaragoza, D. I., & Barquera, R. An HLA map of the world: A comparison of HLA frequencies in 200 worldwide populations reveals diverse patterns for class I and class II. Frontiers in Genetics, 14. (2023).
Rivollat, M. et al. Extensive pedigrees reveal the social organization of a neolithic community. Nature 620, 600–606 (2023).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinf. (Oxford Engl.) 26, 589–595 (2010).
Sánchez-Quinto, F. et al. Genomic affinities of two 7,000-Year-old Iberian Hunter-gatherers. Curr. Biol. 22, 1494–1499 (2012).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, (2021).
Patterson, N. et al. Ancient Admixture in Human History. Genetics 192, 1065–1093 (2012).
Lazaridis, I. et al. Genomic insights into the origin of farming in the ancient Near East. Nature 536, 419–424 (2016).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, (2015).
McVicker, G., Gordon, D., Davis, C. & Green, P. Widespread Genomic Signatures of Natural Selection in Hominid Evolution. PLOS Genetics 5, (2009).
Acknowledgements
Alan Vladimir Godínez Plascencia is a doctoral student from Programa de Doctorado en Ciencias Biomédicas at Universidad Nacional Autónoma de México (UNAM) and received fellowship 781930 from the PNPC PhD fellowship program at CONAHCYT. The implementation of HLA typing software, as well as the computation analyses carried out in this study were enabled by resources in projects naiss2023-22-827 and naiss2023-22-829 provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at UPPMAX, funded by the Swedish Research Council through grant agreement no. 2022-06725. We thank Maria Avila-Arcos and Diego Ortega Del Vecchyo for valuable revisions and comments to improve the quality of this manuscript. We also thank Gabriel Renaud for advice to modify and adapt the gargammel suite.
Author information
Authors and Affiliations
Contributions
F.S.-Q. conceived and designed the study. A.G.P. Processed sequence data processing, performed analyses, generated figures and tables. M.J. Provided access to samples, discussed results, and provided feedback. F.S.-Q., and A.G.P. wrote the paper with input from M.J. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Plascencia, A.G., Jakobsson, M. & Sánchez-Quinto, F. Ancient DNA HLA typing reveals significant shifts in frequency in Europe since the Neolithic. Sci Rep 15, 6161 (2025). https://doi.org/10.1038/s41598-024-82449-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-82449-w
