
Abstract
Loin muscle area (LMA) is an important production trait in pigs and is highly correlated with lean meat percentage. However, the genetic architecture of LMA has not yet been fully elucidated. This study conducted genome-wide association studies (GWAS) and meta-analyses of LMA in Duroc (n = 337), Landrace (n = 662), and Yorkshire pigs (n = 3,176) using imputed whole-genome sequencing to identify new QTLs and candidate genes associated with LMA traits. A total of 108, 34, and 232 significant variants were identified in the Duroc, Landrace, and Yorkshire populations, respectively. The meta-analysis revealed 143 genome-wide significant SNPs and 276 suggestive SNPs, among which 213 were not identified in single population GWAS. Notably, 229 and 413 SNPs were located on SSC16 in the Yorkshire population and meta-analysis, respectively. Based on the 2-LOD drop-off interval, the SSC16 QTL in the Yorkshire population was further narrowed to a 679.835 kb interval (from 32.818 Mb to 33.498 Mb). The most significant variant within this QTL, 16_33228254 (P = 4.45 × 10–9), explained 1.11% phenotypic variance, representing a potential novel locus for LMA. Further bioinformatics analysis determined seven promising candidate genes (NDUFS4, ARL15, FST, ADAM12, DAB2, PLPP1, and SGMS2) with biological processes such as myoblast fusion and positive regulation of transforming growth factor beta receptor signaling pathway. Among them, ARL15 was previously reported in LMA studies, while the other six genes represent novel candidate genes. These findings reveal potential functional genes and pathways associated with LMA, providing valuable insights for future genetic improvement in pigs.
Similar content being viewed by others

Introduction
The Loin muscle area (LMA) is a crucial economic trait in pig carcass characteristics. Numerous studies have shown a significant positive correlation between LMA and lean meat percentage (LMP)1,2, with LMP being a key factor that directly impacts the profitability of the pig industry. Although traditional genetic improvement of carcass and growth-related traits has made some progress in recent decades3,4, significant challenges remain in fully understanding the biological mechanisms underlying these complex traits. If LMA can be improved at the molecular level, it would significantly accelerate the breeding process for this trait. However, a critical step in this process is the identification of key genes or mutation sites that influence the target trait.
Genome-wide association studies (GWAS) have become a powerful tool for uncovering causal variations in complex traits5,6. GWAS has been extensively applied to identify candidate genes affecting growth7,8,9, reproduction10,11,12, and carcass traits13,14 in pigs. However, the effectiveness of GWAS is influenced by several factors, such as the number and coverage of SNPs, genetic background, and the genetic architecture of the traits15,16. Typically, as sample size and SNP density increase, the power of GWAS to detect SNPs associated with phenotypes also increases, reducing false positives17,18. Despite the decreasing cost of resequencing, performing high-coverage sequencing on thousands of animals remains expensive. Tong et al.19 utilized a high-quality WGS-derived haplotype reference panel to impute SNP 50K data to the WGS level, identifying five new SNPs with significant signals in a subsequent GWAS. Therefore, imputing SNP chip genotype data to the WGS level is a more efficient and cost-effective approach for GWAS. Additionally, population stratification is a major factor influencing the power of GWAS20,21. To minimize this issue, single-trait GWAS are often conducted, but this approach leads to smaller sample sizes. Meta-analysis offers a solution by combining data from multiple independent studies. For example, Jiang et al.22 conducted GWAS on reproductive traits across four pig populations with different genetic backgrounds. The results showed that while single-trait GWAS did not yield any significant findings, meta-GWAS identified 11 key loci associated with the target trait. Another study on pig fat traits also demonstrated that cross-breed meta-GWAS identified five shared QTL across different breeds23. Therefore, meta-GWAS is an effective approach for integrating data from multiple populations and for elucidating the genetic architecture of complex traits24.
In this study, we used data from 4,175 pigs from China, including three breeds: Duroc, Landrace, and Yorkshire. This study aimed to identify novel significant SNPs and candidate genes associated with LMA through GWAS and meta-analysis. The results can provide valuable insights into the genetic improvement of pig carcass traits.
Results
Phenotype and heritability
As shown in Table 1, the heritability of LMA across the three breeds ranged from 0.40 to 0.63, indicating that genetic selection can effectively enhance this trait. Additionally, LMA was adjusted to the values at 100 kg body weight to facilitate comparisons between breeds. The coefficients of variation for adjusted 100 kg LMA across the three breeds ranged from 10.01% to 10.81%, suggesting considerable genetic potential for improving LMA. The adjusted 100 kg LMA values for Duroc, Landrace, and Yorkshire pigs were 43.05, 34.87, and 31.53, respectively, with significant differences observed between breeds (P < 0.001) (Table 1).
Single-population GWAS
The animals in this study were sourced from three different pig breeds. Principal component analysis (PCA) was conducted to identify potential population stratification (Fig. 1). The PCA plot shows that Duroc, Landrace, and Yorkshire pigs formed three distinct clusters, indicating that the three populations have relatively independent genetic backgrounds. Consequently, a univariate linear mixed model was used to perform a single-population GWAS analysis for LMA in each population. Additionally, to assess the presence of potential false positive signals in the GWAS results, we calculated the genomic inflation factor (λ). The λ values for each population ranged from 0.996 to 1.04, and the Q-Q plots showed no signs of inflation, indicating no population stratification and that the GWAS results are reliable (Supplementary Fig. S1).

PCA plot of population structure showing the top two principal components. PC1: principal component 1; PC2: principal component 2. The purple dot represents the Duroc pigs; the triangle represents the Landrace pigs, and the rhombus represents the Yorkshire pigs.
