
Abstract
This study explores the differential expression of inflammation and iron metabolism-related genes (IIMRDEGs) in Ischemic Stroke (IS), a major contributor to global morbidity and mortality. Using the Gene Expression Omnibus (GEO) query tool, we integrated gene expression datasets GSE22255 and GSE16561. We identified 56 differentially expressed genes (DEGs), including 42 that were upregulated and 14 downregulated, according to criteria of |logFC| > 0.5 and p < 0.05. An intersection with known IIMRDEGs revealed 16 genes with significant relevance to IS, such as SLC22A4 and DUSP1. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses indicated that these genes are mainly involved in leukocyte chemotaxis and responses to bacterial molecules, in addition to IL-17 and TNF signaling pathways. A protein-protein interaction (PPI) network of 12 IIMRDEGs identified 8 hub genes, including IL7R and ADM, which exhibited significant expression differences (p < 0.001) and potential diagnostic utility with AUC values between 0.7 and 0.9 in ROC curve analysis. Furthermore, immune infiltration analysis showed notable differences in 7 immune cell types between IS and control samples. Our findings advance the understanding of ischemic stroke mechanisms and present potential biomarkers for improving diagnosis and therapeutic strategies.
Similar content being viewed by others

Introduction
IS is a major cause of mortality and long-term disability globally, significantly burdening healthcare systems and societies. It accounts for approximately 87% of all stroke cases worldwide, affecting millions of individuals each year. The pathophysiology of IS involves the obstruction of blood flow to the brain, primarily due to thrombosis or embolism, resulting in neuronal injury and death1. Various risk factors are associated with IS, including hypertension, diabetes, atrial fibrillation, and hyperlipidemia2. Despite advancements in acute management and secondary prevention, the recurrence rate of IS remains high, highlighting the urgent need for further research into its underlying mechanisms and potential therapeutic targets3. Current treatment options for ischemic stroke include intravenous thrombolysis and mechanical thrombectomy. However, thrombolysis is limited by a narrow therapeutic window and carries a risk of bleeding, while thrombectomy necessitates specialized facilities. Moreover, existing preventive measures do not completely eliminate the risk of recurrence, underscoring the need for new therapeutic approaches.
Inflammation and iron metabolism have emerged as critical factors in the pathogenesis of IS. Inflammatory processes play a pivotal role in the onset and progression of IS, contributing to endothelial dysfunction, plaque instability, and thrombus formation4. Key inflammatory markers such as C-reactive protein (CRP), interleukins, and tumor necrosis factor-alpha (TNF-α) have been implicated in IS, with elevated levels correlating with worse outcomes5. Similarly, iron metabolism is intricately linked to oxidative stress and neuroinflammation in IS. Dysregulation of iron homeostasis lead to the generation of reactive oxygen species (ROS), exacerbating neuronal damage and promoting inflammatory responses6. Studies have shown that iron-related proteins, including ferritin, transferrin, and hepcidin, are altered in IS patients, suggesting their potential role as biomarkers and therapeutic targets7.
IIMRDEGs have garnered attention for their involvement in IS. Genes such as HAMP (hepcidin antimicrobial peptide), FTH1 (ferritin heavy chain), and TF (transferrin) are crucial regulators of iron metabolism and have been found to be differentially expressed in IS8. Additionally, genes encoding inflammatory cytokines and chemokines, such as IL6 (interleukin 6), TNF (tumor necrosis factor), and CCL2 (C-C motif chemokine ligand 2), are upregulated in IS, further highlighting the interplay between inflammation and iron metabolism in this condition9. Understanding the differential expression of these genes in IS could provide insights into the molecular mechanisms driving the disease and identify novel targets for therapeutic intervention10.Despite identifying key IIMRDEGs involved in IS, the precise molecular mechanisms and their interconnected roles in disease progression remain poorly understood. Therefore, this study aims to elucidate these interactions to identify potential therapeutic targets for intervention.
Given the critical role of inflammation and iron metabolism in IS, this study aims to investigate the differential expression of IIMRDEGs in IS and elucidate their potential mechanisms in disease progression. We will utilize public gene expression datasets from the GEO database, specifically GSE22255 and GSE16561, to analyze the expression profiles of IIMRDEGs in IS patients. By employing advanced bioinformatics tools and statistical methods, we will identify key DEGs and perform functional enrichment analyses to uncover the biological pathways involved. Additionally, we will construct PPI networks and regulatory networks to identify hub genes and their interactions with transcription factors and microRNAs (miRNAs). The findings from this study could enhance our understanding of IS pathogenesis and pave the way for the development of targeted therapies aimed at modulating inflammation and iron metabolism in IS.
Results
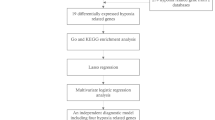
Technology roadmap (Fig. 1)
To provide a clearer description of the entire research process, we have included a technical roadmap in Fig. 1. This study integrated the GSE22255 and GSE16561 datasets and utilized the sva package to remove batch effects. Subsequently, we employed the limma package for differential analysis and identified IIMRDEGs. To further explore the biological functions of these genes, we conducted GO and KEGG enrichment analyses and constructed a PPI network using the STRING database. Additionally, GSEA analysis revealed key signaling pathways, while ROC curve analysis assessed the diagnostic value of the genes. The CIBERSORT algorithm was used to analyze immune cell infiltration. Finally, we constructed a regulatory network to clarify the regulatory roles of key genes. This technical roadmap ensures the systematic and scientific nature of the study, providing a reliable basis for exploring potential biomarkers and mechanisms related to ischemic stroke.

Flow Chart for the Comprehensive Analysis of IIMRDEGs. IS, Ischemic Stroke; GSEA, Gene Set Enrichment Analysis; DEGs, Differentially Expressed Genes; IRGs, Inflammatory-Related Genes; IMRGs, Iron-Metabolism-Related Genes; IIMRGs, Inflammatory&Iron-Metabolism-Related Genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; IIMRDEGs, Inflammatory&Iron-Metabolism-Related Differentially Expressed Genes; PPI, Protein-Protein Interaction Network; ROC, Receiver Operating Characteristic; TF, Transcription Factor.
Merging of cerebral infarction datasets
The R package sva was used to remove batch effects from the IS datasets GSE22255 and GSE16561, resulting in a combined GEO datasets. Subsequently, distribution boxplots (Fig. 2A-B) were created to compare expression values before and after batch effect removal. Additionally, PCA (Principal Component Analysis) plots (Fig. 2C-D) were generated to assess the distribution of low-dimensional features. The results of both the boxplot and PCA demonstrated that the batch effects in the IS datasets were largely eliminated after applying the correction.

