
Abstract
Heavy metal contamination in acidified agricultural soils poses significant environmental and health risks. This study presents the development of a cost-effective dual-functional adsorbent, the attapulgite-oyster shell (ATP–OS) composite, engineered for the simultaneous removal of co-contaminated Cd(II) and Pb(II) ions. The ATP–OS composite was synthesized through thermal treatment at 700 °C, facilitating a synergistic interaction between attapulgite (ATP) and oyster shell (OS) powders. Characterization revealed enhanced dispersibility, increased active site density, and an expanded specific surface area, contributing to superior adsorption capacities of 197 mg g−1 for Cd(II) and 1459 mg g−1 for Pb(II), outperforming conventional adsorbents. Kinetic studies indicated that Pb(II) adsorption occurred at a significantly faster rate than Cd(II), with the adsorption processes aligning well with the pseudo-second-order model, suggesting chemisorption as the primary mechanism. Langmuir isotherm analysis confirmed monolayer adsorption behavior for both metal ions, corroborating the formation of stable PbCO3 and CdCO3 precipitates on the adsorbent surface. The composite demonstrated high adsorption efficiency across a broad pH range, despite significant influences from increased environmental pH and competitive adsorption by co-existing metal ions, underscoring its robustness in complex environmental conditions. In practical applications, ATP–OS demonstrated significant pH-neutralizing abilities, increasing soil pH values by 2.30–3.07 units and effectively reducing the concentrations and bioavailability of Cd(II) and Pb(II) in acidic agricultural soils. This study highlights the ATP–OS composite’s potential as an effective solution for mitigating heavy metal pollution in agricultural settings, contributing to sustainable soil management practices.
Similar content being viewed by others
Introduction
Cadmium (Cd) and lead (Pb) are well-documented persistent environmental pollutants that pose severe risks to human health. Pb(II) predominantly impairs reproductive and nervous systems, while Cd(II) inflicts significant damage to the kidneys and bones1. The entry of these heavy metals into ecosystems primarily occurs via industrial wastewater discharge, which leads to their widespread occurrence in aquatic and terrestrial environments—particularly in areas with intensive industrial operations, including smelting and mining activities2,3. For example, studies in agricultural lands downstream from copper mines in Dexing City, Jiangxi Province, have revealed that concentrations of Cd and Pb in both crops and soils exceed national safety standards in China, thereby raising serious concerns regarding food and water safety4. Consequently, it is imperative and urgent to develop effective remediation technologies targeting the simultaneous contamination by Cd(II) and Pb(II) in acidified agricultural soils.
Current remediation techniques for heavy metal pollution include membrane filtration, adsorption, chemical precipitation, ion exchange, and biodegradation. Among these, adsorption has garnered particular attention due to its simplicity, cost-effectiveness, and high efficiency5. Waste natural oyster shells (OS), which contain approximately 96% calcium carbonate, exhibit high porosity, non-toxicity, and biodegradability, making them widely studied and applied for the adsorption of heavy metals5. Numerous studies have demonstrated the exceptional performance of OS-derived materials in removing Cd(II) from aqueous solutions6. High-temperature calcination significantly enhances the microporous structure and specific surface area of OS, thereby substantially improving its capacity to adsorb Cd(II)7. For instance, Lian et al.8 reported that OS powder calcined at 900 °C exhibited a Cd(II) adsorption capacity 10.6 times greater than that of natural OS8. However, the same study noted that calcined OS powder exhibited significantly lower Pb(II) adsorption capacity compared to its natural form. This reduction in Pb(II) adsorption may be attributed to the decomposition of organic functional groups (e.g., −OH and −COOH) during calcination, which are critical for Pb(II) complexation9, as well as the dominance of CaO formation, which preferentially interacts with Cd(II) through ion exchange and precipitation10. This limitation underscores the challenge of using OS-based materials for Pb(II) remediation in co-contaminated systems.
Parallel research indicates that natural clay minerals such as attapulgite (ATP) exhibit higher adsorption capacities for Pb(II) than Cd(II) due to their abundant Si–O–Si and hydroxyl (− OH) groups, which facilitate strong chemisorption with Pb(II)11,12. However, the inherent aggregation of ATP particles caused by electrostatic and hydrogen bonding interactions severely restricts their dispersibility and accessibility to active sites13,14. To address these limitations, we propose a synergistic integration of ATP and OS through a one-step calcination process. The alkaline components (e.g., CaO) derived from calcined OS can physically disrupt ATP aggregation while functionalizing its surface, thereby enhancing both dispersibility and Pb(II) adsorption efficiency. Simultaneously, ATP provides additional binding sites for Pb(II), compensating for the diminished Pb(II) adsorption capacity of calcined OS15. This dual-functional design not only overcomes the individual shortcomings of ATP and OS but also leverages their complementary strengths to achieve simultaneous Cd(II) and Pb(II) immobilization—a critical gap in existing remediation technologies for co-contaminated acidic soils.