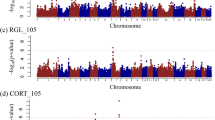
A total of 374 SNPs were identified in this study (Fig. 2B, Table 2 and Supplementary Table S6). In the Duroc pig population, 108 suggestive variants were detected to be associated with LMA. In the Landrace pig population, 34 suggestive variants were detected to be associated with LMA. In the Yorkshire pig population, 192 suggestive and 40 genome-wide variants were detected to be associated with LMA (Fig. 2C and Table 3). Notably, 190 suggestive and 39 genome-wide significant variants were located on SSC16 from 31.98 to 33.49Mb in the Yorkshire population. Additionally, there were no overlapping significant SNPs between the three pig populations (Fig. 3A).

Manhattan plots of GWAS and meta-analysis for LMA in the Duroc, Landrace, Yorkshire pig populations. The solid and dashed lines in the Manhattan plots represent the significant and suggestive thresholds, respectively. Manhattan plot of LMA for (A) Duroc pig (Duroc), (B) Landrace pig (Landrace), (C) Yorkshire pig (Yorkshire), and (D) Meta-analysis, respectively.

Venn plot showing relationships of the identified SNPs in this study. (A) GWAS for Duroc, Landrace, and Yorkshire pig; (B) Single-breed and meta-analysis GWAS.
QTL defined on SSC16 in the Yorkshire population
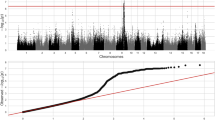
To fine-map the SSC16 QTL associated with LMA, we reanalyzed the GWAS results from the Yorkshire pig population. Based on the 2-LOD drop-off interval, the empirical confidence interval for the QTL was determined to be 679.835 kb from 32,818,583bp to 33,498,418bp (Table 2). Within this QTL region, a total of 225 significant SNPs were identified, with 39 of them reaching genome-wide significance. The top SNP at 33.22 Mb, 16_33228254 (P = 4.45 × 10–9), explained 1.11% of the variation in LMA (Table 2 and Fig. 4). Additionally, we performed ANOVA to analyze the relationship between the top SNP genotypes and adjusted 100 kg LMA. The results showed significant phenotypic differences among the AA, AC, and CC genotypes (P < 0.001). The adjusted 100 kg LMA for the AA genotype was 1.48 cm2 larger than that for the CC genotype (AA: 31.91 cm2, CC: 30.43 cm2) (Fig. 4B,C; Supplementary Table S1). Therefore, this QTL region may represent a strong association with LMA. Furthermore, this QTL region contains two annotated genes: NDUFS4 and ARL15.

Local Manhattan plot and the adjusted 100 kg LMA for different genotypes at the top SNP. (A) Local Manhattan plot (the blue triangle indicates top SNP). The red line represents the peak LOD score (-log P) minus 2 (P = 4.45 × 10–7), while the dark blue line indicates the suggestive significance threshold (P < 1 × 10–6). (B) Adjusted 100 kg LMA for different genotypes at the top SNP. *** indicates significant differences in one-way ANOVA (P < 0.001).
Meta-analysis across population by trait
To improve the statistical power of the GWAS and reduce false positives, we conducted a meta-analysis of the LMA trait GWAS across three pig populations. This study identified 419 significant SNPs through meta-analysis (Fig. 2D, Table 3 and Supplementary Table S6), of which 143 reached genome-wide significance. Compared to individual population GWAS, the meta-analysis detected more SNPs (419 vs. 374), with 213 SNPs not identified in any of the single-population GWAS (Fig. 2D, Table 3, and Fig. 3B). Additionally, the meta-analysis confirmed 206 SNPs previously detected in single-population GWAS, with 79.1% of those SNPs showing lower P-values than in the individual population GWAS (206 SNPs vs. 163). Using linkage disequilibrium analysis, two independent QTLs were precisely mapped within this region (QTL-1: 24,675,594–24,877,580 bp; QTL-2: 32,809,194–34,749,572 bp) (Supplementary Fig.S2). Overall, the meta-analysis improved detection efficiency by identifying SNPs that were missed in single-population GWAS.
Notably, both the meta-analysis and the Yorkshire pig population detected several significant SNPs on SSC16 (meta-analysis: 229; Yorkshire:413). Moreover, there is overlap between the genomic regions of these significant variants (24.67 ~ 34.74 Mb vs. 31.98 ~ 33.49 Mb). These findings further indicate that the QTL on SSC16 may be a critical region influencing pig LMA.
Comparison with previously mapped QTL in pigs
To evaluate whether QTLs associated with LMA traits in this study replicate any previously known QTLs, the pigQTLdb was searched on the basis of SNP and QTL locations. In this study, a total of 587 variants associated with LMA were identified through both single-population and meta-analyses. Among these, 440 SNPs overlapped with findings from previous studies, while 174 SNPs were novel discovered (Supplementary Table S1).
Functional annotation of candidate genes
This study identified a total of 587 SNPs significantly associated with LMA. All significant variants were annotated using VEP. For LMA, 17 candidate genes were identified. KEGG and GO analyses were conducted to highlight the pathways and biological processes associated with LMA in pig (Supplementary Table S2). For LMA, enrichment analysis of the candidate genes using KEGG and GO primarily highlighted pathways related to myoblast fusion and the positive regulation of the transforming growth factor β receptor signaling pathway.