Batch Effects Removal of GSE22255 and GSE16561. (A) Box plot of Combined GEO Datasets distribution before batch removal. (B) Post-batch integrated GEO datasets (combined datasets) distribution box plots. (C) PCA plot of the datasets before debatching. (D) Go to the PCA map of the combined GEO datasets after batch processing. PCA, Principal Component Analysis; IS, Ischemic Stroke. GSE16561 is the IS dataset in light green, and GSE22255 is the IS dataset in light brown.
Cerebral infarction-related inflammation and iron metabolism related differentially expressed genes
The data from the combined GEO datasets were divided into IS samples and control samples, respectively. To analyze the differences in gene expression values between the IS samples and control samples in the combined GEO datasets, the R package limma was used for differential analysis. This analysis yielded a total of 56 DEGs in the integrated GEO datasets (combined datasets) that met the thresholds of |logFC| > 0.5 and p < 0.05. Under these criteria, there were 42 upregulated genes (logFC > 0.5 and p < 0.05) and 14 downregulated genes (logFC < -0.5 and p < 0.05). A volcano plot was created based on the differential analysis results of this dataset (Fig. 3A).

Differential Gene Expression Analysis. (A) Volcano plot of differentially expressed gene analysis of IS samples and control samples in the combined GEO datasets. (B) Venn diagram of DEGs and IIMRGs in the integrated GEO datasets (combined datasets). (C) Heatmap of IIMRDEGs in the integrated GEO datasets (combined datasets). IS, ischemic stroke; DEGs, differentially expressed genes; IIMRGs, inflammation and iron metabolism-related genes; IIMRDEGs, inflammation and iron metabolism-related differentially expressed genes. IS samples are shown in orange, and control samples are shown in light gray. In the heatmap, red represents high expression and blue represents low expression.
In order to obtain the IIMRDEGs, all the previously obtained DEGs with |logFC| > 0.5 and p < 0.05 were compared with the IIMRGs using a Venn diagram (Fig. 3B). A total of 16 IIMRDEGs were identified, which included SLC22A4, DUSP1, MXD1, SLC2A3, C5AR1, ADM, MMP9, PTGS2, ACSL1, CA1, XK, LCN2, IL1B, CCR7, IL7R, and HLA-DQB1. According to the intersection results, the expression differences of IIMRDEGs between different sample groups in the combined GEO datasets were analyzed, and the R package pheatmap was used to create a heatmap to display the analysis results (Fig. 3C).
GO and KEGG enrichment analysis
GO and KEGG enrichment analyses were used to further explore the relationship between the biological process (BP), cellular component (CC), molecular function (MF), and biological pathways (KEGG) of the 16 identified differentially expressed genes (IIMRDEGs) and IS. The 16 IIMRDEGs were analyzed for GO and KEGG enrichment, and the specific results are shown in Table 1. The results indicated that the 16 IIMRDEGs were primarily enriched in the following biological processes: leukocyte chemotaxis, response to molecules of bacterial origin, leukocyte migration, cell chemotaxis, response to glucocorticoids, and others (BP); in cellular components, they were enriched in specific granules, secretory granule lumen, cytoplasmic vesicle lumen, vesicle lumen, and specific granule lumen (CC); and in molecular functions, they included hydrolase activity acting on carbon-nitrogen (but not peptide) bonds, linear amidines, carbonate dehydratase activity, hydro-lyase activity, immune receptor activity, carbon-oxygen lyase activity, and others (MF). Additionally, they were enriched in the IL-17 signaling pathway, Leishmaniasis, TNF signaling pathway, Coronavirus disease (COVID-19), fluid shear stress, and atherosclerosis (KEGG). The results of the GO and KEGG enrichment analyses were visualized using bar and bubble plots (Fig. 4A-B).

GO and KEGG Enrichment Analysis for IIMRDEGs. (A) Bar graph of GO and KEGG enrichment analysis results of IIMRDEGs : BP, CC, MF and KEGG. B. Bubble diagram of GO and KEGG enrichment analysis results of IIMRDEGs : BP, CC, MF and KEGG. GO terms and KEGG terms are shown on the ordinate. C-F. GO and KEGG enrichment analysis results of IIMRDEGs: BP (C), CC (D), MF (E) and KEGG (F). Yellow nodes represent items, blue nodes represent molecules, and the lines represent the relationship between items and molecules. The bubble size in the bubble plot represents the number of genes, while the color of the bubbles indicates the size of the p value. Redder colors correspond to smaller p values, whereas bluer colors correspond to larger p values. The screening criteria for GO and KEGG enrichment analysis were p value < 0.05 and FDR (q value) < 0.25, with the p value correction method being Benjamini-Hochberg (BH).
At the same time, the network diagram of BP, CC, MF, KEGG was drawn according to GO and KEGG enrichment analysis (Fig. 4C-F). The lines show the corresponding molecules and the annotations of the corresponding entries, and the larger the nodes, the more molecules contained in each entry.
Gene set enrichment analysis (GSEA)
To determine the impact of expression levels of all genes in the integrated GEO datasets (combined datasets) on IS, GSEA was used to investigate the expression of all genes and the biological processes involved in the integrated GEO datasets. The link between the affected cellular components and the molecular functions involved is illustrated in (Fig. 5A), with detailed results shown in Table 2. The results indicated that all genes in the combined GEO datasets were significantly enriched in the NF-κB activation through FADD-RIP1 pathway mediated by caspase 8 and 10 (Fig. 5B), TRAF6-mediated NF-κB activation (Fig. 5C), NF-κB pathway (Fig. 5D), NF-κB canonical pathway (Fig. 5E), and other biologically relevant functions and signaling pathways.

GSEA for Combined Datasets. (A) GSEA mountain map presentation of biological functions in the combined GEO datasets. B-E. GSEA showed that all genes were significantly enriched in NF-κB activation through the FADD-RIP1 pathway mediated by caspase 8 and 10 (B), TRAF6-mediated NF-κB activation (C), the NF-κB pathway (D), and the NF-κB canonical pathway (E). The screening criteria for GSEA were p < 0.05 and FDR (q-value) < 0.25, with the p-value correction method being Benjamini-Hochberg (BH).
Construction of PPI network and screening of hub genes
A PPI network of 16 IIMRDEGs was constructed using the STRING database (Fig. 6A). The results of the PPI network showed that 12 IIMRDEGs were related, namely: DUSP1, C5AR1, ADM, MMP9, PTGS2, CA1, XK, LCN2, IL1B, CCR7, IL7R, and HLA-DQB1. Subsequently, the scores of the 12 IIMRDEGs were calculated using the five algorithms from the CytoHubba plug-in of Cytoscape software, and the IIMRDEGs were ranked according to their scores. The five algorithms were as follows: Maximal Clique Centrality (MCC), Density of Maximum Neighborhood Component (DMNC), Degree, Closeness, and Betweenness. Then, the top 10 IIMRDEGs from the five algorithms were used to draw the PPI network, which included the following: MCC (Fig. 6B), DMNC (Fig. 6C), Degree (Fig. 6D), Closeness (Fig. 6E), and Betweenness (Fig. 6F). The color of the circles ranges from red to yellow, representing ratings from high to low. Finally, the intersection of the genes identified by the five algorithms was taken, and a Venn diagram (Fig. 6G) was created for analysis. The intersecting genes from the algorithms were identified as hub genes of IS, and the eight hub genes were: IL7R, LCN2, HLA-DQB1, ADM, PTGS2, IL1B, DUSP1, and MMP9.