This study first employed Fourier-transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS) to comprehensively characterize the structure, morphology, composition, and properties of the synthesized ATP–OS powder. Subsequently, batch experiments were conducted to evaluate the adsorption behavior of ATP–OS powder for Cd(II) and Pb(II) in aqueous solutions, with an in-depth exploration of the synergistic adsorption mechanisms. Finally, soil samples from acidified agricultural fields in Dexing City, Jiangxi Province, contaminated with Cd(II) and Pb(II), were collected to assess the practical remediation efficacy of ATP–OS powder as a dual-functional adsorbent, thereby verifying its feasibility and efficiency under complex environmental conditions.
Materials and methods
Materials
Analytical-grade lead nitrate (Pb(NO3)2) and cadmium nitrate (Cd(NO3)2) were used to prepare stock solutions. Oyster shells (OS) were sourced from Fujian Mata Ecological Technology Co., Ltd., and attapulgite (ATP) was obtained from Gansu Zhongke Qitai Biological Engineering Co., Ltd. All reagents and materials employed in this study were of analytical purity. Deionized water was utilized for preparing all solutions and for rinsing containers. Soil samples were collected from a cadmium and lead-contaminated paddy field located in Xinying Second Village, Xinying Street, Dexing City, Jiangxi Province, China (28.92670 °N, 117.61460 °E).
Synthesis of ATP–OS composite
Prior to calcination, raw OS were ground using an 800Y grinder (Yongkang Boo’s Hardware Products Co., Ltd., China). OS powder (200 mesh) was mixed with ATP powder (200 mesh) at a mass ratio of 9:1. This ratio was selected based on preliminary experiments (see Supplementary Material, Table S1). The resulting mixture was calcined in a muffle furnace at temperatures ranging from 400 to 800 °C for 2 h. The calcined products were designated as ATP–OS 400, ATP–OS 500, ATP–OS 600, ATP–OS 700, and ATP–OS 800, corresponding to their respective calcination temperatures.
Adsorption experiments in aqueous systems
Batch adsorption experiments were conducted to evaluate the Cd(II) and Pb(II) removal performance of ATP–OS composite in aqueous solutions. All experiments were performed at 25 °C using 20 mL of metal solution containing 5 ± 0.05 mg of ATP–OS adsorbent in triplicate. The mixtures were incubated on a rotary shaker at 250 rpm for 24 h. The removal efficiencies of Cd(II) and Pb(II) were evaluated at pH 6 over the 24 h incubation period with initial metal concentrations of 50 mg/L to assess the adsorptive capacities of the different materials. Stock solutions of Pb(II) and Cd(II) were prepared by dissolving lead nitrate (Pb(NO3)2) and cadmium nitrate (Cd(NO3)2) in deionized water to concentrations of 1000 mg/L each. These stock solutions were subsequently diluted to achieve the desired concentrations for batch studies. ATP–OS exhibited high removal efficiencies for both Cd(II) and Pb(II) and was selected for further investigations. Adsorption efficiency (%) and adsorption capacity (Qe) were calculated using the following equations:
where C0 and Ce represent the initial and equilibrium concentrations (mg/L), V is the volume of the solution (mL), and m is the mass of the adsorbent (g).
The influence of pH on the removal efficiencies of Cd(II) and Pb(II) was examined using a 50 mg/L metal solution across a pH range of 1 to 9. An optimal pH of 4 was identified and applied in subsequent batch experiments focusing on single-contaminant systems. This pH value was selected based on two key considerations: (1) The equilibrium pH of Pb(II) solutions stabilized at 3.95 ± 0.12, creating favorable conditions for PbCO3 precipitation and surface complexation while avoiding metal hydroxide precipitation; (2) For Cd(II) systems, pH 4 maintained solution stability with minimal pH fluctuation, ensuring valid kinetic measurements.
Adsorption kinetics were investigated (Supplementary Material) using 50 mg/L solutions of Cd(II) and Pb(II) maintained at pH 4.00 ± 0.04. Samples were taken at designated time intervals and filtered through GE cellulose nylon membrane filters with a pore size of 0.22 µm.
Adsorption isotherms for Cd(II) on ATP–OS were determined (Supplementary Material) by equilibrating Cd(II) solutions at pH 4.00 ± 0.04 across a concentration range of 5 to 300 mg/L for 24 h. Similarly, Pb(II) isotherms were established using Pb(II) concentrations ranging from 20 to 500 mg/L under identical conditions. All solutions were filtered through 0.22 µM membrane filters prior to analysis.
The co-adsorptive behavior of Cd(II) and Pb(II) was evaluated by examining their interactions at varying concentrations and pH levels. The experimental setups included: (1) simultaneous addition of 25 mg/L Cd(II) and 25 mg/L Pb(II) across pH values from 1 to 8 to determine the optimal pH of 4 for subsequent experiments; (2) simultaneous addition of Cd(II) at concentrations of 10, 20, 40, 50, 80, and 100 mg/L with Pb(II) at 10, 30, or 60 mg/L at pH 4.00 ± 0.04; and (3) simultaneous addition of Pb(II) at concentrations of 20, 50, 100, 150, 200, and 250 mg L−1 with Cd(II) at 5, 20, or 50 mg/L at pH 4.00 ± 0.04. These setups were designed to identify optimal conditions for the simultaneous removal of both heavy metals using the selected adsorbent.