Discussion
Single-population GWAS versus meta-analysis of GWAS
In this study, according to the PCA results, three populations came from different genetic backgrounds, so single-population GWAS was performed on LMA. For the eye muscle area trait, 108, 34, and 232 SNP were detected in the GWAS of Duroc, Landrace, and Yorkshire populations, respectively. It is worth noting that no overlapping SNPs were detected for LMA traits in the three populations. Many previous studies have also detected that there are no or only a small number of shared SNPs among different varieties or different populations within the same breed. For example, Jiang et al.25 conducted GWAS on the backfat thickness and body weight of 2,025 Yorkshire pig populations in the United States and the United Kingdom reaching 100 kg age, and no significant SNPs shared by these two populations were detected. Zhou et al.26 conducted a GWAS on body measurement traits (chest circumference, loin circumference, and abdominal circumference) in 4,288 Duroc pigs from the United States and Canada using 50K SNP chips but did not identify any significant SNPs or QTLs shared between the two populations. The above research results may be due to different genetic backgrounds, SNP densities, and other factors among populations. In this study, the Duroc (n = 337) and Landrace (n = 662) populations had smaller sample sizes compared to the Yorkshire population (n = 3,176), resulting in a higher number of significant SNPs detected in Yorkshire (significant SNPs: 232) than in Duroc (significant SNPs: 108) and Landrace (significant SNPs: 34). Therefore, increasing sample size improves the statistical power of GWAS16,27,28. However, most studies have focused on specific breeds, which may limit the applicability of molecular markers across different populations.
A meta-analysis can integrate multiple single-population GWAS results, improve detection efficiency, reduce false positive results, and even identify some new gene loci24,29 .Studies have shown that meta-analysis has better statistical power than mixed populations. Therefore, this study conducted meta GWAS on LMA traits in three populations. The results showed that the significant SNPs detected by meta-analysis were significantly higher than those detected by single-population association analysis. In addition, meta-GWAS analysis also detected some SNPs that were not detected in a single population GWAS, including 105 significant SNPs associated with LMA. This is consistent with previous research findings. For example, Zhou et al.30 conducted single-trait and multi-trait GWAS and meta-GWAS based on the average daily weight gain and lean meat percentage traits of different Duroc populations, and the results showed that meta-GWAS had higher detection efficiency compared to mixed population GWAS. Guo et al.31 conducted single-trait, multi-trait GWAS, and meta-GWAS on nine fat and growth traits of 2,004 pigs from four different populations. The results showed that both multi-trait methods and meta-analysis improved the effectiveness of GWAS. It is worth noting that the direction of the β-effect values for the significant loci in each individual population is largely consistent with the direction of the Z-effect values. Further comparison of the β-values across the three populations revealed that only one β-value direction was inconsistent among the 207 loci (excluding missing loci). This result indicates that there are loci that jointly influence the LMA trait across the three populations. In summary, meta-analysis expanded the sample size by integrating multiple independent studies, improved the testing effectiveness of GWAS, identified some additional SNPs, and also confirmed some SNPs detected in a single population GWAS.
QTL defined on SSC16 in the Yorkshire population
To identify candidate regions associated with LMA, we conducted genome-wide association analyses across three pig populations. Using the 2-LOD drop-off interval method, we identified a QTL strongly associated with LMA on SSC16 in Yorkshire pigs, spanning approximately 679.835 kb (from 32.81 Mb to 33.49 Mb). Additionally, the meta-GWAS analysis identified a QTL on SSC16 that is strongly associated with LMA. Linkage disequilibrium analysis further refined this region, leading to the precise mapping of two independent QTLs (QTL-1: 24,675,594—24,877,580 bp; QTL-2: 32,809,194—34,749,572 bp). Notably, previous studies have reported QTL regions associated with LMA on SSC16 in different crossbred populations, spanning from 24.3 cM to 44.8 cM and 24.3 cM to 92.6 cM32,33, respectively. In this QTL region, Zhuang et al.34, identified a 709 kb QTL associated with LMA (from 33.46 Mb to 34.36 Mb) in two Duroc populations based on 50k SNP chip data. Interestingly, Fan et al.35 also identified an LMA-associated SNP (rs81458278) in the same region using 60k SNP chip data in Yorkshire and Yorkshire × Landrace populations, a finding that was confirmed in our results.
Compared to previous studies, this study utilized imputed resequencing data and a larger sample size, which enhanced the detection power for QTL identification. However, despite narrowing the QTL interval, most of the significant variants were located in non-coding regions, limiting the identification and validation of causal variants. Many GWAS studies have reported that a substantial proportion of significant variants are located in non-coding regions, which often serve as regulatory elements influencing gene expression rather than directly altering protein-coding sequences36,37 These non-coding variants may reside in enhancers, promoters, or other regulatory regions that modulate gene activity through interactions with transcription factors and chromatin structure, ultimately affecting phenotypic traits such as muscle development38. Therefore, future studies will employ integrative multi-omics approaches, such as ATAC-seq for chromatin accessibility, ChIP-seq for histone modifications, and whole-genome bisulfite sequencing for DNA methylation, to elucidate the regulatory mechanisms underlying these variants.
Candidate genes and function analyses
A total of 17 functional genes were located within or near the significant SNP loci identified in this study. Further gene enrichment analysis of these 17 genes revealed seven candidate genes associated with LMA: NDUFS4 (ubiquinol-cytochrome c reductase subunit S4), ARL15 (ADP-ribosylation factor-like GTPase 15), FST (follicle-stimulating hormone), ADAM12 (ADAM metallopeptidase domain 12), DAB2 (DAB adaptor protein), PLPP1 (phospholipid phosphatase 1), and SGMS2 (sphingomyelin synthase 2).