PPI Network and hub genes Analysis. PPI network of IIMRDEGs calculated from the STRING database (A). (B–F) The PPI network of the top 10 IIMRDEGs, calculated using five algorithms from the CytoHubba plug-in, including Maximal Clique Centrality (MCC) (B), Density of Maximum Neighborhood Component (DMNC) (C), Degree (D), Closeness (E), and Betweenness (F). G. Venn diagram of the top 10 IIMRDEGs from the five algorithms of the CytoHubba plug-in. H. GeneMANIA website predicts the interaction network of the eight hub genes with similar functions. The circles in the figure represent the hub genes and genes with similar functions, while the colors corresponding to the lines indicate the interconnected functions.
The interaction network of the 8 hub genes and their functionally similar genes (Fig. 6H) was predicted and constructed using the GeneMANIA website. The lines with different colors represent the co-expression and shared protein domains and other information between them. Among them, there are 8 hub genes and 20 functionally similar proteins. The detailed information is shown in Table S2.
Construction of regulatory network
Firstly, the transcription factors (TFs) that bind to hub genes were obtained from the ChIPBase database, and the mRNA-TF regulatory network was constructed and visualized using Cytoscape software (Fig. 7A). Among them, there were 5 IIMRDEGs and 32 TFs. The specific information is provided in Table S3.

Regulatory Network of IIMRDEGs. (A) mRNA-TF Regulatory Network of hub genes. (B) mRNA-miRNA Regulatory Network of hub genes. Orange is mRNA, green is TF, and blue is miRNA.
Then, the miRNAs related to the hub genes were obtained from the TarBase database, and the mRNA-miRNA regulatory network was constructed and visualized using Cytoscape software (Fig. 7B). Among them, there were 4 hub genes and 30 miRNAs, with the specific information shown in Table S4.
Differential expression verification and ROC curve analysis of hub genes
To explore the expression differences of hub genes in the combined GEO datasets, the group comparison figure (Fig. 8A) presents the analysis results of the expression levels of eight hub genes in IS samples compared to control samples in the integrated GEO datasets. The differential results showed (Fig. 8A) that the expression levels of five hub genes in IS samples and control samples of the combined GEO datasets were highly statistically significant (p < 0.001). These genes are: ADM, DUSP1, IL7R, MMP9, and PTGS2. Two hub genes with statistically significant expression (p < 0.01) in IS samples compared to control samples in the combined GEO datasets were HLA-DQB1 and LCN2. The expression of IL1B in IS samples and control samples in the combined GEO datasets was statistically significant (p < 0.05).

Differential Expression Validation and ROC Curve Analysis. (A) Group comparison plots of hub genes in IS samples and control samples from the combined GEO datasets. ROC curves for hub genes IL7R and LCN2 (B), HLA-DQB1 and ADM (C), PTGS2 and IL1B (D), and DUSP1 and MMP9 (E) in the integrated GEO datasets (combined datasets) are displayed. The symbols denote statistical significance: * indicates p < 0.05 (statistically significant); ** represents p < 0.01 (highly statistically significant); and *** represents p < 0.001 (very highly statistically significant). When AUC > 0.5, it indicates that the expression of the molecule is trending towards promoting the occurrence of the event, and the closer the AUC is to 1, the better the diagnostic effect. AUC demonstrates low accuracy in the range of 0.5 to 0.7, and moderate accuracy in the range of 0.7 to 0.9. TPR, true positive rate; FPR, false positive rate. Light gray represents control samples, while orange represents IS samples.
Finally, the R package pROC was used to draw ROC curves based on the expression levels of hub genes in the integrated GEO datasets (combined datasets). The ROC curves (Fig. 8B-E) showed that the expression levels of the hub genes IL7R, ADM, DUSP1, and MMP9 demonstrated certain accuracy in the classification of IS samples and control samples (0.7 < AUC < 0.9). In contrast, the expression levels of LCN2, HLA-DQB1, PTGS2, and IL1B displayed low accuracy (0.5 < AUC < 0.7) in the classification of IS samples and control samples.
Immune infiltration analysis of cerebral infarction (CIBERSORT)
The infiltration abundance of 22 immune cells in the combined GEO datasets was calculated using the CIBERSORT algorithm. According to the results of the immune infiltration analysis, a bar chart showing the proportion of immune cells in the integrated GEO datasets was created (Fig. 9A). Group comparison plots (Fig. 9B) were then used to illustrate the differences in immune cell infiltration abundance between IS samples and control samples in the integrated GEO datasets. The results indicated that the expression levels of seven immune cells in IS samples and control samples were statistically significant (p < 0.05) in the combined datasets, including: immature dendritic cells, neutrophils, M0 macrophages, CD8 T cells, monocytes, gamma-delta T cells, and activated dendritic cells.

Combined Datasets Immune Infiltration Analysis by CIBERSORT Algorithm. (A,B) The proportion of immune cells in the integrated GEO datasets is shown in a bar graph (A) and a group comparison graph (B). (C) Correlation heatmap of immune cell infiltration abundance in the integrated GEO datasets. (D) Bubble plot illustrating the correlation between hub genes and immune cell infiltration abundance in the integrated GEO datasets. The symbols indicate statistical significance: * represents p < 0.05 (statistically significant); ** represents p < 0.01 (highly statistically significant); and *** represents p < 0.001 (very highly statistically significant). An absolute value of the correlation coefficient (r) below 0.3 indicates weak or no correlation, while a correlation between 0.5 and 0.8 is considered moderate. IS samples are depicted in orange, and control samples are shown in light gray. Blue indicates negative correlation, while red indicates positive correlation, with color depth representing the strength of the correlation.
Next, the correlation results of the infiltration abundance of these seven immune cells in the immune infiltration analysis were displayed using a correlation heat map (Fig. 9C). The results showed that neutrophils and M0 macrophages exhibited the greatest positive correlation (r = 0.363), while neutrophils and CD8 T cells showed the greatest negative correlation (r = -0.466).
Finally, correlation bubble plots were used to demonstrate the relationship between hub genes and immune cell infiltration abundance in the combined GEO datasets (Fig. 9D). The results of the correlation bubble map indicated a significant positive correlation between the hub gene MMP9 and the immune cells (M0 macrophages) (r = 0.535, p < 0.05). There was also a significant negative correlation between the hub gene MMP9 and resting dendritic cells (r = -0.573, p < 0.05).