Remediation experiments in contaminated soils
Soil experiments aimed to validate the practical application of ATP–OS for Cd(II)/Pb(II) immobilization in acidified agricultural soils. Soil samples (pH = 4.8 ± 0.3) were collected from a farmland near a mining area in Dexing City, Jiangxi Province. The soils were air-dried, homogenized, and sieved through a 2-mm mesh. 600 g of soil was placed into 1500 mL rectangular plastic containers with lids. Deionized water was added to achieve a moisture content equal to 60% of the soil’s water-holding capacity. ATP–OS was incorporated into the soil at weight percentages of 2%, 4%, and 6%, designated as 2% ATP–OS, 4% ATP–OS, and 6% ATP–OS, respectively, alongside a control treatment (CK) without the adsorbent. The containers were incubated at 25 °C in a thermostatic incubator. Each treatment, including the control, was replicated three times. Every two days, glass vials were removed for weighing and moisture adjustment to maintain the soil’s water-holding capacity at 60%. The incubation period lasted 60 d.
Measurements
The total concentrations of Cd(II) and Pb(II) were quantified using inductively coupled plasma mass spectrometry (ICP-MS; ICAP RQ, Germany). Morphological analyses of ATP, OS, and ATP–OS were conducted with field emission scanning electron microscopy (FE-SEM; Hitachi Regulus 8100, USA). Structural characterization was performed using FTIR (Thermo Fisher Nicolet iS5, USA). The crystalline structures of ATP, OS, and ATP–OS particles were determined via XRD (Bruker-D8 ADVANCE, Germany). Additionally, XPS analysis was carried out using an ESCALAB 250Xi system (Thermo Fisher, USA).
Soil samples (50 g) were collected from the culture boxes on days 1, 5, 10, 15, 30, and 60 and subsequently air-dried for heavy metal concentration and physicochemical property analyses. The active fractions of Cd and Pb were extracted using the DTPA method and quantified by ICP-MS (ICAP RQ, Germany). Soil pH was determined by shaking a soil-KCl suspension (2.5:1 w/v ratio) for 2 h, followed by measurement with a pH meter. Exchangeable calcium and magnesium were extracted with a 1 mol/L ammonium acetate solution and quantified using atomic absorption spectroscopy (iCETM3000; Thermal Sciences, Inc.).
Results and discussion
Synthesis optimization and structural characterization of ATP–OS
The thermal treatment temperature played a critical role in optimizing the adsorption performance of the ATP–OS composite. Comparative analysis of Cd(II) and Pb(II) removal efficiencies across calcination temperatures (400–800 °C) revealed that ATP–OS synthesized at 700 °C achieved the highest Cd(II) removal efficiency while maintaining robust Pb(II) adsorption capacity (Fig. 1). This temperature was selected as optimal, balancing cost-effectiveness with enhanced adsorption capabilities for both metals. Calcination converted OS into alkaline CaO (via CaCO3 decomposition: CaCO3 → CaO + CO2↑), which reduced ATP’s interparticle electrostatic/hydrogen bonding interactions (Fig. 2). This disruption of ATP’s aggregated needle-like structure (raw ATP) into dispersed irregular morphologies adhered to OS fragments (Fig. 2) increased the composite’s specific surface area and active site accessibility16,17.

Temperature-dependent removal efficiency of Cd(II) and Pb(II) by calcined attapulgite, oyster shell, and composite materials. (a) Attapulgite (ATP). (b) Oyster shell (OS). (c) ATP–OS composite. Experiments used 50 mg/L initial metal concentration at pH 6. Non-calcined controls (CK) are shown for comparison.

Morphological evolution of attapulgite, oyster shell, and composite materials before and after metal adsorption. (a) Attapulgite (ATP). (b) Oyster shell (OS). (c) ATP–OS composite. (d–f) ATP–OS after exposure to Cd(II) (d), Pb(II) (e), and both metals (f).
Structural characterization via SEM–EDX demonstrated that the layered OS structure fragmented into regular rectangular blocks after thermal treatment, while ATP’s needle-like crystals transformed into irregular morphologies adhered to the OS surface (Fig. 2). This morphological evolution enhanced the composite’s porosity and accessibility to adsorption sites. FTIR spectra (Fig. 3a) confirmed the retention of ATP’s Al–Mg–OH (949 cm−1) and silicate (515 cm−1) functional groups, critical for metal coordination, alongside OS-derived carbonate (CO32−, 1,420/878/710 cm−1). XRD (Fig. 3b) revealed CaCO3 as the dominant phase in OS, while ATP–OS exhibited a weak Ca₅(SiO4)2(CO3) peak, indicating Ca2+-silicate interactions during calcination. This phase provides additional binding sites for Cd(II)/Pb(II) via ion exchange (e.g., Ca2+ ↔ Pb2+)18.

