
Abstract
Cowpea is a seed legume, important for food and nutritional security in Africa’s arid and semi-arid zones. Despite its importance, cowpea is experiencing a loss of genetic diversity due to climate change. Therefore, this study aimed to evaluate the genetic variability of 33 cowpea mutant collections using 20 SSR and 13 ISSR markers. This analysis shows an average number of alleles of 5.15 for SSR and 6.7 for ISSR. The highest average genetic distance based on Nei’s index among subgroups was 0.740 and the value of polymorphism information content varied from 0.02 to 0.23 for SSR and from 0.05 to 0.36 for ISSR. Our results revealed high genetic diversity based on the GD (0.126), Fst (0.513), and Shannon information index (0.246). The population structure analysis showed 3 and 4 clusters for ISSR and SSR markers, respectively. Genetic variation, as assessed by analysis of molecular variance, mostly indicates higher genetic diversity within the population than among populations. The hierarchical clustering of combined SSR and ISSR markers grouped the accessions into four groups, supporting the STRUCTURE analysis results. Additionally, the combination of SSR and ISSR provided better information on the level of genetic diversity, and population structure, and was more effective in determining the relationship between cowpea collection. This study enhances our understanding of the genetic organization of our mutant collection which can be used in breeding programs in the era of climate change.
Similar content being viewed by others
Introduction
Genetic diversity is the degree of genetic variability among individuals of a variety or population within a species. It is the primary driver of selection and evolution and the genetic basis for breeders1. Genetic diversity is an essential resource on which humanity depends to increase the resilience and profitability of agriculture2. Consequently, the need for crop genetic diversity to improve food production will increase in the context of global population growth and climate change. According to3, by 2050, more than 820 million people worldwide will suffer from chronic hunger and about two billion people will be malnourished4. To meet this challenge, it is imperative to increase the quantity and improve the quality of crop production by developing outstanding varieties and conserving and maintaining the genetic diversity of these valuable resources for sustainable food security.
In this context, improving cowpea genetic diversity, particularly in the Subsaharan region of Africa where the crop is the most important food legume can be a relevant strategy in response to climate change. Cowpea (Vigna unguiculata (L.) Walp.) is a seed legume crop of great importance for food and nutrition security, especially in arid and semi-arid areas of Africa5. It is a diploid crop (2n = 2x = 22), widely produced in the tropical and subtropical zones of Africa (95.4%), Asia (2.9%), America (1.3%) and very weakly in Europe (0.5%) on 15,190,896 ha with worldwide production estimated at 9,774,865.72 tons (https://www.fao.org/faostat/fr/#data/QCL/visualize).
To improve this production, several conventional and transgenesis breeding programs have been carried out in recent decades to expand the genetic base of the crop in the hope of developing outstanding cultivars. Despite these significant efforts, the genetic base of cowpea is still narrow. This has led to exploring other methods, such as plant mutation breeding. Mutagenesis is one of the main factors that induce genetic diversity in a population, resulting in agro-morphological, yield, and gene expression variation in cowpea6. This genetic diversity can be assessed based on seed storage proteins7, morphological8, biochemical9,10, and DNA markers. Morphological and biochemical markers are limited by their small number and sensitivity to environmental factors11.
Because of these constraints, analysis of genetic diversity in cowpea was carried out in favor of DNA markers such as chloroplast DNA polymorphisms12, random amplified polymorphic DNA (RAPD;13, restriction fragment length polymorphisms (RFLP;14, DNA amplification fingerprinting (DAF;15,16, amplified fragment length polymorphisms (AFLP;17, diversity array technology (DArT;18 inter-simple sequence repeats (ISSR ;6,19, simple sequence repeats (SSR20 and SNP markers21. The major advantage of molecular markers is their unlimited number and wide distribution in the eukaryotic genome. They have been extensively used to study the genetic diversity of several core collections of legume crops such as mungbean22, soybean23, common bean24 pigeon pea25.
In this study, SSR and ISSR markers have been chosen for their efficiency, abundance, random distribution in the genome, high polymorphism, heritability, and traceability, and they do not require sophisticated equipment. SSR markers allowed the characterization of available cowpea collections in Senegal6,13,26,27, Ghana28, and Ethiopia29, and used in crop selection and improvement programs. ISSRs are mostly present in the non-coding region and have also been found between coding genes. These markers are unique and advantageous for DNA fingerprinting due to their presence as satellites in certain chromosomes30. SSR detects genetic diversity at a particular locus, while ISSR examines variation between two nearby SSRs. Thus, the combination of SSR and ISSR data allows the analysis of genetic variability to be extended to a larger part of the genome than either of these markers can do individually.
Therefore, this study aimed to assess the genetic variability in the different mutant lines developed in our Laboratory based on the hypothesis that mutagenesis induces genetic diversity. Assessing the genetic diversity within the cowpea mutants will enhance our understanding of their genetic organization and guide future efforts to develop improved cowpea varieties.
Materials and methods
Plant materials
Healthy seeds of 33 genotypes including 30 mutants31 and their parents namely Bambey 21, Ndout violet pods (VP), and the mutant Me51M4-39M9 from the collection of the “Laboratoire Campus de Biotechnologies Végétales” of the Département de Biologie Végétale at Université Cheikh Anta Diop were used during this study. Bambey 21 is an early flowering and non-photosensitive genotype while Ndout violet pods is a photosensitive variety with large seeds and the mutant Me51M4-39M9 is partially tolerant to nematodes (unpublished results), both are widely cultivated in the region of Thiès (Sénégal).
