
Abstract
In recent years, immune checkpoint inhibitors (ICIs) has emerged as a fundamental component of the standard treatment regimen for patients with head and neck squamous cell carcinoma (HNSCC). However, accurately predicting the treatment effectiveness of ICIs for patients at the same TNM stage remains a challenge. In this study, we first combined multi-omics data (mRNA, lncRNA, miRNA, DNA methylation, and somatic mutations) and 10 clustering algorithms, successfully identifying two distinct cancer subtypes (CSs) (CS1 and CS2). Subsequently, immune-regulated genes (IRGs) and machine learning algorithms were utilized to construct a consensus machine learning-driven prediction immunotherapy signature (CMPIS). Further, the prognostic model was validated and compared across multiple datasets, including clinical characteristics, external datasets, and previously published models. Ultimately, the response of different CMPIS patients to immunotherapy, targeted therapy, radiotherapy and chemotherapy was also explored. First, Two distinct molecular subtypes were successfully identified by integrating immunomics data with machine learning techniques, and it was discovered that the CS1 subtype tended to be classified as “cold tumors” or “immunosuppressive tumors”, whereas the CS2 subtype was more likely to represent “hot tumors” or "immune-activated tumors". Second, 303 different algorithms were employed to construct prognostic models and the average C-index value for each model was calculated across various cohorts. Ultimately, the StepCox [forward] + Ridge algorithm, which had the highest average C-index value of 0.666, was selected and this algorithm was used to construct the CMPIS predictive model comprising 16 key genes. Third, this predictive model was compared with patients’ clinical features, such as age, gender, TNM stage, and grade stage. The findings indicated that this prognostic model exhibited the best performance in terms of C-index and AUC values. Additionally, it was compared with previously published models and it was found that the C-index of CMPIS ranked in the top 5 among 94 models across the TCGA, GSE27020, GSE41613, GSE42743, GSE65858, and META datasets. Lastly, the study revealed that patients with lower CMPIS were more sensitive to immunotherapy and chemotherapy, while those with higher CMPIS were more responsive to radiation therapy and EGFR-targeted treatments. In summary, our study identified two CSs (CS1 and CS2) of HNSCC using multi-omics data and predicted patient prognosis and treatment response by constructing the CMPIS model with IRGs and 303 machine learning algorithms, which underscores the importance of immunotherapy biomarkers in providing more targeted, precise, and personalized immunotherapy plans for HNSCC patients, significantly contributing to the optimization of clinical treatment outcomes.
Similar content being viewed by others

Introduction
Head and neck squamous cell carcinoma (HNSCC) is a type of cancer that affects the upper part of the digestive system, impacting regions such as the mouth, lips, nasal passages, larynx, and pharynx1. Currently, HNSCC is identified as the sixth most prevalent form of cancer worldwide, with an annual report of more than 650,000 new cases and 90,000 deaths2. Estimates suggest that the number of head and neck cancer cases might rise to 856,000 by the year 2035, underscoring the urgent need to address and advance treatment methodologies in the coming years3. Immunotherapy has emerged as an advanced standard treatment for patients with HNSCC, showing significant efficacy. However, the challenge remains in accurately predicting the therapeutic outcomes of immune checkpoint inhibitors (ICIs) for patients at the same TNM staging due to the lack of effective biomarkers4,5.
Over the past decade, research has shown that treatment with ICIs delivers enduring clinical advantages to individuals with late-stage malignancies, such as HNSCC6,7. Specifically, inhibiting the PD-1 signaling receptor restores the cytotoxic activity of tumor-specific CD8 + T cells in the tumor microenvironment (TME), thus reducing the tumors’ ability to evade immune detection and managing disease advancement.As a crucial form of immunotherapy, ICIs have attracted extensive attention from researchers over the years. These inhibitors work by blocking immune checkpoints such as PD-L18, thereby lifting the suppression on T cells and enabling them to more effectively identify and attack cancer cells9. Immune checkpoints serve as safety mechanisms within the immune system, preventing it from mistakenly attacking normal cells. However, cancer cells could utilize these checkpoints to escape immune detection and destruction. Consequently, this therapeutic approach has become a primary treatment for HNSCC in recent years10,11,12.
ICBs offer a revolutionary approach to treating HNSCC. These compounds, especially those focusing on the PD-1/PD-L1 signaling route, have shown effectiveness across multiple cancer forms, such as kidney cancer, lung cancer, and HNSCC13. They have significantly improved one-year survival rates for patients with recurrent metastatic HNSCC to a range of 36% to 57%, and extended median survival time to between 7.7 and 13.0 months14,15. This therapeutic effect is driven by a mechanism known as ICIs, where ICIs reinvigorate T-cell-mediated immune responses against tumor cells by obstructing inhibitory signaling pathwa16. For example, researchers conducted two important trials, KEYNOTE-040 and CheckMate 141, which respectively studied the effects of pembrolizumab and nivolumab on recurrent or metastatic HNSCC therapies that had progressed after previous treatments. Both trials showed an improvement in overall survival (OS) and led to the approval of these therapies for recurrent or metastatic HNSCC17. Nevertheless, clinical trials indicate that only a minority of HNSCC patients (18%) benefit from ICI treatment18,19.
Consequently, there is a pressing need to develop models based on large-scale multi-omics data and advanced methods that can identify and predict biomarkers for patients likely to respond effectively to immunotherapy. In this context, we carried out the following research activities: Firstly, through the integration of machine learning with multi-omics approaches, two molecular subtypes were independently identified and, by analyzing their markers, related immune-regulated genes (IRGs) were discovered. Next, leveraging these IRGs, a combination of 303 machine learning algorithms was utilized to develop a consensus machine learning-driven prediction immunotherapy signature (CMPIS). Subsequently, the prognostic model was validated and compared across multiple datasets, including clinical characteristics, external datasets, and previously published models. Ultimately, it was found that the low-CMPIS patients were more suitable for immunotherapy and chemotherapy. Conversely, patients with high CMPIS were better suited for radiation therapy and EGFR-targeted treatment. These findings indicated that patient stratification based on CMPIS can enhance the accuracy of treatment plan selection.