Structural and chemical characterization of attapulgite (ATP), oyster shell (OS), and their composite (ATP–OS). (a) FT-IR spectra. (b) XRD patterns. (c) XPS spectra.
XPS analysis (Fig. 3c) corroborated the dominance of CaCO3 in the composite, with characteristic peaks for O 1s (529.6 eV), C 1s (284.8 eV), and Ca 2p (347.9 eV). The high-resolution C 1s spectrum revealed contributions from C–C, C–O, and CO32− groups, while O 1s deconvolution highlighted M–O, hydroxyl (–OH, 531.5 eV), and carbonate (CO32−, 533.1 eV) groups (Fig. 4d,e), which directly participate in chemisorption. These structural modifications, driven by calcination, collectively enhanced the composite’s surface reactivity and adsorption capacity.

Structural and compositional characterization of attapulgite (ATP), oyster shell (OS), and their composites (ATP–OS) pre- and post-heavy metal adsorption. (a) FT-IR spectra, (b) XRD patterns, and (c) XPS survey spectra before and after Cd(II)/Pb(II) adsorption. High-resolution (d) C 1s and (e) O 1s XPS spectra.
Compared to previous studies using ATP or OS individually16,18, the ATP–OS composite demonstrated 2.4- and 1.4-fold higher maximum adsorption capacities (Qmax) for Cd(II) and Pb(II), respectively. This enhancement originates from the unique Ca₅(SiO4)2CO3 phase formed during calcination (Fig. 3b), which provides both ion-exchange sites (Ca2+) and coordination groups (SiO44−, CO32−). Such synergistic effects overcome the limitation of pristine ATP’s low Pb-selectivity and OS’s insufficient Cd-adsorption capacity5,6, establishing a new dual-functional soil remediation agents.
Adsorption kinetics and isotherm behavior
The adsorption kinetics of Cd(II) and Pb(II) onto ATP, OS, and the ATP–OS composite were systematically investigated (Fig. 5a,b). All adsorbents exhibited rapid initial adsorption rates for both metals, followed by gradual equilibration. Notably, ATP–OS demonstrated superior kinetic performance: Pb(II) adsorption reached equilibrium within 20 min, significantly faster than ATP (50 min) and OS (120 min) (Fig. 5b). In contrast, Cd(II) adsorption equilibration times were 480 min for ATP and ATP–OS, while OS achieved equilibrium within 60 min (Fig. 5a). The pseudo-second-order model provided the best fit for all materials (R2 > 0.99, Table S2), indicating chemisorption as the dominant mechanism19,20. The higher pseudo-second-order rate constant (K) for ATP–OS compared to individual components highlighted enhanced adsorption kinetics, likely attributed to Pb(II)'s lower hydration free energy and higher reactivity relative to Cd(II)21.

Adsorption kinetics and isotherms of Cd(II) and Pb(II) on ATP, OS, and ATP–OS composites. (a, c) Cd(II) adsorption kinetics (a) and isotherms (c); (b, d) Pb(II) adsorption kinetics (b) and isotherms (d). Experiments were conducted in single-metal systems using attapulgite (ATP), oyster shell (OS), or ATP–OS composites. Qt, adsorption capacity at time t; Qe, equilibrium adsorption capacity; Ce, equilibrium concentration.
Langmuir isotherm analysis revealed monolayer adsorption behavior for both metals, with Qmax of 197.0 mg/g for Cd(II) and 1523 mg/g for Pb(II) on ATP–OS (Fig. 5c,d; Table S3). These values surpassed those of pristine ATP (138.3 mg/g for Cd(II); 154.8 mg/g for Pb(II)) and OS (140.5 mg/g for Cd(II); 1,459 mg/g for Pb(II)), underscoring the composite’s synergistic advantages. The “L”-shaped isotherms further corroborated the Langmuir model, consistent with the formation of stable PbCO3 and CdCO3 precipitates on the adsorbent surface22,23. Remarkably, ATP–OS outperformed conventional adsorbents such as activated carbon and bentonite in heavy metal removal5,6, validating its potential for practical applications.
Equilibrium adsorption capacities increased with initial metal concentrations until saturation, reflecting the limited availability of active sites at higher concentrations. The Freundlich model better described adsorption in co-contaminated systems (Table S4), suggesting multilayer heterogeneous adsorption under competitive conditions. This dual adsorption behavior emphasized ATP–OS’s adaptability to both single- and multi-metal contaminated environments.
pH-dependent adsorption mechanisms
The adsorption performance of ATP–OS for Cd(II) and Pb(II) was critically influenced by solution pH (Figs. 6a,c). For Cd(II), removal efficiencies across all adsorbents showed minimal variation at pH < 2.0 but increased sharply for ATP–OS at pH > 2.0, achieving near-complete removal (≈100%) at pH 4.0 and maintaining stability at higher pH values. Similarly, Pb(II) adsorption capacities of ATP–OS and OS rose rapidly at pH > 2.0, reaching > 95% removal at pH 3.0, while ATP alone exhibited gradual efficiency improvements until pH > 6.0. These trends were attributed to reduced competition from H+ ions and enhanced negative surface charge density at higher pH, promoting electrostatic attraction and metal precipitation24,25.