Methods
DNA genotyping
DNA extraction
Cowpea genomic DNA was isolated from 100 mg of fresh leaves of one-month-old seedlings from one plant per genotype. The leaf was ground in a mortar using a modified method of32. RNA was removed by adding 50 µg/mL RNAse A (CalBiochem), and the tubes were incubated at room temperature for 1 h. After this, the purification and precipitation were carried out according to the protocol described by27. Then, the DNA was washed with 100 µl of 70% ethanol (PROLABO) before drying with a Speed Vac Plus Sc 110 (Savant) for 20 min and dissolved in 100 µl x 0.1 TE (pH8). The DNA concentration and purity were determined using a NanoDrop™ One/OneC Microvolume UV-Vis spectrophotometer (Thermo Scientific) at A260, A280, A260/A280, and A260/A230 and visualized on a 1% agarose gel. All working solutions of the genomic DNA were adjusted to 12.5 ng/µl. Then, the DNA samples were stored at − 20 °C or immediately used for PCR amplification.
SSR amplification of DNA
PCR reactions were performed in a total volume of 20 µL containing 4 µl de 5x HOT FIREPol® Multiplex Mix (Solis Biodyne), 2 µl (0.5 µM) of each Forward and Reverse primers33 (Table 1), 10 µl H2O and 2 µl (25 ng) DNA. Amplification was carried out using a thermal cycler (TECHNE 5 Prime) under the following PCR conditions: initial denaturation at 95 °C for 12 min, followed by 35 cycles of denaturation at 95 °C (30 s), annealing temperature depending on the primer for 60 s, elongation at 72 °C (2 min) and a final extension at 72 °C for 8 min. PCR products were separated by electrophoresis on 10% polyacrylamide gel (PAGE) containing 20 ml acrylamide (40%), 6 ml TBE (10x), 1 ml APS (10%), 34 ml distilled water, and 50 µl TEMED. DNA electrophoresis was performed at 70 V for 12 h 1x TBE buffer and staining with 300 ml of GelRed® Biotium (3X in water) for 1 h and photographed under UV light using (320 nm) Gel Doc system (High-performance UV Transilluminator UVP).
ISSR amplification
Amplifications were performed in a total volume of 20 µL containing 4 µl de 5x HOT FIREPol® Multiplex Mix (Solis Biodyne), 2 µl (0.5 µM) of a single Primer (Table 1), 12 µl H2O and 2 µl (25 ng) DNA. PCR reactions were performed using a thermal cycler (TECHNE 5 Prime) under the following PCR conditions: initial denaturation at 95 °C for 12 min, followed by 35 cycles of denaturation at 95 °C (30 s), annealing temperature depending on the primer for 60 s, elongation at 72 °C (2 min) and a final extension at 72 °C for 8 min. After amplification, the PCR products were separated on 2% agarose gel (Sigma) for 2 h at 70 V in 1x TBE buffer. The gel was finally stained with 300 ml of GelRed® Biotium (3X in water) for 1 h according to the manufacturer’s instructions and photographed under UV light using the Gel Doc system (High-performance UV Transilluminator UVP).
Analysis of genetic diversity and population structure
The clear and reproducible bands were scored as 1 for present and 0 for absent, and this data matrix was subjected to further analysis. Based on the presence or absence of bands, the percentage of polymorphism was calculated, and the genetic variability was estimated using this data matrix.
The genetic diversity was analyzed using GenAlex 6.5 software developed by Peakall and Smouse34 to calculate the Shannon diversity index, number of alleles, heterozygosity (Nei’s index), fixation index, genetic similarity, and private alleles. The genetic variation between and within the groups was assessed by analysis of molecular variance (AMOVA) with the same software. Polymorphic information content (PIC) was calculated using PowerMarker 3.25 software35. This was used to assess the discriminatory power of each marker. The population structure was analyzed using the Bayesian clustering approach implemented in the STRUCTURE 2.3.3 software36. The number of sub-populations was tested from 1 to 10 independent runs (K). Using the admixture model37, each simulation was set to 100,000 burn-in periods, and 10 runs of 200,000 Markov chain Monte Carlo (MCMC) iterations were performed. STRUCTURE software was used to calculate the differentiation indices (Fst). These results were uploaded to the STRUCTURE HARVESTER online software38 to determine the most likely number of sub-populations using the Evanno Δk method39. Individuals were assigned to a cluster using a membership coefficient (q) ≥ 0.7. The genotypes within clusters with membership coefficients (q) < 0.6 were considered genetically admixed. A Neighbour-Joining dendrogram40 was constructed using the inter-individual distance matrix and was calculated using the Jaccard index41,42. This analysis was carried out using the Darwin 6.0 software43.
Results
Genetic polymorphism analysis
In total, 20 SSR primers amplified 103 alleles with an average of 5.15 alleles per primer. In contrast, 13 ISSR primers revealed 79 alleles with an average of 6.18, higher than the results obtained by SSR. The highest number of alleles, 9 and 11, was obtained with primers SSR-6683 and ISSR-UBC 809, respectively. The PIC value ranged from 0.02 for the SSR-6357 primer to 0.23 for the SSR-6277 primer, with an average value of 0.09. However, the ISSR PIC value ranged from 0.05 for the ISSR-UBC 823 primer to 0.36 for the ISSR-HB8 primer, with an average value of 0.20, which was higher than the SSR PIC (Table 1). The percentage of polymorphic markers varies from 20 to 100% for SSRs and from 25 to 100% for ISSR markers.