Materials and methods
Downloading and organizing of publicly available data
We first acquired multi-omics data on HNSCC from the TCGA database, including mRNA, lncRNA, miRNA, DNA methylation, somatic mutations, and clinical data (Supplementary Table S1). Meanwhile, we acquired mRNA and clinical data on four HNSCC cohorts from the GEO database, including GSE27020, GSE41613, GSE42743, and GSE65858 (Supplementary Table S2)20,21,22,23. Finally, mRNA and clinical data were obtained from the GEO database and the R package “IMvigor210CoreBiologies” for four immunotherapy cohorts (GSE78220, GSE91061, GSE135222, and IMvigor210) (Supplementary Tables S3-S6)24,25,26,27,28.
For data from microarrays, we performed a log2 transformation and normalized the expression values to achieve inter-array consistency29. When analyzing RNA-seq data obtained through high-throughput sequencing, researchers commonly rely on TPM values. These values offer a standardized measure of gene expression levels that closely resemble the data output of microarray analysis, thereby facilitating easier comparisons across different samples30. The GSE27020, GSE41613, GSE42743, and GSE65858 datasets were merged into the META dataset using the “ComBat” function, and the validity and stability of the dataset merge were further verified using PCA analysis31. The gene signatures for this study were acquired from the package “IOBR” and previously published literature19,32,33.
Identification of cancer subtypes by multi-omics consensus clustering analysis
We first obtained the expression matrix of immune checkpoint genes (ICGs) from mRNA data. For lncRNA, miRNA, and methylation data, the top 1500 genes with the highest degree of variation were filtered by the “getElitee” function and the prognostic genes were screened by univariate Cox analysis with clinical data (p < 0.05). For the mutation data, the top 5% of most frequently mutated genes were filtered by the “getElitee” function. Next, the above multi-omics data were included in the R package “MOVICS” to continue further analysis34. The optimal number of clusters was determined from the sum of the Clustering Prediction Index (CPI) and gaps-statistics35. Finally, we used 10 clustering algorithms included in the “getMOIC” function to analyze the above multi-omics data and combined the results of the various algorithms with consensus clustering to enhance the robustness of the clusters34.
Validation, molecular characterization, and immune landscape of cancer subtypes
Initially, to ensure the robustness of subtype classifications, we conducted a rigorous validation process by leveraging subtype-specific biomarkers within the META cohort. This validation step allowed us to confirm the reliability and stability of the clustering results obtained. Subsequently, the assessment of the concordance between the consensus clustering approach and the NTP and PAM classifiers was proceeded. By comparing these different classification methods, the aim was to evaluate the consistency and reproducibility of subtype assignments across various analytical frameworks. The PCA, TSNE, and UMAP analysis were also utilized to find the gene distribution of cancer subtypes (CSs)36.
Then, we investigated the association between CSs and marker genes for clinical characteristics and subtypes. The GSVA algorithm was employed to assess the changes in the relevant signaling pathways in different CSs37. The transcriptional regulatory networks, also known as regulons, were meticulously built utilizing the powerful package “RTN”. This comprehensive analysis revealed a network comprising 23 induced or repressed target-associated transcription factors (TFs) intricately linked to the regulatory landscape. Furthermore, our investigation identified 71 candidate regulators intricately associated with cancerous chromatin remodeling processes, shedding light on the intricate regulatory mechanisms underlying cancer progression and gene expression dynamics38.
Finally, based on the R package “IOBR” and previously published literature, we gathered a variety of published signatures associated with tumor microenvironment (TME), T cells, B cells, CAFs, CAFs at the single-cell level, immune suppression, immune exclusion, immune function, immunotherapy, and ICGs, and utilized a standardized approach to compute the enrichment score for individual samples, allowing for a thorough examination of the immunological variances between patients with CSs32,39.
Construction of a consensus machine learning-driven prediction immunotherapy signature
To improve comparison across datasets, we standardized the data in all cohorts with Z scores. The TCGA-HNSCC dataset was regarded as the training set, while the GSE27020, GSE41613, GSE42743, and GSE65858 cohorts were regarded as the test sets. We first take the intersection of the genes in these datasets to obtain the shared genes for subsequent analyses. Second, we conducted univariate Cox analysis in the TCGA-HNSCC, GSE27020, GSE41613, GSE42743, and GSE65858 cohorts. Genes with p < 0.001 for the TCGA-HNSCC cohort, genes with p < 0.01 for the other cohorts, and the same HR orientation were recognized as prognostic immune-regulated genes (PIRGs). We used 10 machine learning algorithms whose parameters were primarily referenced from previous high-quality literature40.These learning algorithms were then ranked and combined to form a combination of 303 models. Then, based on the model of the training set and related parameters, it is used to construct the model of the test set. In the end, we chose the algorithm with the highest C-index value among all the cohorts as the final method for a CMPIS.
Comparison of CMPIS with clinical features and published signatures
First, we obtained the risk score of each sample by StepCox [forward] + Ridge algorithm. Then, we used the function “surv_cutpoint” based on the R package “survminer” to determine the optimal cut point for continuous variables. Finally, this optimal cutpoint was used as a threshold to differentiate patients into low CMPIS and high CMPIS groups. C-index values were employed to compare the stability and accuracy of CMPIS with clinical features and published signatures. Univariate and multivariate Cox analyses were performed to confirm whether CMPIS was an independent prognostic factor. The time-dependent ROC curve was also employed to compare the stability and accuracy of CMPIS with clinical features. To obtain a comprehensive list of previously published signatures, we searched the PubMed database with the keywords “model”, “signature”, and "head and neck squamous cell carcinoma", or "laryngeal squamous cell carcinoma", or "oral squamous cell carcinoma", or "tongue squamous cell carcinoma" as keywords. All studies from 2018 to now were searched, and after screening, a total of 94 previously published models were obtained41.