pH-dependent metal removal efficiency and solution equilibrium pH by attapulgite (ATP), oyster shell (OS), and their composites (ATP–OS) in single-metal systems. a,b, Cd(II) systems showing removal efficiency (a) and final pH (b). (c, d) Pb(II) systems showing removal efficiency (c) and final pH (d). Experimental parameters: 50 mg L−1 initial metal concentration, 24 h contact time.
The equilibrium pH of treated solutions further highlighted ATP–OS’s pH-modulating capability. For Cd(II) solutions with initial pH > 2.0, ATP–OS induced a rapid pH increase (Fig. 6b), surpassing adjustments by ATP or OS alone. In Pb(II) systems, ATP–OS and OS treatments significantly elevated equilibrium pH at initial pH > 2.0, with ATP–OS stabilizing at higher pH levels (≈4.0) compared to other treatments. This alkalization effect stemmed from the composite’s CaCO3-derived alkaline components, which neutralized acidic conditions and facilitated metal carbonate precipitation7,26.
The pH-dependent adsorption behavior of ATP–OS was attributed to the synergistic interplay between alkaline component—driven chemical neutralization and surface hydroxyl group deprotonation (Fig. 6b,d). Specifically, CaO-derived hydroxyl ions (OH−) generated via hydrolysis in acidic media (CaO + 2H+ → Ca2+ + H2O; CaO + H2O → Ca(OH)2 → Ca2+ + 2OH−) neutralized protons, creating an alkaline microenvironment that promoted the precipitation of CdCO3 and PbCO3 (Fig. 4b). Concurrently, surface charge enhancement was evidenced by FTIR spectra showing diminished CaCO3 vibrational bands (1420/878/cm), indicative of carbonate dissolution, and weakened O–H stretching vibrations (3642/cm), suggesting hydroxyl group deprotonation. XPS analysis further corroborated this mechanism, with the proportion of surface –OH groups increasing from 15.05% to 25.48% after Pb(II) adsorption (Fig. 4e), thereby amplifying negative charge density and strengthening electrostatic interactions with cationic Cd2+/Pb2+. These findings collectively demonstrate that alkaline components stabilize acidic environments through pH buffering, while surface charge modification enhances heavy metal immobilization. Future work incorporating zeta potential analysis is warranted to validate the proposed surface charge dynamics.
Competitive adsorption behavior
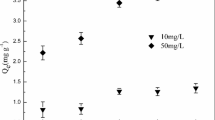
The competitive adsorption behavior of ATP–OS in co-contaminated systems was systematically evaluated (Fig. 7). Compared to single-contaminant systems, ATP–OS exhibited reduced removal efficiencies for both Cd(II) and Pb(II) under co-contamination conditions (Fig. 7a). Notably, the composite displayed a higher affinity for Cd(II) than Pb(II) in such systems, with Cd(II) removal efficiencies significantly surpassing those of Pb(II). However, ATP–OS retains substantial adsorption capacities for both metals (Fig. 7b,c), with Pb(II) capacities remaining stable (~ 1,450 mg/g) even at elevated Cd(II) concentrations. This dual functionality arises from the composite’s heterogeneous adsorption mechanisms: Pb(II) primarily forms stable PbCO3 precipitates, while Cd(II) binds to silicate or ATP–derived sites. Furthermore, practical soil remediation trials (Fig. 8) confirm ATP–OS’s ability to simultaneously reduce bioavailable Cd and Pb, underscoring its applicability in complex environments. As Pb(II) concentrations increased, the adsorption capacity for Cd(II) exhibits a decreasing trend, although the rate of decrease varies across different Pb(II) concentration ranges (Fig. 7b), whereas variations in Cd(II) concentrations had minimal impact on Pb(II) removal (Fig. 7c). This asymmetric competition highlights ATP–OS’s preferential adsorption for Pb(II), likely due to its higher ionic charge density and stronger interactions with functional groups on the adsorbent surface27.

pH-dependent removal performance and competitive adsorption characteristics of attapulgite-oyster shell composites. (a) Pb(II) and Cd(II) removal efficiencies as a function of initial pH (initial concentration: 50 mg/L for each metal). (b) Competitive adsorption behavior: Cd(II) uptake capacity in the presence of varying Pb(II) concentrations at pH 6 (equilibrium time: 24 h). (c) Competitive adsorption behavior: Pb(II) uptake capacity in the presence of varying Cd(II) concentrations at pH 6 (equilibrium time: 24 h).

Soil remediation dynamics under variable attapulgite-oyster shell composites amendment levels. (a) pH evolution, (b) DTPA-extractable Cd, and (c) DTPA-extractable Pb in contaminated farmland soils over consecutive treatment cycles (CK: untreated control).