Population structure analysis
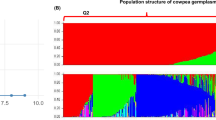
The Bayesian clustering program STRUCTURE revealed that the highest LnP(D) and ∆K values for K = 4 (Fig. 1a) for SSR, K = 3 (Fig. 2a) for ISSR, and K = 4 using the combined SSR and ISSR data (Fig. 3a). Based on a membership probability threshold (Q) of 0.70, established for SSR, the structure plot revealed 4 sub-populations (Fig. 1b). Sub-population 1 (SP 1) consists of 13 genotypes (39.39%), which are mainly Ndout violet pods (1) and their mutants, Bambey 21 (12) and its mutants 13, 14 and 18. SP 2 contains 9 genotypes (27.27%), mainly Me51M4-39M9 mutants (23) and one mutant (16) of Bambey 21. Sub-population 3 (SP 3) contains 12.12% of the genotypes derived exclusively from Ndout VP (6 and 7) and mutant (15 and 19) of Bambey 21. In addition, sub-population 4 (SP 4), which contains 21.21% of the genotypes, is mainly encompassed by Me51-M4-39M9 (23) and its mutants, and a mutant (17) of Bambey 21. The structure analysis based on ISSR marker identified three clusters (K = 3, Fig. 2b). Sub-population 1 (SP 1) encompasses genotypes of Bambey 21 (12) and one admixture (22) between Bambey 21 and Me51-M4-39M9. Sub-population 2 (SP 2) contains only Ndout VP and their mutants, and sub-population 3 (SP 3) contains Me51-M4-39M9 and its mutant. The population structure obtained by combining the ISSR and SSR data matrix is similar to the one obtained by the SSR (Fig. 3b). However, there is a reorganization of the sub-populations regarding membership and genotype number within a sub-population, while the Ndout VP group conserves its sub-clustering.

Population structure of three Cowpea varieties and their mutants using SSR markers. a Graph showing the best value of K = 4. b Bar plot of K = 4 showing 4 populations.

Population structure of three cowpea genotypes and their mutants based on ISSR markers. a Graphical representation of the optimal number of groups in the program STRUCTURE inferred using a structure harvester. b STRUCTURE plot of membership coefficients for all Cowpea genotypes.

Population genetic structure of three cowpea varieties and their mutants using combined SSR and ISSR markers. a Graph showing the best value of K = 4. b Bar plot of K = 4 showing 4 populations.
Genetic diversity
For the assessment of genetic diversity within populations, average intra-population indices which included the number of alleles (Na), number of effective alleles (Ne), Shannon index (I), Expected Heterozygosity (He), Fst, Major Allele Frequency (MAF), Gene diversity (GD) and PIC were calculated (Table 2). The total number of alleles (Na) by population varied between 0.981 in SP 2 to 1.117 in SP 4 with a mean value of 1.044 for SSR markers. The mean value of Na for SSR (1.044) is less high than the one for ISSR (1.371). Meanwhile, in SP 1 and SP 3, the Na per population varies from 1.342 to 1.418 based on ISSR markers. For each locus, the number of effective alleles (Ne) values per population for SSR, ranged from 1.065 in SP 2 to 1.150 in SP 3, and the Ne for ISSR varied between 1.304 in SP 1 to 1.316 in SP 2. In the present study, Shannon’s Information index value ranged from 0.097 to 0.121 for the SSR markers and from 0.241 to 0.254 for the ISSR markers. The Expected Heterozygosity (He) varied from 0.041 in SP 2 and 0.083 in SP 3 based on SSR markers. However, it is higher for ISSR and varies from 0.165 in SP 3 to 0.177 in SP 2. The sub-population obtained by ISSR had the highest mean (43.04%) of the Percentage of Polymorphic Loci (%P), and the SSR sub-population had the lowest %P (20.87%). In addition, the average of diversity indices such as Na, Ne, I, He, and %P of ISSR was higher than those obtained by combining the data matrix of SSR and ISSR markers. The value of Fst obtained by ISSR differs significantly between subpopulations, ranging from 0.235 in SP 1 to 0.651 in SP 2, however, it varied from 0.252 in SP 4 to 0.610 in SP 2 for SSR markers. The mean Fst value revealed by the ISSR was higher (0.510) than the one of the SSR sub-population (0.413). The genotypic frequency revealed by SSR and ISSR were 0.695 and 0.572, respectively. SSR analysis showed high Major Alleles Frequency (MAF) ranging from 0.93 (SP 3) to 0.97 (SP 2), with an average of 0.95. However, the ISSR and SSR markers had an average of 0.82 to 0.92 MAF per locus, respectively. This was lower than the MAF obtained from the subpopulations. The MAF differs among the ISSR sub-populations, ranging from 0.90 in SP 2 and 3 to 0.93 in SP 1. The SSR revealed that the gene diversity (GD) values ranged from 0.048 (SP 2) to 0.085 (SP 3) per sub-population with an overall mean of 0.071. The latter values were lower than those obtained by the ISSR, which ranged from 0.099 (SP 1) to 0.144 (SP 3), with an overall mean of 0.126. Indeed, the overall average of GD per locus is higher for ISSRs (0.244) than SSRs (0.108). Moreover, the average PIC of ISSR (0.104) by sub-population is higher than that obtained by SSR (0.057). Gene diversity value was similar to the one shown by PIC. This result suggests that sub-population revealed by SSR had the least variation than one obtained by ISSR markers.