In this study, a major QTL influencing LMA was identified on SSC16 in the Yorkshire pig population. All significant SNPs within this QTL region were annotated using the VEP, revealing two key genes: NDUFS4 and ARL15. Among these, 86 significant variants were located upstream, downstream, and within the 3’ UTR and intronic regions of the NDUFS4 gene (Table 3 and Supplementary Table S6). GO analysis revealed that this gene is involved in the positive regulation of fibroblast proliferation. Interestingly, studies on intramuscular fat (IMF) deposition have shown that NDUFS4 is more highly expressed in pigs with higher IMF content, suggesting its potential role in muscle metabolism and fat deposition39. Additionally, research has demonstrated that NDUFS4 is one of the key genes associated with mitochondrial function, and mutations or disruptions in this gene can impair energy production, potentially leading to muscle weakness, loss of muscle mass, and growth retardation40,41. Similarly, 155 significant variants were located upstream, downstream, and within the 3’ UTR and intronic regions of the ARL15 gene (Table 3 and Supplementary Table S6). ARL15 has been reported to positively regulate TGF-β family signaling by enhancing the assembly of the Smad4 complex42. TGF-β signaling has been shown to be essential for restricting myoblast fusion in mammals, determining the number of myonuclei and the size of muscle fibers43.
Four significant SNPs on SSC16 were located in the intronic and regulatory regions of the FST gene (Table 3 and Supplementary Table S6). It has been reported that follicle-stimulating hormone binds to several members of the transforming growth factor-β (TGF-β) superfamily, such as myostatin (Mstn) and activin (Act), making it a crucial regulator of skeletal muscle growth and fat deposition. Studies in knockout mice have shown that FST promotes muscle hypertrophy by inhibiting Mstn and Activin while enhancing satellite cell proliferation44. Additionally, Long et al.45 demonstrated that specific expression of FST in the skeletal muscle tissue of transgenic pigs promoted muscle growth and reduced fat deposition. These findings suggest that FST may play a pivotal role in muscle growth and should be considered a strong candidate gene for LMA. Two significant SNPs on SSC14 (14_135344062, 14_135369912) were identified as intronic variants of the ADAM12 gene. GO analysis revealed that this gene is involved in the myoblast fusion process. The growth and development of skeletal muscle depend on the process of myoblast fusion. Myoblasts are precursor cells of skeletal muscle, and during muscle fiber formation, they coordinate their proliferation, migration, and fusion through signaling pathways such as TGF-β/BMP and Wnt/β-catenin, ultimately forming mature skeletal muscle fibers46. Lafuste et al.47 reported that both ADAM12 and α-9β1 integrin are expressed during postnatal human myogenic differentiation, suggesting that ADAM12 participates in regulating mammalian myoblast fusion through its interaction with α-9β1 integrin. Therefore, ADAM12 should be considered a strong candidate gene for LMA. A significant SNP (16_24675594) was identified as an intronic variant of the DAB2 gene. GO analysis revealed that this gene is associated with the positive regulation of the transforming growth factor beta receptor signaling pathway. Shang et al. reported that DAB2 is a positive regulator of early myoblast differentiation. These findings suggest that DAB2 may influence muscle growth and development by regulating early myoblast differentiation, and it should be considered a strong candidate gene for LMA.
Notably, in this study, four significant SNPs on SSC16 were identified as intronic variants of the PLPP1 gene, while one significant SNP on SSC8 was identified as an intronic variant of the SGMS2 gene (Table 4 at the end of the document text file). Term and pathway analyses revealed that both genes are primarily associated with sphingolipid metabolism and sphingomyelin biosynthesis (Fig. 5, Supplementary Table S2). Research has shown that sphingolipids regulate the proliferation, differentiation, growth, and apoptosis of skeletal muscle cells48,49. In another study on age-related muscle dysfunction, it was reported that sphingolipids accumulate in aging skeletal muscle, disrupting muscle function by altering cell membranes and affecting signaling pathways involved in muscle maintenance and regeneration. Further experiments indicated that reducing sphingolipid levels counteracted muscle loss50. These findings suggest that PLPP1 and SGMS2 may play roles in muscle growth and development and should be considered candidate genes. Additionally, the significant terms and pathways identified in this study were related to fat digestion and absorption, adiponectin levels, and visceral fat, suggesting a potential link between LMA and fat traits. It is well known that LMA is a key indicator of LMP. Numerous studies have confirmed that lean meat percentage is positively correlated with muscle mass and negatively correlated with subcutaneous fat51. Therefore, the author speculates that fat may also be one of the factors affecting LMA, and further experimental verification is needed.

Bar plot illustrating the P-values for selected terms related to LMA.
Conclusions
Using imputed resequencing data, we conducted single-trait GWAS and meta-analysis on LMA traits in 4,175 pigs from three populations. A total of 374 significant variants associated with LMA were identified across the three populations, with an additional 213 variants identified through meta-analysis. Notably, a QTL associated with LMA was detected on SSC16 in the Yorkshire population, spanning 679.835 kb. Compared to previous studies, our results significantly narrowed the QTL interval. A series of bioinformatics analyses revealed several functional genes associated with LMA, particularly those involved in regulating muscle growth and myoblast fusion. NDUFS4, ARL15, FST, ADAM12, DAB2, PLPP1, and SGMS2 were identified as the most promising candidate genes for LMA traits in pigs. These findings provide new insights into pig’s genetic architecture of LMA traits. Furthermore, some key variants or QTLs associated with LMA identified in this study may be helpful to for marker-assisted selection in pig breeding.