Kinetic analysis revealed that the pseudo-second-order rate constants (K) for ATP–OS in co-contaminated systems were lower than those in single-metal systems (Table S5), indicating slower adsorption kinetics under competitive conditions. Adsorption isotherm modeling further demonstrated that the Freundlich model provided a better fit (higher R2 values) compared to the Langmuir model (Table S4), suggesting multilayer heterogeneous adsorption on the composite surface. This contrasts with the monolayer adsorption observed in single-metal systems, emphasizing the complexity of adsorption mechanisms in multi-component environments.
Qmax derived from isotherm analysis confirmed that the presence of Pb(II) significantly reduced Cd(II) adsorption capacity, while Cd(II) exerted negligible effects on Pb(II) removal. This competitive hierarchy aligns with the stronger binding affinity of Pb(II) to active sites such as carbonate (CO32−) and hydroxyl (–OH) groups, as evidenced by XPS and FT-IR analyses. Despite competitive interference, ATP–OS maintained robust removal efficiencies across a broad pH range, demonstrating its adaptability to complex environmental scenarios. These findings underscore the composite’s potential for practical applications in soils co-contaminated with multiple heavy metals.
Molecular-level removal mechanisms
The molecular-level mechanisms governing Cd(II) and Pb(II) removal by ATP–OS were elucidated through comprehensive spectroscopic analyses. FT-IR spectra (Fig. 4a) revealed that after Cd(II) adsorption, the O–H stretching vibration at ~ 3642/cm weakened and shifted, while Pb(II) adsorption caused the complete disappearance of the O–H band. These changes indicate the involvement of hydroxyl (–OH) groups in metal binding, likely through coordination or hydrogen bonding9,28. Concurrently, the carbonate (CO32−) peaks at 1420, 878, and 710/cm diminished in intensity after adsorption, suggesting CO32− participation in metal precipitation7,19.
XRD patterns (Fig. 4b) further confirmed the formation of CdCO3 and PbCO3 precipitates, as evidenced by the emergence of distinct diffraction peaks at 2θ = 24.9°, 32.1°, and 43.8° for CdCO3, and 25.9°, 31.5°, and 45.3° for PbCO3. The reduction in calcite (CaCO3) peak intensity post-adsorption corroborated the dissolution of CaCO3 and subsequent release of Ca2+, which facilitated ion exchange with Pb(II) and Cd(II). Notably, Pb(II) exhibited stronger affinity for Ca2+ exchange sites compared to Cd(II), as inferred from the preferential formation of PbCO3 over CdCO318.
XPS analysis provided additional insights into the surface interactions. Deconvolution of the C 1s spectra (Fig. 4d) demonstrated a significant decrease in CO32− peak area (from 39.99% to 31.61% for Cd and 34.90% for Pb), accompanied by an increase in C–C/C=C and C–O contributions, highlighting the role of carbonate and organic functional groups in metal sequestration. In the O 1s spectra (Fig. 4e), the CO32− peak area decreased from 79.68 to 68.71% for Pb(II), while the –OH contribution increased from 15.05 to 25.48%, underscoring the dominance of hydroxyl-mediated chemisorption for Pb(II) (Wu et al. 2014). For Cd(II), the reduction in both CO32− and –OH groups suggested a combination of ion exchange and surface complexation.
High-resolution Cd 3d and Pb 4f spectra (Fig. S1) confirmed the presence of CdCO3 (Cd 3d52 at 405 eV) and Cd(OH)2 (412 eV), as well as Pb–O/Pb–OH complexes (Pb 4f72 at 138.3 eV). The smaller binding energy shifts for CO32− in Pb-laden systems compared to Cd-laden systems indicated stronger electronic interactions between Pb(II) and CaCO3, consistent with its higher ionic potential27. These findings collectively demonstrate that ATP–OS removes Cd(II) and Pb(II) through synergistic mechanisms, including carbonate precipitation, ion exchange, and hydroxyl-mediated chemisorption, with Pb(II) exhibiting preferential binding due to its higher reactivity and charge density.
Soil remediation performance
The practical efficacy of the ATP–OS composite was validated through soil incubation experiments under realistic environmental conditions. As illustrated in Fig. 8, the application of ATP–OS significantly increased the pH of acidified soils by 2.30–3.07 units across varying dosage levels (2%, 4%, and 6% w/w). This pH elevation played a critical role in reducing the solubility and bioavailability of Cd(II) and Pb(II) by promoting their precipitation as stable carbonate phases9,28. During the initial 15 days of treatment, the concentrations of exchangeable Cd and Pb exhibited notable fluctuations, likely due to dynamic interactions between the adsorbent and soil matrix. However, these concentrations stabilized thereafter, demonstrating the composite’s capacity for sustained metal immobilization. Over the 60-day incubation period, ATP–OS treatments reduced exchangeable Cd and Pb levels by an average of 20% and 49%, respectively, compared to the CK, highlighting its robust remediation potential.
The long-term stabilization of exchangeable heavy metals underscores ATP–OS’s ability to alter soil chemistry effectively. The alkaline nature of calcined OS-derived calcium oxide contributed to neutralizing soil acidity, thereby enhancing the formation of CdCO3 and PbCO3 precipitates18. This mechanism aligns with observations from aqueous adsorption studies, where carbonate groups in ATP–OS facilitated chemisorption via ion exchange and surface complexation. Furthermore, the composite’s dual-functional design mitigated particle aggregation, ensuring sustained accessibility of active sites for metal binding even in complex soil environments.