Construction of a nomogram and implementation of enrichment analysis
To improve the clinical utility value of the CMPIS, a nomogram incorporating both clinical features and the CMPIS was developed to predict the survival rates of HNSCC. To elucidate the biological functions and signaling pathway differences among different CMPIS groups, we obtained differentially expressed genes (DEGs) from different CMPIS groups (|log2FC > = 1| and FDR < 0.05) and performed GO and KEGG enrichment analysis29,42,43.
Immunotherapy, radiotherapy, chemotherapy, and EGFR-targeted therapy responses
For the immunotherapy response, we initially assessed the restricted mean survival (RMS) and long-term survival (LTS) of patients to immunotherapy in the IMvigor210 cohort. Similar evaluations were completed in the GSE78220, GSE135222, and GSE91061 cohorts. We also combined the Tracking Tumor Immunophenotype (TIP) method and the Tumor Immune Dysfunction and Exclusion (TIDE) method to evaluate immunotherapy response44,45. In conjunction with immunotherapy, radiotherapy, chemotherapy, and targeted therapy represent quintessential modalities frequently employed within the contemporary clinical sphere. The ssGSEA algorithm was used to calculate two radiotherapy signatures (hypoxia and cell cycle) and EGFR-ligand signatures to predict response to radiotherapy and EGFR-targeted therapy. Finally, the package “oncoPredict” was utilized to predict the sensitivity of patients in different CMPIS patients to conventional chemotherapeutic drugs (Cisplatin, Docetaxel, 5-Fluorouracil, and Gemcitabine) and EGFR-targeted therapeutic agents (Osimertinib, Lapatinib, and Sapitinib)46.
Statistical analysis
For comparisons of two groups, the normally distributed variables were tested by unpaired Student’s t tests, and the non-normally distributed variables were tested by the Wilcoxon rank-sum test. A two-sided Fisher’s exact test was performed for the contingency tables. The log-rank test was used to compare differences in Kaplan–Meier survival analysis between different groups. All statistical analyses and figures were performed in R v.4.3.3.
Results
Identification of cancer subtypes by immune multi-omics consensus clustering analysis
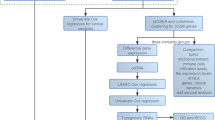
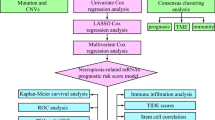
The research workflow illustrated the methodology employed in this study (Fig. 1). Prior to and after the processing of batch effects, PCA was utilized to validate the validity and stability of the dataset integration (Figures S1A and S1B). Through the application of ten multi-omics ensemble clustering algorithms, we autonomously identified two distinct subtypes, with the determination of the subtype count being meticulously guided by the Composite Predictability Index (CPI), gap statistical analysis, and silhouette score (Fig. 2A and B). Subsequently, the clustering outcomes were amalgamated utilizing a consensus ensemble approach, highlighting discernible molecular expression patterns across transcriptomes (including ICGs, lncRNA, and miRNA), epigenetic methylation, and somatic mutations (Fig. 2C–E). Notably, CS1 demonstrated the most favorable OS (p < 0.001; Fig. 2F).

The research workflow.

Identification of cancer subtypes by immune multi-omics consensus clustering analysis. (A) The CPI and gap statistical analysis of the immune multi-omics clusters. (B) The sample similarity of each subgroup was assessed by calculating the silhoutte score. (C) Visualization of immune multi-omics consensus clustering analysis. (D) Consensus heatmap for CSs. (E) Consensus clustering matrix based on the 10 multi-omics algorithms. (F) Kaplan–Meier analysis of CSs in the TCGA cohort.
Validation, molecular characterization, and immune landscape of cancer subtypes
Based on the marker genes for each CSs, we identified 1500 genes that were specifically up-regulated for each CSs as classifiers and immune-regulated genes (IRGs) (Supplementary Table S7). These genes were subsequently validated in the META cohort to reinforce the stability of the subtypes (Fig. 3A). In line with these findings, it was observed that CS1 in the META cohort exhibited the most favorable prognosis (p < 0.001; Fig. 3B). Furthermore, the concordance of CSs with NTP and PAM algorithms was evaluated (p < 0.001; Fig. 3C–E). The robustness of the classification was substantiated by the outcomes derived from PCA, tSNE, and UMAP approaches, which unveiled notable distinctions among the various subtypes (Fig. 3F–H).

Validation of cancer subtypes. (A) Molecular subtype validation in the META cohort based on IRGs by the NTP algorithm. (B) Kaplan–Meier analysis of CSs in the META cohort. (C) The consistency between CSs and NTP in the TCGA cohort. (D) The consistency between CSs and PAM in the TCGA cohort. (E) The consistency between NTP and PAM in the META cohort. (F–H) The PCA, tSNE, and UMAP methods unveiled notable distinctions among the CSs.
To investigate the association between CSs and clinical features, we performed a heat map, which showed that CS1 was associated with higher mortality, older age, and a higher clinicopathologic stage (Fig. 4A). To explore the differences in signaling pathways between CSs, we used the GSVA algorithm to analyze the KEGG pathways that differed in each CS (Supplementary Table S8). CS1 was notably enriched in pathways related to the extracellular matrix, coagulation system, and TGF-beta signaling pathway. CS2 was significantly enriched in pathways related to the cell cycle, immune-related signaling, and metabolism-related signaling. To investigate transcriptome differences, we analyzed underlying regulators related to TFs and chromatin remodeling in HNSCC (Fig. 4B). RXRA, EGFR, HIF1A, PGR, and RARA regulators were significantly activated in CS1, whereas ESR2, FOXA1, PPARG, RARB, RXRB, FGFR3, ERBB2, and ERBB3 were specifically enriched in CS2 (Fig. 4C). The diverse range of regulatory activities linked to oncogenic chromatin remodeling underscores the potential for distinctive patterns of differential regulation among the CSs. This observation suggests that epigenetically-driven transcriptional networks could serve as crucial distinguishing factors for these molecular subtypes.