Materials and methods
Ethics approval and consent to participate
In this study, all procedures involving animals followed the guidelines for the Care and Use of Laboratory Animals. The Animal Care and Use Committee of the Sanya Institute, Hainan Academy of Agricultural especially approved this study. The phenotypic data and ear tissue samples of pigs used in this study were provided by the at the Xinda Livestock Company Ltd., Henan, China, and written informed consent was obtained before data collection from this company. The study was carried out in compliance with the ARRIVE guidelines.
Animals and phenotype collected
This study collected LMA data from 4,175 pigs (337 Duroc, 662 Landrace, and 3,176 Yorkshire) between 2016 and 2022 in the Xinda Livestock Company Ltd., Henan, China. During the feeding period, all three breeds were raised under consistent feeding conditions and management practices to minimize the influence of non-genetic factors. Experienced researchers used an Aloka 500V SSD ultrasound device (Corometrics Medical Systems, USA) to collect LMA and backfat thickness (BF) phenotypes from the 10th to 11th rib of the pigs. This diagnostic ultrasound system, along with its transducer, provided high-resolution images, and LMA and BF were determined using specialized computer software. Additionally, to compare LMA across different populations, we adjusted traits to the LMA at a body weight of 100 kg. A one-way analysis of variance (ANOVA) was then performed using the R function to evaluate the differences in adjusted 100 kg LMA among populations52. The adjustment formula is described as follows:
where A and B is different for sex and breed, and the value is as follows Supplementary Table S5
Genotyping and quality control
Genomic DNA was extracted from ear tissue using the standard phenol–chloroform method. In this study, all animals were still raised until elimination after phenotypic measurement and ear tissue collection. These animals were not anesthetized during the ear tissue collection. The quality of the DNA was assessed via UV spectrophotometry (A260/280) and gel electrophoresis. Genotyping of 4,645 pigs was performed using a 50K chip, which includes 51,315 SNPs across the genome. Quality control (QC) was conducted using Plink v1.90 software53 (Cambridge, MA, USA) to exclude markers not meeting the following criteria: (1) individual genotype call rate below 95%, (2) SNP genotype call rate below 90%, (3) minor allele frequency (MAF) > 0.05, and deviations from Hardy–Weinberg equilibrium (P < 10E − 6). SNPs located on sex chromosomes and unplaced genomic regions were excluded from the study. After quality control, 37,006 SNPs were retained for subsequent imputation. We increased genotype data to the whole genome resequencing level through a filling strategy, with a reference population of 947 resequencing samples from 30 varieties19. Genotype imputation was performed using Beagle software (version 5.4)54. After imputation, the QC standard is consistent with the chip-SNP genotype data, and qualified SNPs were used for subsequent analysis (Supplementary Table S3).
Population structure analysis
Principal component analysis (PCA) for all SNPs was conducted using GCTA software (v1.93)55 to assess population structure. We used the R package ggplot256 to plot the PCA graph of the first two principal components.
Single-population GWAS
GWAS was conducted using GEMMA software to analyze three breeds, employing a univariate linear mixed model57. Prior to GWAS, the genomic relationship matrix (GRM) between individuals was estimated using GEMMA. The matrix form was applied to the following statistical model:
where \({\varvec{y}}\) refers to the vector of phenotype value; \({\varvec{W}}\) refers to the vector of phenotype value; including factors such as top five eigenvectors of PCA, sex, live weight, year-season, and batch; \(\boldsymbol{\alpha }\) is the vector of corresponding coefficients including the intercept; \({\varvec{X}}\) is the vector of marker genotypes; \({\varvec{\beta}}\) is the corresponding effect size of the marker; \({\varvec{u}}\) an n × 1 vector of random effects \({\varvec{u}}\sim {{\varvec{M}}{\varvec{V}}{\varvec{N}}}_{{\varvec{n}}}(0,{\varvec{\lambda}}{{\varvec{\tau}}}^{-1}{\varvec{K}})\); and \(\varepsilon\) is the vector of random residuals, with \({\varvec{\varepsilon}}\sim {{\varvec{M}}{\varvec{V}}{\varvec{N}}}_{{\varvec{n}}}(0,{{\varvec{\tau}}}^{-1}{\varvec{l}}{\varvec{n}})\); \(\lambda\) refers to the ratio between the two variance components; \({{\varvec{\tau}}}^{-1}\) is the variance of the residual errors; \({\varvec{K}}\) is a known n × n relatedness matrix calculated in previous step; and I is a known n × n relatedness matrix calculated in previous step. \({{\varvec{M}}{\varvec{V}}{\varvec{N}}}_{{\varvec{n}}}\) denotes the n-dimensional multivariate normal distribution. The SNP-based heritability (\({h}_{SNP}^{2}\)) for LMA, adjusted 100kg LMA was calculated using GCTA v1.93.2 beta software55 based on the GRM between individuals. The restricted maximum likelihood method (REML) was used to fit the single trait animal model, and the top five classifiers of PCA, sex, live weight, year-season, and batch were used as covariates. Additionally, we calculated the genomic inflation factor (λ). We also created quantile–quantile (QQ) plots using the R package rMVP58 to evaluate the GWAS results.