Discussion
IS stands as a major cause of death and long-term disability globally, with an estimated incidence rate of around 15 million cases annually and a mortality rate of approximately 11% within the first 30 days. This highlights the urgent need for research into its underlying mechanisms and potential therapeutic targets. Recent studies have emphasized the critical roles of inflammation and iron metabolism in the pathophysiology of IS11; yet, specific molecular mechanisms and potential biomarkers remain inadequately explored. Addressing these gaps, our study aimed to identify DEGs related to these pathways in IS, thereby contributing to a better understanding of the disease and the identification of new diagnostic targets.
In our study, we identified 16 DEGs linked to inflammation and iron metabolism in IS. Through integrative analyses, including GO and KEGG enrichment, we found that these DEGs participate in essential biological processes and pathways such as immune response, oxidative stress, and iron homeostasis. Our findings align with previous research that highlights the roles of inflammatory mediators and iron-related genes in IS12. To further explore the interactions among these identified genes, we constructed a PPI network using the CytoHubba plugin. This analysis revealed eight hub genes, including IL7R and ADM, which showed statistically significant expression differences in the integrated GEO datasets. The diagnostic potential of these hub genes was validated through ROC curve analysis, which produced AUC values ranging from 0.7 to 0.9, suggesting their potential as biomarkers for IS.
We conducted a differential expression analysis of GEO datasets and identified 56 genes that met the criteria of |logFC| > 0.5 and p-value < 0.05. Notably, 16 of these DEGs are involved in inflammation and iron metabolism, such as SLC22A4, DUSP1, MXD1, and IL1B.
SLC22A4, also known as OCTN1, is an organic cation transporter linked to various autoimmune diseases. Numerous studies have established associations between polymorphisms in SLC22A4 and conditions such as rheumatoid arthritis (RA) and Crohn’s disease (CD) across diverse populations13,14. Notably, an intronic SNP within a RUNX1 binding site of SLC22A4 has been recognized as a significant genetic risk factor for RA, particularly in the Japanese population15. Furthermore, SLC22A4 is highly expressed in inflammatory tissues, suggesting its involvement in immune responses. In the context of IS, recent studies have identified SLC22A4 as an immune-related signature gene, with increased expression observed in astrocytes within affected brain regions16. This aligns with our findings, which indicate that SLC22A4 contributes to the inflammatory processes following IS, potentially positioning it as a promising therapeutic target for modulating immune responses to improve patient outcomes. Additionally, we are investigating SLC22A4’s role in iron metabolism, given that iron homeostasis is vital for neuronal function and survival after a stroke. Our study not only corroborates previous research but also highlights the significance of SLC22A4 in understanding the molecular mechanisms underlying IS.
The identified DEGs warrant further exploration into their specific expression patterns across various tissues or cell types. For instance, genes such as IL1B and MMP9 are well-known for their roles in inflammatory responses, particularly in neuronal tissues during ischemic conditions17. Understanding whether these genes show varied expression in glial cells versus neurons could elucidate their distinct contributions to neuroinflammation. Additionally, identifying tissue-specific expression profiles help in pinpointing the cellular sources of these inflammatory mediators, further unraveling their roles in the pathophysiology of cerebral ischemia18.
Studies have shown that the immediate response to ischemia involves a surge in inflammatory cytokines, followed by a more complex regulatory network as the condition progresses19. Investigating how the expression levels of our identified DEGs evolve over time could provide insights into their functional relevance at different stages of disease progression. For example, while some genes play a role in early inflammatory responses, others be more involved in the later stages of recovery or secondary injury20.
The possibility of utilizing these DEGs as potential biomarkers for early diagnosis and prognosis in ischemic stroke is a promising avenue for future research. The identification of specific genes that reliably indicate the onset of stroke or predict outcomes could revolutionize clinical approaches. For example, elevated levels of certain inflammatory markers have been associated with poorer prognoses in stroke patients21. Therefore, analyzing the expression levels of our identified DEGs in patient samples could validate their utility as biomarkers, ultimately aiding in timely interventions and tailored therapeutic strategies.
The enrichment analysis of 16 DEGs related to IIMRDEGs in IS showed significant involvement in key biological processes and pathways, including leukocyte chemotaxis, response to bacterial molecules, and the IL-17 and TNF signaling pathways. These pathways are crucial for understanding the mechanisms of inflammation and immune response in ischemic stroke.
The IL-17 signaling pathway plays a key role in the pathogenesis of ischemic stroke by mediating inflammation. IL-17 A, a pro-inflammatory cytokine predominantly produced by Th17 cells, has been shown to exacerbate brain injury by promoting the infiltration of neutrophils and the release of other pro-inflammatory cytokines22. Studies have shown that IL-17 A is involved in both the acute and chronic phases of ischemic stroke, contributing to neuronal damage and impairing recovery23. Furthermore, the IL-17 signaling pathway has been implicated in the regulation of astrocytic responses post-stroke, which are critical for neurogenesis and synaptic plasticity24.Similarly, the TNF signaling pathway is another critical inflammatory pathway activated in IS. TNF-α, a central cytokine in this pathway, is known to induce apoptosis and exacerbate inflammation, thereby contributing to the extent of brain damage following an ischemic event25. The pathway’s activation leads to the upregulation of various inflammatory mediators, including interleukins and chemokines, which further propagate the inflammatory response26.
Identifying these pathways highlights the need to focus on inflammatory processes when managing IS. Therapeutic strategies that adjust IL-17 and TNF signaling reduce inflammatory damage. This could lead to better outcomes for IS patients. For example, interventions that block IL-17 A or its receptor have shown promise in reducing infarct size and enhancing neurological function in preclinical stroke models27. Similarly, researchers are investigating TNF-α inhibitors for their potential neuroprotective effects in stroke therapy28. The presence of IIMRDEGs in the IL-17 and TNF signaling pathways emphasizes the significant role that inflammation plays in IS29. Understanding these pathways provides valuable insights into the molecular mechanisms driving IS and opens up new avenues for therapeutic intervention aimed at modulating inflammatory responses to improve patient outcomes.
From the PPI network, we identified eight hub genes: IL7R, LCN2, HLA-DQB1, ADM, PTGS2, IL1B, DUSP1, and MMP9. These genes serve as critical nodes within the network and indicate their roles as important regulatory molecules in IS. Previous studies have established connections between several of these hub genes and IS. For instance, IL1B has been recognized for its involvement in the inflammatory response following ischemic events, linking it to neuronal damage and recovery30. Likewise, DUSP1, a dual specificity phosphatase, modulates MAPK pathways, which play a key role in cellular responses to stress and inflammation; it has also been associated with protective effects during cerebral ischemia31. Moreover, PTGS2, as a key enzyme in prostaglandin synthesis, is frequently upregulated in IS and contributes to the inflammatory cascade following ischemic injury32. Our findings not only support the existing literature connecting these hub genes to IS but also underscore their potential significance as therapeutic targets, enhancing our understanding of the molecular mechanisms underlying the disease.