The Nei index distance and genetic identity matrices were estimated using Nei (1978) in overall SSR markers (Table 3). The sub-populations SP 2 and SP 3 showed the highest genetic distance (0.115) while the highest genetic similarity was observed between SP 1 and SP 4 (0.980). This is followed by SP 1 and SP 3 with a similarity of 0.960. To assess the genetic diversity within the populations, the Nei index, was calculated using the ISSR (Table 4). The highest genetic identity and distance were observed between the sub-populations SP 2 and SP 3 (0.816) and SP 1 and SP 2 (0.313), respectively. The high genetic similarity observed in this study may have resulted from close parental genetic distance. Furthermore, the genetic distance between genotypes in each sub-population revealed the existence of considerable genetic diversity in the studied materials. Similarly, a high degree of similarity was obtained between the sub-populations derived from the combined ISSR and SSR data matrix (Table 5). The analysis of the genetic identity showed that the Ndout VP and Bambey21 subgroups were closer to each other than they were to Me51-M4-39M9 (Table 6).
Analysis of molecular variance in the populations
The AMOVA performed on the four sub-populations identified by STRUCTURE based on SSR marker revealed that within the population was higher (62%), than among the population (38%) as shown in Fig. 4a. In fact, analysis of molecular variance was also carried out on the identified groups by STRUCTURE using ISSR markers partitioned that 51% of the total variation was found among populations, and 49% was found within populations (Fig. 4b). AMOVA was performed also using the combined ISSR and SSR matrix data (Fig. 4c) according to the sub-population generated by Structure. Based on the AMOVA results, most of the genetic variation (81%) was found within population and only a small part (19%) among populations. AMOVA was calculated in the parent subgroup (Ndout violet pods, Bambey 21, and Me51-M4-39M9) with the combined SSR and ISSR data matrix and 79% of the genetic variation was observed within the population and 21% among the population (Fig. 4d).

The molecular variance analysis within and among populations (a) SSR, (b) ISSR, (c) Combining SSR and ISSR data matrix, and d: parental subgroup.
Phylogenetic analysis of Cowpea genotypes based on SSR and ISSR
Based on their molecular profiles using informative SSR and ISSR markers, the 33 cowpea genotypes were clustered into 3 (A-C) and 2 (A-B) main groups, respectively. The clusters revealed by SSR (Fig. 5) encompassed different genotypes. Cluster A contained 33.33% genotypes, mostly from SP2, followed by SP1 (10) and SP4 (22), and cluster B included most of the lines (51.51%). The genotypes of this latter cluster consisted of the majority of genotypes from SP 1, one from SP 4 (17), and those from SP 3 (6, 7, 15, and 19). Cluster C had 15.15% of all genotypes and only from SP 4, except genotype 22. The phylogenetic tree obtained with ISSR markers (Fig. 6) showed that group A was the most diverse with 66.67% of the genotypes with two sub-populations, 2 and 3. Group B consisted of 33.33% genotypes exclusively sub-population 1 including Ndout violet pods and its mutants.

Genetic relationship among 33 Cowpea genotypes using Darwin software (version 6.0.17) generated from 20 SSR markers.

Genetic relationship among 33 Cowpea’s genotypes using Darwin software (version 6.0.17) generated from 13 ISSR markers.
Genetic relationship based on combined ISSR and SSR markers
The dendrogram generated from the combined SSR and ISSR matrix (Fig. 7) showed four distinct clusters from A to D, which was higher than the number of groups obtained by SSR and ISSR. Group A contained 27.28% of genotypes, mostly the mutants of Me51-M4-39M9 and one mutant (16) of Bambey 21. Group B clustered most of the mutants of Bambey 21, including Me51-M4-39M9 (23) and its two mutants 24 and 25. Cluster C encompassed the most of genotypes (42.42%). This cluster included Ndout violet pods and its mutants, and 3 mutants of Bambey 21 (12, 13, and 14). This cluster is followed by Cluster D, with a minority of 6.06%, which includes two Bambey 21 genotypes (18 and 19).

Phylogenetic tree among 33 cowpeas using combined SSR and ISSR markers.
Discussion
This study assessed genetic diversity using 20 SSR, 13 ISSR, and a combined data matrix of these markers to characterize 33 cowpea genotypes. This approach plays a critical role in genetic resources management by reducing redundancy and establishing a core collection that can be screened for traits of interest. Furthermore, the success of a crop breeding and conservation program depends primarily on the availability of genetic diversity and the characterization of that diversity20.
Polymorphism analysis
The analysis of genetic polymorphisms includes an assessment of the polymorphism information content (PIC) and the percentage of polymorphic markers. It determines the ability of a genetic marker to detect polymorphisms, which is essential for marker selection and genetic diversity analysis44. Moreover, based on45 scores, mean PIC values greater than or equal to 0.5 are highly informative, those ranging from 0.25 to 0.50 are moderately informative, and those less than 0.25 are slightly informative. In this study, the PIC of ISSR shows large variability between 0.05 and 0.36 (Table 1), which is higher than SSR markers (0.02–0.23). The average PIC value of 0.20 obtained in this analysis indicated that the ISSRs are moderately informative. The lowest value of PIC was similar to the one reported by27,46,47, and48. In this paper, the SSR and ISSR markers revealed a high percentage of polymorphism, and it may be helpful for DNA genomic fingerprinting. Furthermore, in this study, the mean number of amplified SSR and ISSR loci ranged from 5.15 to 6.07, which suggests a very high allelic richness. This result is similar to those reported by13,27, and49 on Senegalese cowpea varieties and50 on a Burkina Faso collection. This allelic variability may be explained by the nature of markers and the germplasm origin. In this study, the variability observed among our genotypes suggests that gamma irradiation is efficient for genetic variation inducing and introducing new alleles into a germplasm. Taheri et al.51 reported a variation in the number of loci in Curcuma alismatifolia exposed to gamma irradiation.