Meta-analysis of GWAS
Meta-analysis was conducted using METAL software59 to combine the results of single-breed GWAS analyses. METAL converts the effect sizes and P-values observed for each SNP in each population into Z-scores. In this study, METAL combined the results of the two single-population GWASs by calculating the pooled inverse-variance-weighted β-coefficients, standard errors, and Z-scores, and the formulas were as follows:
where \({{\varvec{\beta}}}_{{\varvec{i}}}\) is the β-coefficients for study \({\varvec{i}}\); \({\varvec{S}}{\varvec{E}}\) corresponds to the standard errors for study \({\varvec{i}}\). In the single-population GWAS and meta-analysis, the Bonferroni correction method is typically used to determine the significance threshold, specifically set at 0.05/N, where N represents the number of SNPs. However, Bonferroni’s correction is overly stringent60. Additionally, we used the FDR method to determine the significance threshold; however, it is too lenient and prone to generating more false positive results (FDR = 0.01) (Supplementary Fig. S3). Assuming an equal number of independent haplotype blocks between pigs and humans, the human GWAS studies suggest using 5.0 × 10–8 as the significant threshold61,62. Therefore, in this study’s single-population GWAS and meta-analysis, we respectively set 5 × 10–8 and 1 × 10–6 as the genome-wide significance and suggestive thresholds to correct for false positives arising from multiple testing63. Manhattan plots were generated using the R package rMVP.
QTL detection
To detect QTLs associated with LMA, the 2-LOD drop-off interval method was used. As described in previous studies, one unit of -log (P-value) approximately corresponds to one unit of the log of the odds ratio (LOD). All SNPs with LOD scores higher than the peak LOD score (-log P) of two within each SSC were retained64. Furthermore, To further analyze the effect of the QTL on LMA, we performed ANOVA using the R ano() function to compare adjusted 100kg LMA among individuals with different genotypes at the most significant loci within the QTL region.
Functional annotation of candidate genes
Annotation of genes nearest to significant SNPs was performed using the Variant Effect Predictor (VEP) module from the Ensembl database (http://ensembl.org/Sus_scrofa/Info/Index). To explore candidate genes involved in pathways and biological processes, KEGG (Kyoto Encyclopedia of Genes and Genomes) and GO (Gene Ontology) analyses were conducted using KOBAS 3.0 (http://kobas.cbi.pku.edu.cn/kobas3)65. To enhance the reliability of gene function annotation, a more comprehensive and well-annotated human database was used as the reference for functional enrichment analysis. The significance of enriched pathways was assessed with Fisher’s exact test, with a threshold of P < 0.05.
Data availability
The datasets generated and/or analyzed in this study are not publicly available since the test populations are consisted of the nucleus herd of the pig breeding company, but are available from the corresponding author on reasonable request.
References
Lopez, B. I. M., Song, C. & Seo, K. Genetic parameters and trends for production traits and their relationship with litter traits in Landrace and Yorkshire pigs. Anim. Sci. J. 89, 1381–1388. https://doi.org/10.1111/asj.13090 (2018).
Lopez, B. I., Viterbo, V., Song, G. W. & Seo, K. S. Estimation of genetic parameters and accuracy of genomic prediction for production traits in Duroc pigs. Czech J. Anim. Sci. 64, 160–165. https://doi.org/10.17221/150/2018-Cjas (2019).
Martinez-Montes, A. M. et al. Deciphering the regulation of porcine genes influencing growth, fatness and yield-related traits through genetical genomics. Mamm. Genome 28, 130–142. https://doi.org/10.1007/s00335-016-9674-3 (2017).
Zeng, Q. & Du, Z. Q. Advances in the discovery of genetic elements underlying longissimus dorsi muscle growth and development in the pig. Anim. Genet. 54, 709–720. https://doi.org/10.1111/age.13365 (2023).
Tan, X. et al. Research progress and applications of genome-wide association study in farm animals. Anim. Res. One Health 1, 56–77. https://doi.org/10.1002/aro2.14 (2023).
Visscher, P. M. et al. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 101, 5–22. https://doi.org/10.1016/j.ajhg.2017.06.005 (2017).
Shi, L. Y. et al. Integrating genome-wide association studies and population genomics analysis reveals the genetic architecture of growth and backfat traits in pigs. Front. Genet. https://doi.org/10.3389/fgene.2022.1078696 (2022).
Guo, Y. M. et al. A genome-wide association study identifies genomic loci associated with backfat thickness, carcass weight, and body weight in two commercial pig populations. J. Appl. Genet. 58, 499–508. https://doi.org/10.1007/s13353-017-0405-6 (2017).
Cai, Z. X., Christensen, O. F., Lund, M. S., Ostersen, T. & Sahana, G. Large-scale association study on daily weight gain in pigs reveals overlap of genetic factors for growth in humans. BMC Genom. https://doi.org/10.1186/s12864-022-08373-3 (2022).
Li, M. Y. et al. Genome-wide association study reveals the candidate genes for reproduction traits in Yunong black pigs. Anim. Genet. 54, 403–407. https://doi.org/10.1111/age.13294 (2023).
Iso-Touru, T., Uimari, P., Elo, K., Sevon-Aimonen, M. L. & Sironen, A. Identification of copy number variations and candidate genes for reproduction traits in Finnish pig populations. Agr. Food Sci. 31, 149–159. https://doi.org/10.23986/afsci.116081 (2022).
Zhang, L. G. et al. Genome-wide association studies and runs of homozygosity to identify reproduction-related genes in Yorkshire pig population. Genes-Basel 14, 2133. https://doi.org/10.3390/genes14122133 (2023).
Qiu, Y. B. et al. Identification of candidate genes associated with carcass component weights in commercial crossbred pigs through a combined GWAS approach. J. Anim. Sci. https://doi.org/10.1093/jas/skad121 (2023).
Wang, H. Y. et al. Genome-wide identification of quantitative trait loci and candidate genes for seven carcass traits in a four-way intercross porcine population. BMC Genom. https://doi.org/10.1186/s12864-024-10484-y (2024).