Molecular characterization of cancer subtypes. (A) A heat map showed that CS1 was associated with higher mortality, older age, and a higher clinicopathologic stage. (B) CS1 was notably enriched in pathways related to the extracellular matrix, coagulation system, and TGF-beta signaling pathway. CS2 was significantly enriched in pathways related to the cell cycle, immune-related signaling, and metabolism-related signaling. (C) RXRA, EGFR, HIF1A, PGR, and RARA regulators were significantly activated in CS1, whereas ESR2, FOXA1, PPARG, RARB, RXRB, FGFR3, ERBB2, and ERBB3 were specifically enriched in CS2.
Using the R package “IOBR”, we found that T cells and B cells were significantly enriched in CS2, suggesting an immune-activated state, while CAFs were mainly enriched in CS1, suggesting an immune-suppressed state (Fig. 5A–E). Immune suppression signatures and immune exclusion signatures were significantly enriched in CS1, while immune function signatures, immunotherapy signatures, and ICGs were significantly enriched in CS2 (Fig. 6A–E). These results suggested that CS1 was more likely to be a “cold tumor” or “immunosuppressive tumor”, while CS2 was more probably classified as a “hot tumor” or "immune-activated tumor".

Immune cell infiltration of cancer subtypes. (A-E) T cells and B cells were significantly enriched in CS2, suggesting an immune-activated state, while CAFs were mainly enriched in CS1, suggesting an immune-suppressed state.

Immunotherapy of cancer subtypes. (A-E) Immune suppression signatures and immune exclusion signatures were significantly enriched in CS1, while immune function signatures, immunotherapy signatures, and ICGs were significantly enriched in CS2.
Development of a consensus machine learning-driven prediction immunotherapy signature
Firstly, univariate Cox analysis was used to screen PIRGs whose expression was markedly associated with OS from the IRGs in the TCGA and META sets. Subsequently, models were constructed based on each of the 303 algorithms and the average C-index value of each model across all cohorts was computed to evaluate the predictive power (Fig. 7A and Supplementary Table S9). The highest average C-index (0.666) was achieved by the StepCox [forward] + Ridge algorithm, and a CMPIS was constructed using this algorithm, which was constructed with 16 hub PIRGs (CD6, CD5, EPHX3, MAP4K1, CALML5, KLRB1, PDGFA, WNT7A, CHGB, CAMK2N1, DKK1, PTX3, STC2, HOXA1, EFNB2, and GSTO1). Figure 7B showed the coefficients of individual PIRGs in the CMPIS, and Fig. 7C showed the results of individual PIRGs in the univariate Cox analysis in the TCGA and META sets. Finally, we computed CMPIS scores for each sample in all cohorts and found that the high CMPIS patients had a worse clinical prognosis in the TCGA, GSE27020, GSE41613, GSE42743, GSE65858, and META cohorts (Fig. 7D).

Construction of a consensus machine learning-driven prediction immunotherapy signature. (A) Models were constructed based on each of the 303 algorithms, and the average C-index value was calculated for each model across all cohorts. The highest average C-index (0.666) was achieved by the StepCox [forward] + Ridge algorithm. (B) The coefficients of individual PIRGs in the CMPIS. (C) The PIRGs in the univariate Cox analysis in the TCGA and META cohorts. (D) Survival analysis of different CMPIS groups in the TCGA, GSE27020, GSE41613, GSE42743, GSE65858, and META cohorts.
Comparison of CMPIS with clinical characteristics and published signatures
Traditional clinical features have historically served as predictors of patient prognosis. In our study, the predictive efficacy of these conventional clinical features has been evaluated and compared with the CMPIS. The C-index confirmed the superior stability and accuracy of the CMPIS compared to clinical features (Fig. 8A). Considering the clinical prospect of CMPIS, we validated CMPIS as an independent prognostic factor by univariate and multivariate Cox analysis (Fig. 8B and C). Moreover, the Area Under ROC Curve (AUC) value for the CMPIS at 1, 3, 5, and 10 years was greater than 0.65, surpassing that of the clinical features and indicating high reliability (Fig. 8D). In order to conduct a thorough comparative analysis between the CMPIS and other relevant signatures, we systematically reviewed literature spanning from 2018 to the present (Supplementary Table S10). This meticulous search process led to the incorporation of a total of 94 published signatures for our comparative evaluation. These signatures were associated with different biological functions and processes, including immune, immune cell, necroptosis, hypoxia, autophagy, pyroptosis, metastasis, metabolism, ferroptosis, stemness, cell component, cuproptosis, methylation, and alternative splicing. The results indicated that the CMPIS showed better C-index performance than almost 94 models in the TCGA, GSE27020, GSE41613, GSE42743, GSE65858, and META cohorts (Fig. 9).

Comparison of the CMPIS with clinical features. (A) The C-index of CMPIS was higher than the clinical features. (B and C) Univariate and multivariate Cox analyses for the CMPIS and clinical features. (D) The AUC values for CMPIS at 1, 3, 5, and 10 years were greater than 0.65, exceeding the clinical features.

Comparison of CMPIS with published signatures. The CMPIS showed better C-index performance than almost 94 models in the TCGA, GSE27020, GSE41613, GSE42743, GSE65858, and META cohorts.
Construction of a nomogram and implementation of enrichment analysis
To enhance the clinical utility of the CMPIS, we devised a nomogram that combines the CMPIS with pertinent clinical characteristics (Fig. 10A). Furthermore, the calibration curve analysis illustrated that the nomogram’s predictive accuracy aligns closely with real-world outcomes (Fig. 10B). This integration not only facilitates more precise prognostication but also supports informed decision-making in patient management. This validation underscored the reliability and practicality of the nomogram as a predictive tool in the clinical setting. In total, we obtained 759 DEGs by differential expression analysis, with 436 marker genes in the low CMPIS group and 323 marker genes in the high CMPIS group (Supplementary Table S11). The marker genes in the low CMPIS group were significantly associated with immune cell activation, immune cell differentiation, immune-related signaling pathways, and immune checkpoint pathways (Fig. 10C and D, Supplementary Tables S12 and S13), whereas the marker genes in the high CMPIS group were significantly associated with a variety of tumor-related pathways, cancer invasion and metastasis, and cytokine pathways (Fig. 10E and F, Supplementary Tables S14 and S15).Significant differences in biological functions and molecular pathways were found between different CMPIS groups, which might contribute to their different prognoses and treatment responses.