Genetic diversity
The heterozygosity expected (He) is used to evaluate the likelihood that two randomly selected alleles from the population will be different and is commonly used in population genetics. It helps to understand the genetic variability and overall diversity within a population. The present study showed that the average expected heterozygosity of ISSR was 0.170, superior to that of combined ISSR and SSR (0.065), followed by SSR (0.063). In the final analysis, ISSR showed intermediate genetic diversity based on the He, whereas SSR showed low genetic diversity. The low expected heterozygosity values obtained by SSR are similar to those reported by46, while our intermediate values are closer to the ones of 28,49. The Fst scale described by52 states that if 0 < Fst < 0.05 there is little differentiation, if 0.05 < Fst < 0.15 there is moderate differentiation, if 0.15 < Fst < 0.25 there is significant differentiation, and when FST > 0.25 there is a very significantly difference. In this study, the Fst indices were greater than 0.25, indicating strong differentiation between the sub-populations. The fixation index (Fst) obtained in the current study was much higher than the ones reported by49 in the Senegalese germplasm and by47 while assessing IITA Cowpea Genetic Resources Center. Our findings were higher than those of53 who reported a Fst value of 0.423 between populations. Again, our results revealed that gamma irradiations are effective to introduce genetic variation into germplasm. The ISSR Shannon information index (I) mean (0.246), showed a high genetic diversity between populations. However, in this study, Nei’s observed genetic distance across sub-populations was low while the genetic identity was higher. Similar findings have been reported by54,55 among cowpea from USDA Vigna germplasm collection and worldwide collection of cowpea landraces and African ancestral wild cowpeas. The low genetic distances observed among these sub-populations may reflect the early domestication bottleneck maintained by cowpea’s inherent self-pollination mechanism28. This study shows that ISSR was richer in gene diversity (GD) than SSR and these results were similar to those reported by48,56. These findings suggest that gamma irradiation induced more variabilities in the inter Simple sequence Repeat regions than within the SSR. Thus, we can speculate that the SSR regions are more susceptible to homologous DNA recombination than ISSR regions, leading to low diversity. This genetic diversity indicates that mutations have caused major changes in the genome of these cowpea lines57.
Analysis of molecular variance in the studied populations
In addition, an analysis of molecular variance (AMOVA) was performed to test the degree of differentiation among and within the cowpea genotypes. The results of AMOVA showed that most of the variance occurred within populations, accounting for 81%, 79%, and 62% of the total variation for the combined matrix data, parental subgroup, and SSR markers, respectively, whereas 19%, 21%, and 38% of the variation were due to differences among populations (Fig. 4). The results indicated that the diversity within populations (intra-population diversity) was much greater than the diversity among populations (inter-population diversity). These results are close to those reported by58 on Bambara groundnut and by49 on Cowpea. Those findings revealed the difference between and within populations and their heterogeneous status. However, the results of the ISSR showed that the genetic differentiation was moderate within populations (49%) and high among populations (51%). This is in addition to the report of59 on cowpea accessions from China’s national GenBank (Beijing) using SSR markers.
Population structure analysis and genetic relationship
The population structure analysis grouped the cowpea genotypes into four (4) gene pools (clusters 1, 2, 3, and 4) based on the peak of the delta K for SSR and combined ISSR and SSR matrix data. Cluster analysis based on polymorphic molecular markers is an improved and more reliable method for establishing relationships between individuals. Genetic similarity dendrograms revealed three groups based on SSR data. From a sub-population analysis perspective, the different groups in the dendrogram contain genotypes from different sub-populations. This conflicts with the work of27, who analyzed the genetic diversity of cowpea cultivars in Senegal using microsatellites, where only 2 groups were identified. In contrast in our studies, 3 groups have been identified which demonstrate the ability of gamma irradiation to induce significant variations in cowpea. The observed clustering patterns differ from our population structure (K = 4). In this study, ISSR markers also revealed three sub-populations (K optimal = 3) identified by STRUCTURE analysis. These results confirm the present findings of the study of6 cowpea mutant lines’ genetic diversity using ISSR markers. However, the number of gene pools (K = 3) obtained by ISSR using the Structure software differed from the number of clusters (2) in the phylogenetic tree generated by DarWin software. The software resolution power can explain this difference. The phylogenetic tree obtained from the combined matrix ISSR and SSR data shows 4 distinct groups. Again, this clustering agreed with the results obtained by STRUCTURE (K = 4). This result showed that the combination of markers provided better information on the level of genetic diversity and population structure and was more effective in determining the relationship between cowpea varieties. To the best of our knowledge, this is the first study to evaluate the genetic diversity of cowpea mutants using combined SSR and ISSR markers. Combining ISSR and SSR data matrix allowed us to assign a cluster membership. According to60, the combination of molecular markers in genetic diversity studies is intended to increase the accuracy and deepen the analysis. This combined approach allowed us to explore diversity in a wide genomic region for detecting genetic variation.