We analyzed the interactions among the identified hub genes and found that many of them directly interact and share regulatory targets. For instance, IL7R and IL1B modulate lymphocyte activation and differentiation, potentially enhancing the immune response during a stroke33. Additionally, LCN2 regulates MMP9 expression, linking iron metabolism to matrix remodeling crucial for the post-stroke inflammatory response34. These interactions illustrate a complex regulatory network where hub genes collaboratively influence the pathophysiology of IS. These hub genes are considered core nodes due to their significant roles in key biological processes; for example, IL7R is vital for T cell function and adaptive immunity after ischemic events. MMP9 facilitates immune cell migration to injury sites and contributes to inflammation35. Similarly, ADM promotes vasodilation and nerve protection, underscoring the interplay between the nervous and vascular systems in IS36. Identifying these eight hub genes enhances our understanding of IS mechanisms and paves the way for future experimental validation. Exploring these interactions could yield important therapeutic targets for IS intervention, ultimately improving patient outcomes.
Our study identified significant changes in key immune cell types in IS samples, particularly Neutrophils and regulatory T cells. These findings illustrate dynamic alterations in the immune microenvironment critical for understanding IS pathophysiology and immune interactions during stroke development. During the acute phase, Neutrophils increase significantly, contributing to tissue damage and inflammation through the release of pro-inflammatory cytokines37. In contrast, the later stages are marked by an upsurge in regulatory T cells, which are essential for promoting tissue repair and modulating the inflammatory response38. This shift in immune cell populations suggests that targeting specific cell types at different stages could enhance treatment outcomes. For instance, MMP9, which is involved in extracellular matrix remodeling, may activate M0 macrophages and further amplify inflammatory responses39. The hub gene DUSP1 also plays a crucial role in regulating T cell activation, indicating that these hub genes together might significantly influence immune responses in IS40. Our findings highlight the potential for tailored immunotherapy strategies. Enhancing the function of regulatory T cells could mitigate inflammation, while targeted activation of specific macrophage populations may support recovery. Additionally, therapies aimed at modulating dendritic cell activity could improve antigen presentation and adaptive immune responses41. By focusing on Neutrophils and regulatory T cells, our research underscores the importance of the immune microenvironment in IS pathology, providing valuable insights for the development of innovative therapeutic strategies to improve patient outcomes.
The key genes identified in this study, such as IL7R and ADM, may hold significant value in the clinical diagnosis and treatment of ischemic stroke. IL7R has demonstrated diagnostic potential in studies of other immune-related diseases, while ADM, due to its role in vascular biology, could serve as a target for future therapeutic interventions. Future research could focus on developing biomarker detection methods based on these genes, such as measuring their expression levels in blood samples to improve the early diagnosis rate of ischemic stroke. Additionally, drug development targeting these relevant genes may provide novel treatment options for patients. To further validate the clinical application of these genes, we plan to incorporate larger clinical cohorts in our subsequent research and conduct a deeper exploration of the underlying mechanisms.
Despite the comprehensive approach and rigorous analysis employed in this study, several limitations should be acknowledged. Firstly, the study relies solely on bioinformatics and computational methods without incorporating wet lab experiments, which could provide more direct evidence of the biological mechanisms involved. Secondly, the sample size from the datasets GSE22255 and GSE16561 is relatively small, which limit the generalizability of the findings. Thirdly, the study lacks clinical validation, which is crucial for translating these findings into potential diagnostic or therapeutic applications. Additionally, the integration of multiple datasets introduces the possibility of batch effects, despite the use of sva package for batch correction. These factors influence the robustness and reproducibility of the results.
Conclusion
In conclusion, this study provides valuable insights into the differential expression of inflammation and iron metabolism-related genes in ischemic stroke. By integrating data from multiple sources and employing a variety of bioinformatics tools, we identified key differentially expressed genes and constructed a comprehensive regulatory network involving transcription factors and miRNAs. The findings from GO and KEGG enrichment analyses, as well as the PPI network, highlight the potential biological functions and pathways involved in the disease. Furthermore, the immune infiltration analysis offers a deeper understanding of the immune landscape in ischemic stroke. Although there are limitations, the results of this study lay a foundation for future research and potential clinical applications. Future studies should aim to validate these findings through experimental and clinical approaches to fully elucidate the mechanisms and therapeutic targets for ischemic stroke.
Materials and methods
Data download
Using the R package GEOquery42 (Version 2.70.0) downloaded from the GEO database43 (https://www.ncbi.nlm.nih.gov/geo), we accessed the cerebral infarction (Ischemic Stroke, IS) datasets GSE2225544 and GSE1656145. This revision clarifies the use of the software and improves the flow of the sentence.The samples in datasets GSE22255 and GSE16561 were all from Homo sapiens, with blood as the tissue source. The chip platform for dataset GSE22255 is GPL570, while that for dataset GSE16561 is GPL6883. Please see Table 3 for specific information. Among them, dataset GSE22255 contained 20 cerebral infarction (IS) samples and 20 control samples. Dataset GSE16561 contains 39 cerebral infarction (IS) samples and 24 control samples. All IS samples and control samples were included in this study.We selected two publicly available datasets (GSE22255 and GSE16561) from the GEO database. The selection criteria were based on the following considerations: (1) Both datasets comprise whole blood samples, ensuring biological consistency between cases and controls; (2) The combination of these datasets provides a total of 103 samples (59 IS cases and 44 controls), enhancing the statistical power of the study(PMID: 38637126, PMID: 36818726); (3) These datasets have been widely utilized in previous ischemic stroke research, further validating their relevance. To address potential batch effects, we applied the ComBat algorithm for normalization and verified the effect through PCA plots.
The GeneCards database46 (https://www.genecards.org/) is a collection of inflammation and iron metabolism-related genes (Inflammatory and Iron Metabolism Related Genes, IIMRGs). The GeneCards database provides comprehensive information on human genes. We used the terms ‘Inflammatory’ and ‘Iron Metabolism’ as search keywords, respectively. For inflammation-related genes (IRGs), only those that were ‘Protein Coding’ and had a ‘Relevance Score > 12’ were retained, resulting in a total of 214 IRGs. Similarly, for iron metabolism-related genes (IMRGs), the same criteria were applied, yielding a total of 743 IMRGs.
In addition, we used ‘Inflammatory’ as a keyword on the PubMed website(https://pubmed.ncbi.nlm.nih.gov/) to search for published literature47,48,49 related to inflammation, identifying a total of 264 IRGs. We then used ‘Iron Metabolism’ as a keyword on PubMed to identify a total of 14 IMRGs from the published literature50,51,52. After merging and de-duplicating the inflammation-related genes, a total of 426 IRGs were retained. Applying a similar process to the iron metabolism-related genes, we obtained a total of 743 IMRGs after combined de-duplication. Finally, by merging the IRGs and IMRGs, we obtained a total of 1,123 IIMRGs, with detailed information provided in Table S1.