Construction of a nomogram and implementation of enrichment analysis. (A) A nomogram combining the CMPIS with pertinent clinical characteristics to enhance the clinical utility of the CMPIS. (B) The calibration curve analysis illustrated that the nomogram’s predictive accuracy aligns closely with real-world outcomes. (C and D) The marker genes in the low CMPIS group were significantly associated with immune cell activation, immune cell differentiation, immune-related signaling pathways, and immune checkpoint pathways. (E and D) The marker genes in the high CMPIS group were significantly associated with a variety of tumor-related pathways, cancer invasion and metastasis, and cytokine pathways.
Different CMPIS patients are suited for different treatment approaches
To comprehensively evaluate the impact of the CMPIS in the context of HNSCC immunotherapy, a systematic analysis was undertaken. The study accounted for the delayed clinical effects often associated with immunotherapy by assessing the RMS at 12 and 24 months, along with examining LTS differences 6 months post-treatment in the IMvigor210 cohort (Fig. 11A and B). Notably, the group with lower CMPIS scores exhibited superior prognostic outcomes, suggesting a more pronounced benefit from immunotherapy. Furthermore, the distribution of CMPIS scores across patients with varying response levels revealed a significantly lower CMPIS score in the responder group compared to the nonresponder group (Fig. 11C). Subsequently, our results were validated across multiple immunotherapy cohorts with prognostic data. Consistently, lower CMPIS scores correlated with improved prognostic outcomes post-immunotherapy in the GSE78220 cohort (Fig. 11D) and GSE135222 cohort (Fig. 11E), as well as better immunotherapy responses in the GSE91061 cohort (Fig. 11F). Moreover, the study delved into the potential immunological mechanisms by calculating the TIP score, revealing significant differences in priming and activation, trafficking of immune cells to tumors, and infiltration of immune cells into tumors among patients with low CMPIS scores (Fig. 11G). Additionally, the TIDE algorithm was employed to assess patient responsiveness to immunotherapy, demonstrating higher responsiveness in the low CMPIS group (Fig. 11H). Notably, the low CMPIS group exhibited higher Micro-Satellite Instability (MSI) scores and Dysfunction scores, while the high CMPIS group displayed elevated Exclusion scores (Figs. 10K–11I).

Evaluation of immunotherapy response. (A) The RMS time difference by 12 and 24 months after treatment. (B) The LTS difference after 6 months of treatment. (C) A significantly lower CMPIS score in the responder group compared to the nonresponder group. (D-F) Lower CMPIS scores correlated with improved prognostic outcomes post-immunotherapy in the GSE78220 cohort and GSE135222 cohort, as well as better immunotherapy responses in the GSE91061 cohort. (G) Significant differences in priming and activation, trafficking of immune cells to tumors, and infiltration of immune cells into tumors among patients with low CMPIS scores. (H) Higher responsiveness in the low CMPIS group. (I-K) The low CMPIS group exhibited higher MSI scores and Dysfunction scores, while the high CMPIS group displayed elevated Exclusion scores.
In conjunction with immunotherapy, radiotherapy, chemotherapy, and targeted therapy represent quintessential modalities frequently employed within the contemporary clinical sphere. Two radiotherapy-related biomarkers (hypoxia and cell cycle) were significantly enriched in the high CMPIS patients, suggesting that the high CMPIS patients were more suitable for radiation therapy (Fig. 12A). Three commonly used chemotherapeutic drugs (Cisplatin, 5-Fluorouracil, and Gemcitabine) were more sensitive in the low CMPIS patients, suggesting that the low CMPIS patients were better suited for chemotherapy (Fig. 12B). EGFR ligand scores were higher in the high CMPIS patients, and the three EGFR-targeted therapeutic agents were more sensitive in the high CMPIS patients, suggesting that the high CMPIS patients were more suitable for EGFR-targeted therapy (Fig. 12C).

Evaluation of radiotherapy, chemotherapy, and EGFR-targeted therapy responses. (A) Two radiotherapy-related biomarkers (hypoxia and cell cycle) were significantly enriched in the high CMPIS patients. (B) Three commonly used chemotherapeutic drugs (Cisplatin, 5-Fluorouracil, and Gemcitabine) were more sensitive in the low CMPIS patients. (C) EGFR ligand scores were higher in the high CMPIS patients, and the three EGFR-targeted therapeutic agents were more sensitive in the high CMPIS patients.
Our study indicated that the low CMPIS patients exhibited greater suitability for immunotherapy and chemotherapy. Conversely, those in the high CMPIS group demonstrated a higher suitability for radiotherapy and EGFR-targeted therapy. These findings suggested that stratifying patients based on CMPIS can enhance the precision of treatment selection, potentially leading to improved clinical outcomes.
Disscussion
The treatment of HNSCC encompasses a variety of disciplines, including surgery, radiation therapy, and cytotoxic chemotherapy47,48. However, the recent emergence of immunotherapy has significantly changed both treatment procedures and outcomes. Known for its enduring efficacy, minimal side effects, and broad applicability, immunotherapy has shown outstanding performance in the treatment of HNSCC. Research has demonstrated that immunotherapy significantly improves the prognosis of recurrent metastatic HNSCC49. New immunotherapy-based treatments were also being applied to locally advanced cases. Furthermore, numerous ongoing trials are investigating the use of new immunotherapeutic agents, including durvalumab, atezolizumab, avelumab, tremelimumab, monalizumab, and others50. These findings underscored the efficacy of immunotherapy in the treatment of HNSCC. Looking ahead, it is highly probable that this therapeutic approach will become the predominant method of treatment.