These results corroborate previous studies on calcium-based adsorbents, which emphasize their role in reducing heavy metal mobility through pH modulation and precipitation7. The marked decrease in bioavailable Cd and Pb underscores ATP–OS’s suitability for large-scale soil remediation in agricultural regions impacted by acidic and co-contaminated conditions. Notably, the ATP–OS composite demonstrates competitive advantages in both adsorption capacity and economic feasibility when compared to existing calcium-based and clay mineral adsorbents. As shown in Table S3, ATP–OS exhibits superior maximum adsorption capacities for Pb(II) (1459 mg g−1) and Cd(II) (197 mg g−1), outperforming most reported materials including biogenic CaCO3, geological CaCO329, and magnetic porous polymers30. While direct cost comparisons are limited in literature, the simplified one-step calcination synthesis (700 °C, 2 h) using natural attapulgite and waste oyster shells offers significant economic benefits. This contrasts with energy-intensive methods like hydrothermal synthesis of MCCR-350 (requiring 24 h processing) or complex composites involving activated carbon31. The absence of chemical modifiers and the utilization of abundant agricultural/industrial byproducts position ATP–OS as a cost-effective solution for large-scale soil remediation. Future investigations should focus on evaluating the long-term stability and regeneration potential of ATP–OS under diverse environmental scenarios to optimize its practical deployment.
Conclusion
This study successfully developed a cost-effective dual-functional adsorbent, the ATP–OS composite, tailored for the simultaneous removal of co-contaminated Cd(II) and Pb(II) from acidified agricultural soils. The ATP–OS composite demonstrated superior adsorption capacities, achieving maximum values of 197 mg/g for Cd(II) and 1,523 mg/g for Pb(II). These capacities surpass those of conventional adsorbents such as activated carbon and bentonite, underscoring ATP–OS’s competitive advantage in heavy metal remediation. The enhanced adsorptive performance is attributed to the synergistic interactions between ATP and OS, optimized through thermal treatment at 700 °C, which facilitated structural transformations and the formation of additional active sites. Kinetic and isotherm analyses revealed efficient chemisorption mechanisms and robust adsorption capacities even in the presence of competing metal ions. The practical application of ATP–OS in soil treatment demonstrated significant reductions in exchangeable Cd and Pb levels, highlighting its potential for sustainable agricultural practices. Future research should explore the regeneration and long-term stability of ATP–OS in various environmental conditions to further validate its applicability in large-scale soil remediation projects.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Godiya, C. B., Cheng, X., Li, D., Chen, Z. & Lu, X. Carboxymethyl cellulose/polyacrylamide composite hydrogel for cascaded treatment/reuse of heavy metal ions in wastewater. J. Hazard. Mater. 364, 28–38 (2019).
Zhao, X., Li, M., Zhai, F., Hou, Y. & Hu, R. Phosphate modified hydrochars produced via phytic acid-assisted hydrothermal carbonization for efficient removal of U(VI), Pb(II) and Cd(II). J. Environ. Manag. 298, 113487 (2021).
Du, B. et al. Environmental and human health risks from cadmium exposure near an active lead-zinc mine and a copper smelter, China. Sci. Total Environ. 720, 137585 (2020).
Zhou, Z. et al. Cadmium contamination in soils and crops in four mining areas, China. J. Geochem. Explor. 192, 72 (2018).
Tamjidi, S. & Abolhasan, A. A review of the application of sea material shells as low cost and effective bio-adsorbent for removal of heavy metals from wastewater. Environ. Sci. Pollut. Res. 27, 31105–31119 (2020).
Kim, W. & Singh, R. Modified oyster waste shells as a value-added sorbent for lead removal from water. Bull. Environ. Contam. Toxicol. 108, 518–523 (2022).
Bi, D. et al. Conversion of oyster shell waste to amendment for immobilising cadmium and arsenic in agricultural soil. Bull. Environ. Contam. Toxicol. 105, 277–282 (2020).
Lian, W. et al. Influence of pyrolysis temperature on the cadmium and lead removal behavior of biochar derived from oyster shell waste. Bioresour. Technol. Rep. 15, 100709 (2021).
Wu, Q., Chen, J., Clark, M. & Yu, Y. Adsorption of copper to different biogenic oyster shell structures. Appl. Surf. Sci. 311, 264–271 (2014).
Habte, L. et al. Removal of Cd(II) and Pb(II) from wastewater via carbonation of aqueous Ca(OH)2 derived from eggshell. Process Saf. Environ. Prot. 141, 278–286 (2020).
Jiang, M. Q., Jin, X. Y., Lu, X. Q. & Chen, Z. L. Adsorption of Pb(II), Cd(II), Ni(II) and Cu(II) onto natural kaolinite clay. Desalination 252, 33–39 (2010).