Tang, H. & He, Z. Advances and challenges in quantitative delineation of the genetic architecture of complex traits. Quant. Biol. 9, 168–184. https://doi.org/10.15302/j-qb-021-0249 (2021).
Lappalainen, T., Li, Y. I., Ramachandran, S. & Gusev, A. Genetic and molecular architecture of complex traits. Cell 187, 1059–1075. https://doi.org/10.1016/j.cell.2024.01.023 (2024).
Yan, G. R. et al. Imputation-based whole-genome sequence association study rediscovered the missing QTL for lumbar number in Sutai pigs. Sci. Rep. https://doi.org/10.1038/s41598-017-00729-0 (2017).
Wang, X. Q. et al. GWAS of reproductive traits in large white pigs on chip and imputed whole-genome sequencing data. Int. J. Mol. Sci. 23, 13338. https://doi.org/10.3390/ijms232113338 (2022).
Tong, X. K. et al. Accurate haplotype construction and detection of selection signatures enabled by high quality pig genome sequences. Nat. Commun. https://doi.org/10.1038/s41467-023-40434-3 (2023).
Yan, S., Sha, Q. & Zhang, S. Control for population stratification in genetic association studies based on GWAS summary statistics. Genet. Epidemiol. 46, 604–614. https://doi.org/10.1002/gepi.22493 (2022).
Akond, Z., Ahsan, M. A., Alam, M. & Mollah, M. N. H. Robustification of GWAS to explore effective SNPs addressing the challenges of hidden population stratification and polygenic effects. Sci. Rep. 11, 13060. https://doi.org/10.1038/s41598-021-90774-7 (2021).
Jiang, Y., Tang, S., Xiao, W., Yun, P. & Ding, X. A genome-wide association study of reproduction traits in four pig populations with different genetic backgrounds. Asian-Australas. J. Anim. Sci. 33, 1400–1410. https://doi.org/10.5713/ajas.19.0411 (2020).
Zeng, H. et al. Meta-analysis of genome-wide association studies uncovers shared candidate genes across breeds for pig fatness trait. BMC Genom. 23, 786. https://doi.org/10.1186/s12864-022-09036-z (2022).
Evangelou, E. & Ioannidis, J. P. A. Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet. 14, 379–389. https://doi.org/10.1038/nrg3472 (2013).
Jiang, Y. et al. A genome-wide association study of growth and fatness traits in two pig populations with different genetic backgrounds. J. Anim. Sci. 96, 806–816. https://doi.org/10.1093/jas/skx038 (2018).
Zhou, S. et al. Genome-wide association analysis reveals genetic loci and candidate genes for chest, abdominal, and waist circumferences in two Duroc pig populations. Front. Vet. Sci. 8, 807003. https://doi.org/10.3389/fvets.2021.807003 (2021).
Spencer, C. C. A., Su, Z., Donnelly, P. & Marchini, J. Designing genome-wide association studies: Sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. https://doi.org/10.1371/journal.pgen.1000477 (2009).
Ziyatdinov, A. et al. Estimating the effective sample size in association studies of quantitative traits. G3-Genes Genom. Genet. https://doi.org/10.1093/g3journal/jkab057 (2021).
de Bakker, P. I. W. et al. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 17, R122–R128. https://doi.org/10.1093/hmg/ddn288 (2008).
Zhou, S. P. et al. A meta-analysis of genome-wide association studies for average daily gain and lean meat percentage in two Duroc pig populations. BMC Genom. https://doi.org/10.1186/s12864-020-07288-1 (2021).
Guo, Y. M. et al. Genome-wide detection of genetic markers associated with growth and fatness in four pig populations using four approaches. Genet. Sel. Evol. https://doi.org/10.1186/s12711-017-0295-4 (2017).
Cho, I. C. et al. Genome-wide QTL analysis of meat quality-related traits in a large F2 intercross between Landrace and Korean native pigs. Genet. Sel. Evol. 47, 7. https://doi.org/10.1186/s12711-014-0080-6 (2015).
Choi, I. et al. Identification of carcass and meat quality QTL in an F(2) Duroc x Pietrain pig resource population using different least-squares analysis models. Front. Genet. 2, 18. https://doi.org/10.3389/fgene.2011.00018 (2011).
Zhuang, Z. W. et al. Meta-analysis of genome-wide association studies for loin muscle area and loin muscle depth in two Duroc pig populations. Plos One 14, e0218263. https://doi.org/10.1371/journal.pone.0218263 (2019).
Fan, B. et al. Genome-wide association study identifies Loci for body composition and structural soundness traits in pigs. PLoS ONE 6, e14726. https://doi.org/10.1371/journal.pone.0014726 (2011).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195. https://doi.org/10.1126/science.1222794 (2012).
Long, E., Williams, J., Zhang, H. & Choi, J. An evolving understanding of multiple causal variants underlying genetic association signals. Am. J. Hum. Genet. https://doi.org/10.1016/j.ajhg.2025.01.018 (2025).
Cano-Gamez, E. & Trynka, G. From GWAS to function: Using functional genomics to identify the mechanisms underlying complex diseases. Front Genet 11, 424. https://doi.org/10.3389/fgene.2020.00424 (2020).
Chen, Q. M. et al. Molecular characterization and expression analysis of NDUFS4 gene in of Laiwu pig. Mol. Biol. Rep. 40, 1599–1608. https://doi.org/10.1007/s11033-012-2208-5 (2013).