The R package sva53 (Version 3.50.0) was used to debatching the Datasets GSE22255 and GSE16561 to obtain the Combined GEO datasets. Among them, the Combined Datasets included 59 cerebral infarction (IS) samples and 44 Control samples. The sva package is widely used to identify and remove batch effects from high-throughput datasets, such as microarray gene expression data. In our study, we applied the ComBat algorithm within the sva package to correct for batch effects between the datasets GSE22255 and GSE16561. This algorithm adjusts for systematic differences between batches by modeling the batch effects and normalizing the data accordingly. Finally, the integrated GEO Datasets (Combined Datasets) were standardized by R package limma54 (Version 3.58.1), and the annotation probes were standardized and normalized. The expression matrices before and after removing batch effect were subjected to Principal Component Analysis (PCA) to verify the effect of removing batch effect55. PCA is a method of data dimensionality reduction, which extracts the feature vectors (components) of data from high-dimensional data. To transform the data into low-dimensional data and display these features in 2D or 3D graphs.
Differentially expressed genes related to cerebral infarction-related inflammation and iron metabolism
Based on the sample grouping from the combined GEO datasets, we categorized the samples into cerebral ischemic stroke (IS) samples and control samples, performing differential gene expression analysis with the R package limma (Version 3.58.1). We set the thresholds for identifying differentially expressed genes (DEGs) at |logFC| > 0.5 and p < 0.05. Genes with logFC > 0.5 and p < 0.05 were considered upregulated DEGs, while those with logFC < -0.5 and p < 0.05 were classified as downregulated DEGs, using the Benjamini-Hochberg (BH) method for p-value adjustment. The differential analysis results were visualized with volcano plots created using the R package ggplot2 (Version 3.4.4). We selected the filtering threshold of |logFC| > 0.5 and p < 0.05 to maximize the inclusion of potential DEGs during preliminary screening, allowing us to detect subtle gene expression changes that may be significant in ischemic stroke pathophysiology. A lenient threshold helps identify key genes associated with disease progression, providing a solid basis for further analysis. Additionally, similar studies in the biomedical field have adopted comparable thresholds to avoid overlooking biologically relevant gene expression changes.
To obtain the IIMRDEGs associated with IS, all DEGs with |logFC| > 0.5 and p < 0.05 obtained from the differential analysis in the combined GEO datasets were compared with IIMRGs, and a Venn diagram was drawn. IIMRDEGs were identified, and a heatmap was created using the R package pheatmap (Version 1.0.12).
GO and pathway KEGG enrichment analysis
GO analysis56 is a common method for large-scale functional enrichment studies, encompassing Biological Processes (BP), Cellular Components (CC), and Molecular Functions (MF). KEGG57 is a widely used database that stores information on genomes, biological pathways, diseases, and drugs. We performed GO and KEGG pathway enrichment analyses of IIMRDEGs using the R package clusterProfiler58 (Version 4.10.0). The criteria for item screening were p < 0.05 and an FDR (q-value) < 0.25, with the p-value correction method being the Benjamini-Hochberg (BH).
Gene set enrichment analysis (GSEA)
GSEA59 is a method used to assess the distribution trend of genes within a predefined gene set, based on a ranked gene table correlated with phenotypes, thereby determining their contribution to the phenotype. In this study, genes from the combined GEO datasets were first ranked by log2 fold change (logFC) value. Then, the R package clusterProfiler (version 4.10.0) was used to perform GSEA on all genes in the integrated GEO datasets (combined datasets). The parameters used in GSEA were as follows: the seed was set to 2020, the number of computations was 1000, the minimum number of genes contained in each gene set was 10, and the maximum number was 500. Gene sets were accessed through the Molecular Signatures Database (MSigDB)60 using the c2 gene set “Cp. All. V2022.1. Hs. Symbols.” The GMT [all canonical pathways] (3050) was used for GSEA. The screening criteria for GSEA were p-value < 0.05 and false discovery rate (FDR) < 0.25, and the p-value correction method used was Benjamini-Hochberg (BH).
Protein-protein interaction (PPI) network and hub gene screening
The PPI is known as the protein-protein interaction network, which is composed of proteins that interact with each other, participating in biological signaling, gene expression regulation, all aspects of life processes such as energy and substance metabolism and cell cycle regulation. Systematic analysis of the interaction of proteins in biological systems is of great significance for understanding the working principle of proteins in biological systems, understanding the reaction mechanism of biological signals and energy and substance metabolism under special physiological conditions such as diseases, and understanding the functional relationship between proteins.
The STRING database61 (https://string-db.org) is a database that searches for interactions between known and predicted proteins. In this study, the STRING database was applied based on differentially expressed genes related to IIMRDEGs, with a minimum interaction coefficient greater than 0.40 (minimum required interaction score: medium confidence (0.40)) used as the standard to construct the PPI network related to IIMRDEGs. The closely connected local regions in the PPI network represent molecular complexes with specific biological functions. Genes that interact with other genes in the PPI network were selected for subsequent analysis.
In the CytoHubba62 plug-in of Cytoscape63 software, five algorithms were applied: Maximal Clique Centrality (MCC), Density of Maximum Neighborhood Component (DMNC), Degree, Closeness, and Betweenness. We calculated the scores of IIMR DEGs within the PPI network and selected the top 10 IIMR DEGs based on these scores. Finally, the results from the five algorithms were compared, and a Venn diagram was created to analyze the intersection genes, which were identified as exosome-related hub genes.
GeneMANIA database64 (https://genemania.org) was used to generate hypotheses about gene functions, analyze gene lists, and prioritize genes for functional analysis. Given a list of query genes, GeneMANIA finds functionally similar genes using a large set of genomics and proteomics data. In this mode, it weights each functional genomic dataset according to the predicted value of the query. Another use of GeneMANIA is gene function prediction. Given a query gene, GeneMANIA finds genes that are likely to share functions with it, based on how the gene interacts with it. We predicted the functionally similar genes of hub genes through GeneMANIA’s online website to construct a PPI network.
Construction of regulatory network
Transcription factors (TFs) control gene expression through their interactions with hub genes at the post-transcriptional stage. By using the ChIPBase database (http://rna.sysu.edu.cn/chipbase)65 to retrieve transcription factors (TFs), we conducted an analysis of transcription factor (TF) regulation of hub genes (hub genes). Cytoscape software was used to visualize the mRNA-TF Regulatory Network.
In addition, miRNAs play an important regulatory role in the process of biological development and evolution. They regulate a variety of target genes, and the same target gene can also be regulated by multiple miRNAs. To analyze genetic variations associated with inflammation & iron metabolism (IIMRDEGs) and the relationship between miRNAs, we accessed the TarBase66 database (http://www.microrna.gr/tarbase) to identify hub genes that are associated with microRNAs. The mRNA-miRNA Regulatory Network was visualized by Cytoscape software.