Conclusion
We investigated the genetic diversity among 33 cowpea genotypes using 20 and 13 informative SSRs and ISSRs markers, respectively. This is a crucial step for the management, genetic improvement and conservation of cowpea germplasm. Generally, it can be asserted that the combined microsatellite and inter-microsatellite data can be very powerful, so they could be efficient and accurate tools for detecting genetic diversity in cowpea germplasm. To broaden the genetic base of crops, induced mutation is a powerful tool for creating genetic variability. This assertion supported our research that revealed significant genetic variability in cowpea mutants’ germplasm. This study has allowed us to understand the structure and genetic diversity of the mutant collection that contains a wide range of genotypes harboring interesting traits. This collection can be used in breeding programs, genetic resources conservation, and food security, especially in the era of climate change.
Data availability
The data are available in the Plant Molecular Biology Reporters Journal using this link (https://doi.org/10.1007/s11105-024-01473-2) and from the corresponding authors upon request.
References
Bhandari, H., Bhanu, A. N., Srivastava, K., Singh, M. & Shreya, H. A. Assessment of genetic diversity in crop plants-an overview. Adv. Plants Agric. Res. 7, 279–286. https://doi.org/10.15406/apar.2017.07.00255 (2017).
Salgotra, R. K. & Chauhan, B. S. Genetic diversity, conservation, and utilization of plant genetic resources. Genes 14, 174. https://doi.org/10.3390/genes14010174 (2023).
FAO. (2019). https://www.fao.org/faostat/fr/#home
Delaunay, D. & Guengant, J. P. Le dividende démographique en Afrique subsaharienne (2019).
Ndiaye, A., Gauthier, P. & Sembène, M. Genetic discrimination of two cowpea (Vigna unguiculata (L.) Walp) Bruchid (Coleoptera, Chrysomelidae, Bruchinae): Callosobruchus maculatus (F.) and Bruchidius atrolineatus (Pic). Int. J. Plant. Anim. Environ. Sci. 1, 196–201 (2011).
Diouf, M., Diallo, S., Badiane, F. A., Diack, O. & Diouf, D. Development of new cowpea (Vigna unguiculata) mutant genotypes, analysis of their agromorphological variation, genetic diversity and structure. BIOCELL 45, 345–362. https://doi.org/10.32604/biocell.2021.013706 (2021).
Fotso, M., Azanza, J. L., Pasquet, R. & Raymond, J. Molecular heterogeneity of cowpea (Vigna unguiculata Fabaceae) seed storage proteins. Plant. Syst. Evol. 191, 39–56. https://doi.org/10.1007/bf00985341 (1994).
Ehlers, J. & Hall, A. Cowpea (Vigna unguiculata L. walp). Field Crops Res. 53, 187–204. https://doi.org/10.1016/s0378-4290(97)00031-2 (1997).
Panella, L. & Gepts, P. Genetic relationships within Vigna unguiculata (L.) Walp. based on isozyme analyses. Genet. Resour. Crop Evol. 39, 71–88. https://doi.org/10.1007/bf00051226 (1992).
Pasquet, R. S. Classification infraspécifique des formes spontanées de Vigna unguiculata (L.) Walp. (Fabaceae) a partir de données morphologiques. Bull. Jard Bot. Natl. Belg. Bull. Van Natl. Plantentuin Van Belg. 62, 127–173. https://doi.org/10.2307/3668271 (1993).
Dikshit, H. K. et al. Genetic differentiation of Vigna species by RAPD, URP and SSR markers. Biol. Plant. 51, 451–457. https://doi.org/10.1007/s10535-007-0095-8 (2007).
Vaillancourt, R. & Weeden, N. Chloroplast DNA polymorphism suggests Nigerian center of domestication for the cowpea, Vigna unguiculata (Leguminosae). Am. J. Bot. 79, 1194–1199. https://doi.org/10.1002/j.1537-2197.1992.tb13716.x (1992).
Diouf, D. & Hilu, K. W. Microsatellites and RAPD markers to study genetic relationships among cowpea breeding lines and local varieties in Senegal. Genet. Resour. Crop Evol. 52, 1057–1067. https://doi.org/10.1007/s10722-004-6107-z (2005).
Fatokun, C. A., Danesh, D., Young, N. D. & Stewart, E. L. Molecular taxonomic relationships in the genus Vigna based on RFLP analysis. TAG Theor. Appl. Genet. Theor. Angew Genet. 86, 97–104. https://doi.org/10.1007/bf00223813 (1993).
Spencer, M. M., Gueye, M., Diouf, D., Ndiaye, M. A. & Gresshoff, P. DNA-based relatedness of cowpea [Vigna unguiculata (L.) Walp.] genotypes using DNA amplification fingerprinting. Physiol. Mol. Biol. Plants 6 (2000).
Simon, M. V., Benko-Iseppon, A. M., Resende, L. V., Winter, P. & Kahl, G. Genetic diversity and phylogenetic relationships in Vigna Savi germplasm revealed by DNA amplification fingerprinting. Genome 50, 538–547. https://doi.org/10.1139/g07-029 (2007).
Coulibaly, S., Pasquet, R. S., Papa, R. & Gepts, P. AFLP analysis of the phenetic organization and genetic diversity of Vigna unguiculata L. Walp. reveals extensive gene flow between wild and domesticated types. Theor. Appl. Genet. 104, 358–366. https://doi.org/10.1007/s001220100740 (2002).
Ketema, S. et al. DArTSeq SNP-based markers revealed high genetic diversity and structured in Ethiopian cowpea [Vigna unguiculata (L.) Walp] germplasms. PLoS ONE. https://doi.org/10.1371/journal.pone.0239122 (2020).