Immunotherapy has been associated with more durable outcomes and fewer, less severe, or at least more manageable adverse effects compared to traditional cancer treatments. This innovative approach leveraged the body’s inherent immune response to identify and attack cancerous cells, thus enhancing the specificity and efficacy of treatment while decreasing collateral damage to healthy tissues50. Additionally, the combination of immunotherapy with other treatment approaches, including vaccines and chemotherapy, remained a subject of particular interest50. Cancer vaccines have the potential to boost the immune system’s capacity for identifying and combating cancerous cells, while chemotherapy can reduce tumor burden and alter the tumor milieu in ways that make it more susceptible to immune attack. The integration of these modalities aims to create a synergistic effect, thereby enhancing the overall therapeutic outcome51.
In the realm of HNSCC treatment, numerous clinical trials are exploring the integration7 of immunotherapy with other therapeutic modalities and pharmacotherapy to enhance treatment efficacy and safety52. For example, studies have demonstrated that integrating radiation therapy with immunotherapy offers a promising method for patients with locally advanced, recurrent, or metastatic HNSCC49. These researches offer mechanistic insights into the synergies and therapeutic benefits of such combinations. Several phase II/III clinical trials suggest that augmenting radiation therapy with immunotherapy is a safe option53. Researchers have also examined various immunotherapy regimens and alternatives for patients who are not eligible for immunotherapy. Discussions have focused on using alternative systemic treatments (such as EGFR inhibitors or different forms of immunotherapy) to reduce treatment intensity, including the possibility of excluding simultaneous chemotherapy for patients with a low risk51. In conclusion, a biological framework has been established to investigate the potential interactions between radiation therapy and systemic treatments, particularly emphasizing immunotherapy. A retrospective analysis of G/GEJ adenocarcinoma patients receiving a combination of PD-1 inhibitors and chemotherapy between October 2017 and May 2022 revealed significant findings. Specifically, patients with a PD-L1 CPS of ≥ 5 significantly benefited from this treatment, showing enhanced response rates and prolonged progression-free survival54. Therefore, we foresee that the coming decade will present numerous opportunities for the development of enhanced prognostic methods for personalized and concurrent treatments. This progress would facilitate the personalized choice of the best order and mix of cytotoxic chemotherapy, specific drugs, and immune therapy7, customized to enhance advantages and reduce adverse effects51.
In this study, we successfully identified two distinct molecular subtypes using ten integrated multi-omics clustering algorithms. Through the application of ten machine learning methods to analyze multi-omics data, significant enrichment of immune suppression and immune rejection features was found in the CS1 subtype, while in the CS2 subtype, significant enrichment of immune function, immune therapy features, and ICGs was shown.This suggested that the CS1 subtype was more likely to be a “cold tumor” or "immune-suppressive tumor", while the CS2 subtype was more likely to be a “hot tumor” or "immune-activated tumor". Subsequently, by analyzing the markers of these subtypes, we identified their IRGs, which we referred to as key features. Based on these IRGs, we employed a combination of over 303 machine learning algorithms and utilized the StepCox [forward] + Ridge algorithm to construct a prognostic model named CMPIS, consisting of 16 key genes (CD6, CD5, EPHX3, MAP4K1, CALML5, KLRB1, PDGFA, WNT7A, CHGB, CAMK2N1, DKK1, PTX3, STC2, HOXA1, EFNB2, and GSTO1). Next, the CMPIS model was validated and compared across various datasets, including clinical characteristics, external datasets, and previously published models. Significant biological and molecular pathway differences among different CMPIS groups were observed, which might explain their distinct prognostic and treatment response patterns.A systematic analysis of the impact of CMPIS on immunotherapy in HNSCC revealed that the low CMPIS patients demonstrated better prognosis, indicating a greater benefit from immunotherapy. In conclusion, we validated and compared our results using clinical characteristics, external datasets, and published models, ultimately concluding that patients with lower CMPIS scores were more suitable for immunotherapy and chemotherapy, while those with higher CMPIS scores were better suited for radiation therapy and EGFR-targeted treatment. These findings underscored the importance of patient stratification based on CMPIS in enhancing the accuracy of treatment selection and potentially improving clinical treatment outcomes.
Given the current challenges in the field of immunotherapy, including the lack of effective immune markers, poor clinical efficacy, and the inability to accurately determine patient sensitivity to treatment, there is an urgent need to develop models based on large-scale multi-omics data and advanced algorithms to identify and predict biomarkers that may effectively respond to immunotherapy. Numerous researchers were dedicated to this goal, focusing on predicting and identifying key biomarkers. For instance, S.Trebeschi et al. utilized artificial intelligence technology to characterize lesions based on preprocessed contrast-enhanced CT data, successfully developing and validating a non-invasive machine learning biomarker that can distinguish between responders and non-responders to immunotherapy. Their study concluded that the radiological characteristics of lesions could serve as non-invasive biomarkers for assessing response to immunotherapy and might improve patient stratification in neoadjuvant and palliative treatments55. Additionally, other researchers have emphasized the importance of predictive biomarkers in guiding patients’ choices of monotherapy or combination therapy and in developing alternative treatment strategies56. Michaël Duruisseaux et al. discussed currently available and promising biomarkers, highlighting the need for predictive biomarkers of the efficacy of immune checkpoint inhibitors57. Furthermore, studies have recognized CCDC71L as an independent prognostic marker for HNSCC, with high expression related to ICGs, oncogene mutations, and markers of genomic heterogeneity, indicating its significant role in the progression of HNSCC58. These research findings are highly consistent with our study conclusions, further validating the reliability of our research and underscoring the critical role of immunotherapy biomarkers in the treatment of HNSCC. By comparing our research results with the aforementioned existing literature, we can verify and confirm the reliability of our findings, demonstrating the value and significance of our research.