Differential expression verification and ROC curve analysis of hub genes
In order to further explore the expression differences of hub genes between IS samples and control samples in the combined GEO datasets, a group comparison map was drawn based on the expression levels of hub genes. Finally, the R package pROC67 (Version 1.18.5) was used to plot the ROC Curve of the hub genes and calculate the Area Under the Curve (AUC) value. To evaluate the diagnostic effect of the expression of hub genes on the occurrence of IS. The AUC of the ROC curve was generally between 0.5 and 1. The closer the AUC is to 1, the better the diagnostic performance. When AUC was between 0.5 and 0.7, the accuracy was low, when AUC was between 0.7 and 0.9, the accuracy was moderate, and when AUC was above 0.9, the accuracy was high.
Immune infiltration analysis of cerebral infarction (CIBERSORT)
CIBERSORT68 is based on linear support vector regression to deconvolute the transcriptome expression matrix, estimating the composition and abundance of immune cells in a mixture. The CIBERSORT algorithm, combined with the LM22 feature gene matrix, filtered out data with immune cell enrichment scores greater than zero, resulting in a specific immune cell infiltration matrix for the combined datasets. A proportion bar chart was created for display. Subsequently, the R package ggplot2 (Version 3.4.4) was used to generate group comparison plots to illustrate the expression differences of LM22 immune cells between IS samples and control samples in the integrated GEO datasets. Subsequently, the immune cells with significant differences in the two groups were screened for subsequent analysis, and the correlation between immune cells was calculated based on Spearman algorithm. The R package pheatmap (Version 1.0.12) was used to draw the correlation heatmap to .show the correlation analysis results of immune cells themselves. The correlation between hub genes and immune cells was calculated based on Spearman algorithm, and the results with p value < 0.05 were retained. The R package ggplot2 (Version 3.4.4) was used to draw correlation bubble plot to show the correlation analysis results of hub genes and immune cells.
Statistical analysis
All data processing and analysis in this article were based on R software (Version 4.2.2). If not otherwise specified, the statistical significance of normally distributed variables was estimated by independent Student’s t-test for comparisons of continuous variables between two groups. Mann-Whitney U test (Wilcoxon Rank Sum test) was used to analyze the differences between variables that were not normally distributed. Kruskal-Wallis test was used for the comparison among three or more groups. Spearman correlation analysis was used to calculate the correlation coefficient between different molecules. All statistical p-values were two-sided unless otherwise noted, with a p-value of less than 0.05 considered statistically significant.
Data availability
Data is provided within the manuscript and supplementary information files.
References
Kim, K. & Lee, J. H. Risk factors and biomarkers of ischemic stroke in cancer patients. J. Stroke. 16, 91–96 (2014).
Moerch-Rasmussen, A. et al. Recurrent ischemic stroke is associated with the burden of risk factors. Acta Neurol. Scand. 133, 289–294 (2016).
Berntsson, J. et al. Risk of stroke in patients with atrial fibrillation is associated with stroke in siblings: a nationwide study. J. Am. Heart Assoc. 9, e014132 (2020).
Suanprasert, N. & Tantirithisak, T. Impact of risk factors for recurrent ischemic stroke in Prasat neurological Institute. J. Med. Assoc. Thai. 94, 1035–1043 (2011).
Karakurum Goksel, B. et al. Subclinical hypothyroidism, hyperhomocysteinemia and dyslipidemia: investigating links with ischemic stroke in Turkish patients. Neurol. Res. 29, 871–876 (2007).
Tsai, C. F. et al. Risk factors for ischemic stroke and its subtypes in Chinese vs. Caucasians: systematic review and meta-analysis. Int. J. Stroke. 10, 485–493 (2015).
Cabral, N. L. et al. High five-year mortality rates of ischemic stroke subtypes: A prospective cohort study in Brazil. Int. J. Stroke. 14, 491–499 (2019).
Kono, T. et al. Cancer-associated ischemic stroke is associated with elevated D-dimer and fibrin degradation product levels in acute ischemic stroke with advanced cancer. Geriatr. Gerontol. Int. 12, 468–474 (2012).
Huang, S., Yin, L., Xu, Y., Zou, C. & Chen, L. The homocysteine associated variant rs548987 of SLC17A3 confers susceptibility to ischemic stroke in Chinese population. J. Neurol. Sci. 370, 78–81 (2016).
Artto, V. et al. Stroke mimics and intravenous thrombolysis. Ann. Emerg. Med. 59, 27–32 (2012).
Jickling, G. C. et al. MicroRNA expression in peripheral blood cells following acute ischemic stroke and their predicted gene targets. PLoS One. 9, e99283 (2014).
Stamova, B. et al. Gene expression profiling of blood for the prediction of ischemic stroke. Stroke 41, 2171–2177 (2010).
Yamada, R. et al. SLC22A4 and RUNX1: identification of RA susceptible genes. J. Mol. Med. 82, 558–564 (2004).
Martínez, A. et al. Role of SLC22A4, SLC22A5, and RUNX1 genes in rheumatoid arthritis. J. Rheumatol. 33, 842–846 (2006).
Tokuhiro, S. et al. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat. Genet. 35, 41–348 (2003).
Lu, D. et al. Investigating the ID3/SLC22A4 as immune-related signatures in ischemic stroke. Aging 15, 14803–14829 (2023).
Sanz, P. & Serratosa, J. M. Neuroinflammation and progressive myoclonus epilepsies: from basic science to therapeutic opportunities. Expert Rev. Mol. Med. 22, e4 (2020).
Zeng, J. et al. The mechanism of microglia-mediated immune inflammation in ischemic stroke and the role of natural botanical components in regulating microglia: A review. Front. Immunol. 13, 1047550 (2023).
Mo, Y., Sun, Y. Y. & Liu, K. Y. Autophagy and inflammation in ischemic stroke. Neural Regen Res. 15, 1388–1396 (2020).
Dagonnier, M. et al. Hyperacute changes in blood mRNA expression profiles of rats after middle cerebral artery occlusion: towards a stroke time signature. PLoS One. 13, e0206321 (2018).
Zhang, X. G. et al. Inflammatory markers as independent predictors for stroke outcomes. Brain Behav. 11, e01922 (2021).
Lin, Y. et al. Critical role of astrocytic interleukin-17 A in post-stroke survival and neuronal differentiation of neural precursor cells in adult mice. Cell. Death Dis. 7, e2273 (2016).
Swardfager, W. et al. Interleukin-17 in post-stroke neurodegeneration. Neurosci. Biobehav Rev. 37, 436–447 (2013).
Xu, H. et al. Molecular Mechanism of Epimedium Extract against Ischemic Stroke Based on Network Pharmacology and Experimental Validation. Oxid. Med. Cell. Longev 2022, 3858314 (2022).