Igwe, D. O. et al. Assessment of genetic diversity in Vigna unguiculata L. (Walp) accessions using inter-simple sequence repeat (ISSR) and start codon targeted (SCoT) polymorphic markers. BMC Genet. 18, 98. https://doi.org/10.1186/s12863-017-0567-6 (2017).
Dagnon, Y. D. et al. Genetic diversity and structure of cowpea [Vigna unguiculata (L.) Walp.] accessions from Togo using SSR markers. PLoS ONE https://doi.org/10.1371/journal.pone.0252362 (2022).
Gumede, M. T., Gerrano, A. S., Amelework, A. B. & Modi, A. T. Analysis of genetic diversity and structure of cowpea (Vigna unguiculata (L.) Walp) genotypes using single nucleotide polymorphism markers. Plants. https://doi.org/10.3390/plants11243480 (2022).
Sokolkova, A. et al. Genome-wide association study in accessions of the mini-core collection of mungbean (Vigna radiata) from the World Vegetable Gene Bank (Taiwan). BMC Plant. Biol. 20, 363. https://doi.org/10.1186/s12870-020-02579-x (2020).
Jeong, N. et al. Korean soybean core collection: Genotypic and phenotypic diversity structure and genome-wide association study. PLoS ONE. https://doi.org/10.1371/journal.pone.0224074 (2019).
Özkan, G. et al. Determining genetic diversity and structure of common bean (Phaseolus vulgaris L.) Landraces from Türkiye using SSR markers. Genes 13, 1410. https://doi.org/10.3390/genes13081410 (2022).
Kimaro, D., Melis, R., Sibiya, J., Shimelis, H. & Shayanowako, A. Analysis of Genetic Diversity and Structure of Pigeonpea [Cajanus cajan (L.) Millsp] Accessions Using SSR Markers. Plants 9, 1643. https://doi.org/10.3390/plants9121643 (2020).
Kouakou, C. K. et al. Diversité génétique des variétés traditionnelles de niébé [Vigna unguiculata (L.) Walp.] au Sénégal: étude préliminaire (2007).
Badiane, F. A. et al. Genetic relationship of cowpea (Vigna unguiculata) varieties from Senegal based on SSR markers. Genet. Mol. Res. GMR 11, 292–304. https://doi.org/10.4238/2012.february.8.4 (2012).
Asare, A. T. et al. Assessment of the genetic diversity in cowpea (Vigna unguiculata L. Walp.) germplasm from Ghana using simple sequence repeat markers. Plant. Genet. Resour. 8, 142–150. https://doi.org/10.1017/s1479262110000092 (2010).
O’Neill, R., Snowdon, R. & Köhler, W. Genetics aspects of biodiversity. In Progress in Botany: Genetics Physiology Systematics Ecology (eds Esser, K., Lüttge, U., Beyschlag, W. & Hellwig, F.) 115–137 (Springer, 2003). https://doi.org/10.1007/978-3-642-55819-1_7.
Samarina, L. S. et al. Transferability of ISSR, SCoT and SSR markers for Chrysanthemum × Morifolium Ramat and genetic relationships among commercial russian cultivars. Plants 10, 1302. https://doi.org/10.3390/plants10071302 (2021).
Diallo, S. et al. Development of Cowpea (Vigna unguiculata) mutant lines for dissecting resilience to drought through physiological and molecular crosstalk analysis. Plant. Mol. Biol. Rep. https://doi.org/10.1007/s11105-024-01473-2 (2024).
Fulton, T. M., Chunwongse, J. & Tanksley, S. D. Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant. Mol. Biol. Rep. 13, 207–209. https://doi.org/10.1007/bf02670897 (1995).
Chen, H. et al. De novo transcriptomic analysis of cowpea (Vigna unguiculata L. Walp.) for genic SSR marker development. BMC Genet. 18, 65. https://doi.org/10.1186/s12863-017-0531-5 (2017).
Peakall, R. & Smouse, P. E. GenAlEx 6.5: genetic analysis in Excel. genetic software for teaching and research—an update. Bioinformatics 28, 2537. https://doi.org/10.1093/bioinformatics/bts460 (2012).
Liu, K. & Muse, S. V. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21, 2128–2129. https://doi.org/10.1093/bioinformatics/bti282 (2005).
Pritchard, J. K., Stephens, M., Rosenberg, N. A. & Donnelly, P. Association mapping in structured Am. J. Hum. Genet. 67, 170. https://doi.org/10.1086/302959 (2000).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 164, 1567–1587. https://doi.org/10.1093/genetics/164.4.1567 (2003).
Earl, D. A. & VonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. https://doi.org/10.1007/s12686-011-9548-7 (2012).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Saitou, N. & Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. https://doi.org/10.1093/oxfordjournals.molbev.a040454 (1987).
Jaccard, P. Lois de distribution florale dans la zone alpine. Bull. Soc. Vaudoise Sci. Nat. 38, 69–130 (1902).
Jaccard, P. The Distribution of the Flora in the Alpine Zone.1. New. Phytol. 11, 37–50. https://doi.org/10.1111/j.1469-8137.1912.tb05611.x (1912).
Perrier, X. & Jacquemoud-Collet, J. P. DARwin software 2015 (2006).
Serrote, C. M. L., Reiniger, L. R. S., Silva, K. B., Rabaiolli, S. M. & Stefanel, C. M. dos S. Determining the polymorphism information content of a molecular marker. Gene 726. https://doi.org/10.1016/j.gene.2019.144175 (2020).
Botstein, D., White, R. L., Skolnick, M. & Davis, R. W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 32, 314. https://doi.org/10.1073/pnas.83.19.7353 (1980).