Kushwaha, A., Rani, R. & Patra, J. Adsorption kinetics and molecular interactions of lead [Pb(II)] with natural clay and humic acid. Int. J. Environ. Sci. Technol. 17, 1325–1334 (2020).
Huang, D., Wang, W., Xu, J. & Wang, A. Mechanical and water resistance properties of chitosan/poly(vinyl alcohol) films reinforced with attapulgite dispersed by high-pressure homogenization. Chem. Eng. J. 210, 166–173 (2012).
Feng, Y. et al. Simple fabrication of easy handling millimeter-sized porous attapulgite/polymer beads for heavy metal removal. J. Colloid Interface Sci. 502, 52–61 (2017).
Frost, R., Kristóf, J. & Horváth, E. Controlled rate thermal analysis of sepiolite. J. Therm. Anal. Calorim. 98, 423–428 (2009).
Dai, L. et al. Calcium-rich biochar from the pyrolysis of crab shell for phosphorus removal. J. Environ. Manag. 198, 70–77 (2017).
Huang, R. et al. Removal of Cd(II) and Pb(II) from aqueous solution by modified attapulgite clay. Arab. J. Chem. 13, 4994–5003 (2020).
Santamaria-Perez, D. et al. Post-tilleyite, a dense calcium silicate-carbonate phase. Sci. Rep. 9, 7898 (2019).
Sun, T. et al. Crayfish shell biochar for the mitigation of Pb contaminated water and soil: Characteristics, mechanisms, and applications. Environ. Pollut. 271, 116308 (2021).
Chen, J., Feng, J. & Yan, W. Influence of metal oxides on the adsorption characteristics of PPy/metal oxides for methylene blue. J. Colloid Interface Sci. 475, 26–34 (2016).
Cheng, T., Lee, M., Ko, M., Ueng, T. & Yang, S. The heavy metal adsorption characteristics on metakaolin-based geopolymer. Appl. Clay Sci. 56, 90 (2012).
Foo, K. Y. & Hameed, B. H. Insights into the modeling of adsorption isotherm systems. Chem. Eng. J. 156, 2–10 (2010).
Wu, J. et al. A novel modified method for the efficient removal of Pb and Cd from wastewater by biochar: Enhanced the ion exchange and precipitation capacity. Sci. Total Environ. 754, 142150 (2021).
Xie, Y. et al. Adsorption behavior and mechanism of Mg/Fe layered double hydroxide with Fe3O4-carbon spheres on the removal of Pb(II) and Cu(II). J. Colloid Interface Sci. 536, 440–449 (2019).
Paranavithana, G. et al. Adsorption of Cd2+ and Pb2+ onto coconut shell biochar and biochar-mixed soil. Environ. Earth Sci. 75, 1–10 (2016).
Chu, D. H. et al. CO2 mineralization into different polymorphs of CaCO3 using an aqueous-CO2 system. RSC Adv. 3, 21722–21729 (2013).
Gan, D. et al. Bioinspired functionalization of MXenes (Ti3C2TX) with amino acids for efficient removal of heavy metal ions. Appl. Surf. Sci. 504, 144603 (2020).
Chen, Y. Y. et al. Performance and mechanism of simultaneous removal of Cd(II) and Congo red from aqueous solution by hierarchical vaterite spherulites. Appl. Surf. Sci. 444, 224–233 (2018).
Liu, X. et al. Biogenic and geological calcium carbonate as adsorbents for the removal of lead(II) from aqueous solutions. J. Environ. Sci. 83, 1–9 (2019).
Zhu, H. et al. Magnetic porous polymers prepared via high internal phase emulsions for efficient removal of Pb2+ and Cd2+. ACS Sustain. Chem. Eng. 6, 5206–5214 (2018).
Wang, Y. et al. Magnetic chitosan - clay composite for efficient removal of Pb(II) and Cd(II) from aqueous solutions. J. Clean. Prod. 266, 121917 (2020).
Acknowledgements
This work was supported by the National Key Research and Development Program of China, Project No. 2023YFD190110503, the National Natural Science Foundation of China, Project No.42167004 and 32160752.
Author information
Authors and Affiliations
Contributions
Hongyan Zhu: Data curation, Investigation, Methodology, Validation, Visualization, Writing-original draft. Yangping Wen: Conceptualization, Funding acquisition, Supervision, Visualization, Writing-review and editing. Qingyin Shang: Formal analysis, Funding acquisition, Visualization, Writing-review and editing. Xinyu Chen: Formal analysis, Methodology, Visualization. Yuzhen Huang: Formal analysis, Methodology, Visualization. Peiyue Liu: Formal analysis, Methodology, Visualization. Chengcheng Wang: Funding acquisition, Validation, Visualization. Xiuxia Yang: Conceptualization, Funding acquisition, Project administration, Supervision, Writing-review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, H., Wen, Y., Shang, Q. et al. Attapulgite/oyster shell composite reduces cadmium and lead bioavailability in acidic agricultural soils through synergistic adsorption and pH regulation. Sci Rep 15, 15609 (2025). https://doi.org/10.1038/s41598-025-00302-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-00302-0