Zhou, Z. et al. Silencing of PTGS2 exerts promoting effects on angiogenesis endothelial progenitor cells in mice with ischemic stroke via repression of the NF-κB signaling pathway. J. Cell. Physiol. 234, 23448–23460 (2019).
Wang, X. et al. Is immune suppression involved in the ischemic stroke?? A study based on computational biology. Front. Aging Neurosci. 14, 830494 (2022).
Wu, C. et al. Multi-omics approaches for the Understanding of therapeutic mechanism for Huang-Qi-Long-Dan granule against ischemic stroke. Pharmacol. Res. 205, 107229 (2024).
Li, J. M. et al. Exploring the Potential Mechanism of Shennao Fuyuan Tang for Ischemic Stroke Based on Network Pharmacology and Molecular Docking. Evid. Based Complement. Alternat. Med 2021, 6015702(2021).
Li, X. et al. IL-17 receptor-based signaling and implications for disease. Nat. Immunol. 20, 1594–1602 (2019).
Zhang, B. et al. SQSTM1-dependent autophagic degradation of PKM2 inhibits the production of mature IL1B/IL-1β and contributes to LIPUS-mediated anti-inflammatory effect. Autophagy 16, 1262–1278 (2020).
Saheb Sharif-Askari, F. et al. SARS-CoV-2 attenuates corticosteroid sensitivity by suppressing DUSP1 expression and activating p38 MAPK pathway. Eur. J. Pharmacol. 908, 174374 (2021).
Jiang, H. et al. Essential genes Ptgs2, Tlr4, and Ccr2 regulate neuro-inflammation during the acute phase of cerebral ischemic in mice. Sci. Rep. 13, 13021 (2023).
Li, Z. et al. Identifying the pattern of immune related cells and genes in the peripheral blood of ischemic stroke. J. Transl Med. 18, 296 (2020).
Włodarczyk, L. et al. Circulating serum VEGF, IGF-1 and MMP-9 and expression of their genes as potential prognostic markers of recovery in Post-Stroke Rehabilitation-A prospective observational study. Brain Sci. 13, 846 (2023).
Barr, T. L. et al. Blood-brain barrier disruption in humans is independently associated with increased matrix metalloproteinase-9. Stroke 41, e123–128 (2010).
Kirişci, M. et al. Protective effects of adrenomedullin on rat cerebral tissue after transient bilateral common carotid artery occlusion and reperfusion. Braz J. Cardiovasc. Surg. 35, 314–322 (2020).
Hu, S. et al. Identification of novel biomarkers and immune infiltration characteristics of ischemic stroke based on comprehensive bioinformatic analysis and machine learning. Biochem. Biophys. Rep. 37, 101595 (2020).
Simats, A. & Liesz, A. Systemic inflammation after stroke: implications for post-stroke comorbidities. EMBO Mol. Med. 14, e16269 (2022).
Kim, I. S., Yang, W. S. & Kim, C. H. Physiological properties, functions, and trends in the matrix metalloproteinase inhibitors in Inflammation-Mediated human diseases. Curr. Med. Chem. 30, 2075–2112 (2023).
Chuang, H. C. & Tan, T. H. MAP4K family kinases and DUSP family phosphatases in T-Cell signaling and systemic lupus erythematosus. Cells 8, 1433 (2019).
Zheng, Y. et al. T cell interactions with microglia in immune-inflammatory processes of ischemic stroke. Neural Regen Res. 20, 1277–1292 (2025).
Davis, S. & Meltzer, P. S. GEOquery: a Bridge between the gene expression omnibus (GEO) and bioconductor. Bioinformatics 23, 1846–1847 (2007).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 41, D991–D995 (2013).
Krug, T. et al. TTC7B emerges as a novel risk factor for ischemic stroke through the convergence of several genome-wide approaches. J. Cereb. Blood Flow. Metab. 32, 1061–1072 (2012).
Barr, T. L. et al. Genomic biomarkers and cellular pathways of ischemic stroke by RNA gene expression profiling. Neurology 75, 1009–1014 (2010).
Stelzer, G. et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinformatics 54,1.30.1–1.30.33 (2016).
Han, Y. et al. A novel inflammatory Response-Related gene signature improves High-Risk survival prediction in patients with head and neck squamous cell carcinoma. Front. Genet. 13, 767166 (2022).
Zhai, W. Y. et al. A novel Inflammatory-Related gene signature based model for risk stratification and prognosis prediction in lung adenocarcinoma. Front. Genet. 12, 798131 (2021).
Gu, X. et al. Hub Genes, Diagnostic Model, and Predicted Drugs Related to Iron Metabolism in Alzheimer’s Disease. Front. Aging Neurosci. 14, 949083 (2022).
Mou, Y. et al. The landscape of Iron Metabolism-Related and methylated genes in the prognosis prediction of clear cell renal cell carcinoma. Front. Oncol. 10, 788 (2022).
Niu, R. et al. A stratification system of ferroptosis and iron-metabolism related LncRNAs guides the prediction of the survival of patients with esophageal squamous cell carcinoma. Front. Oncol. 12, 1010074 (2022).
Yao, J. et al. Characterization of a ferroptosis and iron-metabolism related LncRNA signature in lung adenocarcinoma. Cancer Cell. Int. 21, 340 (2021).
Leek, J. T. et al. The Sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Ben Salem, K. Ben Abdelaziz, A. Principal component analysis (PCA). Tunis Med. 99, 383–389 (2021).
Mi, H. et al. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–d426 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Yu, G. et al. ClusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287 (2012).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 102, 5545–5550 (2005).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–d613 (2019).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Chin, C. H. et al. CytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8(Suppl 4), S11 .
Franz, M. et al. GeneMANIA update 2018. Nucleic Acids Res. 46, W60–w64 (2018).
Zhou, K. R. et al. ChIPBase v2.0: decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 45, D43–d50 (2017).
Vlachos, I. S. et al. DIANA-TarBase v7.0: indexing more than half a million experimentally supported MiRNA:mRNA interactions. Nucleic Acids Res. 43, D153–159 (2015).
Robin, X. et al. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform. 12, 77 (2011).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 12, 453–457 (2015).
Author information
Authors and Affiliations
Contributions
Shengwu Wang designed the project. Shengwu Wang and Yunbo Chen analyzed the data. Youcai Bi, Chao Yan, and Xuemei Li conducted a literature search and interpreted the data. All authors took part in drafting, revising or critically editing the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, S., Li, X., Bi, Y. et al. The impact of inflammation and iron metabolism on gene expression alterations in ischemic stroke: a bioinformatics approach. Sci Rep 15, 15233 (2025). https://doi.org/10.1038/s41598-025-00369-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-00369-9
Keywords
This article is cited by
-
When Heroes Fall: Reduced Expression of Heat-Resistant Obscure Proteins in Ischemic Stroke
NeuroMolecular Medicine (2025)