Ali, Z. et al. Assessing the genetic diversity of cowpea [Vigna unguiculata (L.) Walp.] accessions from Sudan using simple sequence repeat (SSR) markers. Afr. J. Plant. Sci. 9, 293–304. https://doi.org/10.5897/ajps2015.1313 (2015).
Fatokun, C. et al. Genetic diversity and structure of a mini-core subset from the world cowpea (Vigna unguiculata (L.) Walp.) germplasm collection. Sci. Rep. 8, 16035. https://doi.org/10.1038/s41598-018-34555-9 (2018).
Seo, E. et al. Structure and genetic diversity in Korean Cowpea germplasm based on SNP markers. Plants 9, 1190. https://doi.org/10.3390/plants9091190 (2020).
Sarr, A. et al. Genetic diversity and structure analyses of wild relatives and cultivated cowpea (Vigna unguiculata (L.) Walp.) from Senegal using simple sequence repeat markers. Plant. Mol. Biol. Rep. 39, 112–124. https://doi.org/10.1007/s11105-020-01232-z (2021).
Sawadogo, M., Ouedraogo, J. T., Gowda, B. S. & Timko, M. P. Genetic diversity of cowpea (Vigna unguiculata L. Walp) cultivars in Burkina Faso resistant to Striga gesnerioides. Afr. J. Biotechnol. 9, 8146–8153. https://doi.org/10.5897/ajb10.1175 (2010).
Taheri, S., L Abdullah, T., Ahmad, Z. & A Abdullah, N. Effect of acute gamma irradiation on curcuma alismatifolia varieties and detection of DNA polymorphism through SSR marker. BioMed. Res. Int. https://doi.org/10.1155/2014/631813 (2014).
Wright, S. Evolution and the Genetics of, 4: Variability within and among Natural 4. Univ. Chic. Press. https://doi.org/10.1126/science.207.4427.173 (1984).
Gbedevi, K. M. et al. Genetic diversity and structure of cowpea [Vigna unguiculata (L.) Walp.] germplasm collected from togo based on DArT markers. Genes 12, 1451. https://doi.org/10.3390/genes12091451 (2021).
Wang, M. L., Barkley, N. A., Gillaspie, G. A. & Pederson, G. A. Phylogenetic relationships and genetic diversity of the USDA Vigna germplasm collection revealed by gene-derived markers and sequencing. Genet. Res. 90, 467–480. https://doi.org/10.1017/s0016672308009889 (2008).
Huynh, B. L. et al. Gene pools and the genetic architecture of domesticated cowpea. Plant. Genome 6 https://doi.org/10.3835/plantgenome2013.03.0005 (2013).
Mafakheri, K., Bihamta, M. R. & Abbasi, A. R. Assessment of genetic diversity in cowpea (Vigna unguiculata L.) germplasm using morphological and molecular characterisation. Cogent Food Agric. 3, 1327092. https://doi.org/10.1080/23311932.2017.1327092 (2017).
Olasupo, F. O., Ilori, C. O., Stanley, E. A., Owoeye, T. E. & Igwe, D. O. Genetic analysis of selected mutants of cowpea (Vigna unguiculata [L.] Walp) using simple sequence repeat and rcbL markers. Am. J. Plant. Sci. 9, 2728–2756. https://doi.org/10.4236/ajps.2018.913199 (2018).
Minnaar-Ontong, A., Gerrano, A. S. & Labuschagne, M. T. Assessment of genetic diversity and structure of Bambara groundnut [Vigna subterranea (L.) verdc.] landraces in South Africa. Sci. Rep. 11, 7408. https://doi.org/10.1038/s41598-021-86977-7 (2021).
Chen, H. et al. Genetic diversity and a structure analysis of accessions in the Chinese cowpea [Vigna unguiculata (L.) Walp.] germplasm collection. Crop J. https://doi.org/10.1016/j.cj.2017.04.002 (2017).
Ramesh, P. et al. Advancements in molecular marker technologies and their applications in diversity studies. J. Biosci. 45, 123. https://doi.org/10.1007/s12038-020-00089-4 (2020).
Acknowledgements
We are grateful to IAEA for supporting the project funding. A special mention to Mr. Thuloane Bernard TSEHLO, Dr. Shoba Sivasankar, Dr. Isaac Kofi Bimpong, and Dr. Fatma SARSU, the Programme Management Officer, the Head of Plant Breeding and Genetics, and Technical Officers, respectively, for their advice and support. I would also like to express my gratitude to the “Centre d’excellence Africain en Agriculture pour la Sécurité Alimentaire et nutritionnelle (CEA-AGRISAN)” for their financial support and the anonymous reviewers for their valuable comments.
Funding
This work was supported by the International Atomic Energy Agency under RAF5076 and RAF5083 projects.
Author information
Authors and Affiliations
Contributions
SD, FAB and DD designed the study. SD and DD developed mutant populations. SD and BAK performed DNA extraction and amplification. SD and DD performed data analyses using Structure software, GenAlex, PowerMarker and DarWin software. SD, FAB, ID and DD drafted the manuscript. All authors contributed to the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Diallo, S., Badiane, F.A., Kabkia, BN. et al. Genetic diversity and population structure of cowpea mutant collection using SSR and ISSR molecular markers. Sci Rep 14, 31833 (2024). https://doi.org/10.1038/s41598-024-83087-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-83087-y
Keywords
This article is cited by
-
Genetic diversity of Brassica oleracea using agromorphological and simple sequence repeat (SSR) markers
Genetic Resources and Crop Evolution (2026)