While our findings hold promise, we acknowledge several limitations in our study. Firstly, the difficulty in obtaining fresh clinical samples impeded our ability to conduct functional validations with fresh tumor samples. Secondly, as our research was retrospective in nature, further validation through multicenter prospective cohort studies is essential to confirm our findings. Additionally, our dataset was relatively small and predominantly comprised non-metastatic patients, potentially introducing bias in our experimental outcomes. Our proposed model may be more suitable for predicting the prognosis and guiding treatment for non-metastatic patients, which could limit its generalizability. Furthermore, a notable limitation was the lack of basic experiments, including cell and animal studies, to explore the functional expression of genes.
Conclusion
In summary, we identified two CSs (CS1 and CS2) of HNSCC using multi-omics data and predicted patient prognosis and treatment response by constructing the CMPIS model with IRGs and 303 machine learning algorithms, finding that low CMPIS patients were more sensitive to immunotherapy and chemotherapy, while high CMPIS patients were more sensitive to radiotherapy and EGFR-targeted therapies, which underscored the importance of immunotherapy biomarkers in providing more targeted, precise, and personalized immunotherapy plans for HNSCC patients, significantly contributing to the optimization of clinical treatment outcomes.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Wu, Z. H., Yun, T. & Cheng, Q. Data mining identifies six proteins that can act as prognostic markers for head and neck squamous cell carcinoma. Cell Transplant. 29, 2138922012 (2020).
Torre, L. A. et al. Global cancer statistics, 2012. Ca. Cancer. J. Clin. 65, 87–108 (2015).
Shield, K. D. et al. The global incidence of lip, oral cavity, and pharyngeal cancers by subsite in 2012. Ca. Cancer. J. Clin. 67, 51–64 (2017).
Wang, Q., Zhao, Y., Wang, F. & Tan, G. A novel immune signature predicts immunotherapy responsiveness and reveals the landscape of the tumor immune microenvironment in head and neck squamous cell carcinoma. Front. Genet. 13, 1051051 (2022).
Zhao, X. & Cui, L. A robust six-mirna prognostic signature for head and neck squamous cell carcinoma. J. Cell. Physiol. 235, 8799–8811 (2020).
Chow, L. Head and neck cancer. N. Engl. J. Med. 382, 60–72 (2020).
Gourd, E. Concurrent chemotherapy improves outcomes in Hnscc. Lancet Oncol. 19, e343 (2018).
Lipson, E. J. & Drake, C. G. Ipilimumab: An Anti-Ctla-4 antibody for metastatic melanoma. Clin. Cancer Res. 17, 6958–6962 (2011).
La-Beck, N. M., Jean, G. W., Huynh, C., Alzghari, S. K. & Lowe, D. B. Immune checkpoint inhibitors: New insights and current place in cancer therapy. Pharmacotherapy. 35, 963–976 (2015).
Velcheti, V. & Schalper, K. Basic overview of current immunotherapy approaches in cancer. Am. Soc. Clin. Oncol. Educ. Book. 35, 298–308 (2016).
De Felice, F. et al. Immune check-point in cervical cancer. Crit. Rev. Oncol. Hematol. 129, 40–43 (2018).
Chai, Q. Q., Du, J. Y., Zhu, J. & Wu, B. The differences in the safety and tolerability of immune checkpoint inhibitors as treatment for non-small cell lung cancer and melanoma: Network meta-analysis and systematic review. Front. Pharmacol. 10, 1260 (2019).
Wakabayashi, G. et al. Development and clinical applications of cancer immunotherapy against Pd-1 signaling pathway. J. Biomed. Sci. 26, 96 (2019).
Ferris, R. L. et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 375, 1856–1867 (2016).
Burtness, B. et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (Keynote-048): A randomised, open-label, Phase 3 study. Lancet. 394, 1915–1928 (2019).
Leach, D. R., Krummel, M. F. & Allison, J. P. Enhancement of antitumor immunity by Ctla-4 blockade. Science. 271, 1734–1736 (1996).
Cohen, E. et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (Keynote-040): A randomised, open-label, phase 3 study. Lancet. 393, 156–167 (2019).
Kartha, V. K. et al. Functional and genomic analyses reveal therapeutic potential of targeting beta-catenin/Cbp activity in head and neck cancer. Genome Med. 10, 54 (2018).
Chu, G., Ji, X., Wang, Y. & Niu, H. Integrated multiomics analysis and machine learning refine molecular subtypes and prognosis for muscle-invasive urothelial cancer. Mol. Ther. Nucleic Acids. 33, 110–126 (2023).
Fountzilas, E. et al. Identification and validation of a multigene predictor of recurrence in primary laryngeal cancer. Plos One. 8, e70429 (2013).
Zhao, Y. et al. Comprehensive analysis of tumor immune microenvironment characteristics for the prognostic prediction and immunotherapy of oral squamous cell carcinoma. Front. Genet. 13, 788580 (2022).
Lohavanichbutr, P. et al. A 13-gene signature prognostic of Hpv-negative Oscc: Discovery and external validation. Clin. Cancer Res. 19, 1197–1203 (2013).
Wichmann, G. et al. The role of Hpv Rna transcription, immune response-related gene expression and disruptive Tp53 mutations in diagnostic and prognostic profiling of head and neck cancer. Int. J. Cancer. 137, 2846–2857 (2015).
Hugo, W. et al. Genomic and transcriptomic features of response to anti-Pd-1 therapy in metastatic melanoma. cell. 165, 35–44 (2016).
Riaz, N. et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 171, 934–949 (2017).
Jung, H. et al. Dna methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 10, 4278 (2019).
Kim, J. Y., Choi, J. K. & Jung, H. Genome-wide methylation patterns predict clinical benefit of immunotherapy in lung cancer. Clin. Epigenetics. 12, 119 (2020).
Mariathasan, S. et al. Tgfbeta attenuates tumour response to Pd-L1 blockade by contributing to exclusion of T cells. Nature. 554, 544–548 (2018).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic. Acids. Res. 43, e47 (2015).
Wagner, G. P., Kin, K. & Lynch, V. J. Measurement of Mrna abundance using Rna-Seq data: Rpkm measure is inconsistent among samples. Theory Biosci. 131, 281–285 (2012).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The Sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 28, 882–883 (2012).
Zeng, D. et al. Iobr: Multi-omics immuno-oncology biological research to decode tumor microenvironment and signatures. Front. Immunol. 12, 687975 (2021).
Yin, J. et al. Head and neck squamous cell carcinoma subtypes based on immunologic and hallmark gene sets in tumor and non-tumor tissues. Front. Surg. 9, 821600 (2022).
Lu, X., Meng, J., Zhou, Y., Jiang, L. & Yan, F. Movics: An R package for multi-omics integration and visualization in cancer subtyping. Bioinformatics. 36, 5539–5541 (2021).
Chalise, P. & Fridley, B. L. Integrative clustering of multi-level ’omic data based on non-negative matrix factorization algorithm. Plos One. 12, e176278 (2017).
Fan, X. et al. A new marker constructed from immune-related lncrna pairs can be used to predict clinical treatment effects and prognosis: In-depth exploration of underlying mechanisms in Hnscc. World J. Surg. Oncol. 21, 250 (2023).
Hanzelmann, S., Castelo, R. & Guinney, J. Gsva: Gene set variation analysis for microarray and Rna-Seq data. Bmc Bioinformatics. 14, 7 (2013).
Lu, X. et al. Multi-omics consensus ensemble refines the classification of muscle-invasive bladder cancer with stratified prognosis, tumour microenvironment and distinct sensitivity to frontline therapies. Clin. Transl. Med. 11, e601 (2021).
Yin, J. et al. Immune-related lncrna signature for predicting the immune landscape of head and neck squamous cell carcinoma. Front. Mol. Biosci. 8, 689224 (2021).
Liu, Z. et al. Machine learning-based integration develops an immune-derived lncrna signature for improving outcomes in colorectal cancer. Nat. Commun. 13, 816 (2022).
Yin, J. et al. Identification of molecular classification and gene signature for predicting prognosis and immunotherapy response in Hnscc using cell differentiation trajectories. Sci. Rep. 12, 20404 (2022).
Wu, T. et al. Clusterprofiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation (Camb.) 2, 100141 (2021).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 32, 2847–2849 (2016).
Fu, J. et al. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med. 12, 21 (2020).
Xu, L. et al. Tip: A web server for resolving tumor immunophenotype profiling. Cancer Res. 78, 6575–6580 (2018).
Maeser, D., Gruener, R. F. & Huang, R. S. Oncopredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 22, (2021).
Caudell, J. J. et al. Nccn Guidelines(R) Insights: Head and neck cancers, Version 1.2022. J. Natl. Compr. Cancer Netw. 20, 224–234 (2022).
Chen, S. et al. Review of biomarkers for response to immunotherapy in Hnscc microenvironment. Front. Oncol. 13, 1037884 (2023).
Okano, S. Immunotherapy for Head and neck cancer: Fundamentals and therapeutic development. Auris Nasus Larynx. 51, 684–695 (2024).
Vallianou, N. G. et al. Immunotherapy in head and neck cancer: Where do we stand?. Curr. Oncol. Rep. 25, 897–912 (2023).
Vasudevan, H. N. & Yom, S. S. Combining systemic therapy with radiation: Head and neck cancer treatments in an era of targeted agents and immunotherapy. J. Natl. Compr. Cancer Netw. 18, 907–913 (2020).
Hecht, M. et al. Induction chemoimmunotherapy followed by Cd8+ immune cell-based patient selection for chemotherapy-free radioimmunotherapy in locally advanced head and neck cancer. J. Immunother. Cancer. 10, (2022).
Gong, H. et al. Camrelizumab-based induction chemoimmunotherapy in locally advanced stage hypopharyngeal carcinoma: Phase II clinical trial. Nat. Commun. 15, 5251 (2024).
Sun, Y. T. et al. Clinical outcomes and biomarker exploration of first-line Pd-1 inhibitors plus chemotherapy in patients with low Pd-L1-expressing of gastric or gastroesophageal junction adenocarcinoma. Cancer. Immunol. Immunother. 73, 144 (2024).
Trebeschi, S. et al. Predicting response to cancer immunotherapy using noninvasive radiomic biomarkers. Ann. Oncol. 30, 998–1004 (2019).
Tray, N., Weber, J. S. & Adams, S. Predictive biomarkers for checkpoint immunotherapy: Current status and challenges for clinical application. Cancer Immunol. Res. 6, 1122–1128 (2018).
Duruisseaux, M., Lize-Dufranc, C., Badoual, C. & Bibeau, F. Predictive biomarkers of efficacy of checkpoint blockade inhibitors in cancer treatment. Ann. Pathol. 37, 46–54 (2017).
Zhang, Y. et al. Ccdc71L as a novel prognostic marker and immunotherapy target via lipid metabolism in head and neck squamous cell carcinoma. J. Stomatol. Oral Maxillofac. Surg. 125, 101799 (2024).
Acknowledgements
Tanks to all the authors for their contributions to this study, especially to T.J for her guidance and assistance. Correspondence and requests for materials should be addressed to T.J.
Funding
Supported by Sichuan Science and Technology Program (2024YFHZ0320, 2024NSFC0722, 2024JDHJ0041). The China Postdoctoral Science Foundation (No. 2023M730378). Xinglin Scholar” Scientific Research Promotion Plan of Chengdu University of TCM (BSH2023025).
Author information
Authors and Affiliations
Contributions
T.J., J.Y., and M.G. conceived and performed the bioinformatics analysis. Z.Z., P.Z., and S.W. collected data. L.Z. and L.X. performed the model. L.Z., Y.Z., and Z.Z. co-wrote the paper. T.J., J.Y., and Y.Z. undertook a manuscript review. All authors contributed to and revised the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yin, J., Xu, L., Wang, S. et al. Integrating immune multi-omics and machine learning to improve prognosis, immune landscape, and sensitivity to first- and second-line treatments for head and neck squamous cell carcinoma. Sci Rep 14, 31454 (2024). https://doi.org/10.1038/s41598-024-83184-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-83184-y