Bris, C. et al. Novel NDUFS4 gene mutation in an atypical late-onset mitochondrial form of multifocal dystonia. Neurol. Genet. 3, e205. https://doi.org/10.1212/NXG.0000000000000205 (2017).
Lamont, R. E. et al. A novel NDUFS4 frameshift mutation causes Leigh disease in the Hutterite population. Am. J. Med. Genet. A 173, 596–600. https://doi.org/10.1002/ajmg.a.37983 (2017).
Shi, M. et al. Arl15 upregulates the TGFbeta family signaling by promoting the assembly of the Smad-complex. Elife https://doi.org/10.7554/eLife.76146 (2022).
Girardi, F. et al. TGFbeta signaling curbs cell fusion and muscle regeneration. Nat. Commun. 12, 750. https://doi.org/10.1038/s41467-020-20289-8 (2021).
Gilson, H. et al. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am. J. Physiol.-Endoc. M 297, E157–E164. https://doi.org/10.1152/ajpendo.00193.2009 (2009).
Long, K. R. et al. Transcriptomic analysis elucidates the enhanced skeletal muscle mass, reduced fat accumulation, and metabolically benign liver in human follistatin-344 transgenic pigs. J. Integr. Agr. 21, 2675–2690. https://doi.org/10.1016/j.jia.2022.07.014 (2022).
Lehka, L. & Redowicz, M. J. Mechanisms regulating myoblast fusion: A multilevel interplay. Semin. Cell Dev. Biol. 104, 81–92. https://doi.org/10.1016/j.semcdb.2020.02.004 (2020).
Lafuste, P. et al. ADAM12 and alpha9beta1 integrin are instrumental in human myogenic cell differentiation. Mol. Biol. Cell 16, 861–870. https://doi.org/10.1091/mbc.e04-03-0226 (2005).
Tan-Chen, S., Guitton, J., Bourron, O., Le Stunff, H. & Hajduch, E. Sphingolipid metabolism and signaling in skeletal muscle: from physiology to physiopathology. Front. Endocrinol. (Lausanne) 11, 491. https://doi.org/10.3389/fendo.2020.00491 (2020).
Green, C. D., Maceyka, M., Cowart, L. A. & Spiegel, S. Sphingolipids in metabolic disease: The good, the bad, and the unknown. Cell Metab. 33, 1293–1306. https://doi.org/10.1016/j.cmet.2021.06.006 (2021).
Laurila, P. P. et al. Sphingolipids accumulate in aged muscle, and their reduction counteracts sarcopenia. Nat. Aging 2, 1159–1175. https://doi.org/10.1038/s43587-022-00309-6 (2022).
Knecht, D. & Duziński, K. The effect of sex, carcass mass, back fat thickness and lean meat content on pork ham and loin characteristics. Arch. Anim. Breed. 59, 51–57 (2016).
Team, R. C. R: A Language and Environment for Statistical Computing, https://www.R-project.org/ (2024).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. https://doi.org/10.1086/519795 (2007).
Browning, B. L., Zhou, Y. & Browning, S. R. A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 103, 338–348. https://doi.org/10.1016/j.ajhg.2018.07.015 (2018).
Yang, J. A., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. https://doi.org/10.1016/j.ajhg.2010.11.011 (2011).
Hadley, W. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, 2016).
Zhou, X. & Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821-U136. https://doi.org/10.1038/ng.2310 (2012).
Lilin, Y. et al. rMVP: Memory-Efficient, Visualize-Enhanced, Parallel-Accelerated GWAS Tool. (2023).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191. https://doi.org/10.1093/bioinformatics/btq340 (2010).
Pe’er, I., Yelensk, R., Altshuler, D. & Daly, M. J. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol. 32, 381–385. https://doi.org/10.1002/gepi.20303 (2008).
Johnson, R. C. et al. Accounting for multiple comparisons in a genome-wide association study (GWAS). BMC Genom. 11, 724. https://doi.org/10.1186/1471-2164-11-724 (2010).
McCarthy, M. I. et al. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 9, 356–369. https://doi.org/10.1038/nrg2344 (2008).
Yan, G. R. et al. An imputed whole-genome sequence-based GWAS approach pinpoints causal mutations for complex traits in a specific swine population. Sci. China Life Sci. 65, 781–794. https://doi.org/10.1007/s11427-020-1960-9 (2022).
Ma, J. W. et al. A splice mutation in the gene causes high glycogen content and low meat quality in pig skeletal muscle. PLoS Genet. 10, e1004710. https://doi.org/10.1371/journal.pgen.1004710 (2014).
Bu, D. C. et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 49, W317–W325. https://doi.org/10.1093/nar/gkab447 (2021).
Funding
This research was funded by the STI 2030-Major Projects (Grant NO.2023ZD0404501), the Academician Workstation (Grant NO. YSPTZX202304 & HAAS2024KJCX05). The funders played no role in study design, data collection and analysis, data interpretation, and manuscript writing.
Author information
Authors and Affiliations
Contributions
W.S. Xin and S.P. Zhou conceived and designed the experiments and methodology. D.D. Duan, M.Y. Li, C.M. Qiao, H. Zhou and J.Y. Han performed the experiments. Z.Y. Wang and S.P. Zhou analyzed the data. Z.Y. Wang and S.P. Zhou wrote the original draft, W.S. Xin contributed to manuscript review and editing, supervision, funding acquisition, and data curation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhenyu, W., Mengyu, L., Dongdong, D. et al. A meta-analysis of genome-wide association studies revealed significant QTL and candidate genes for loin muscle area in three breeding pigs. Sci Rep 15, 18758 (2025). https://doi.org/10.1038/s41598-025-00819-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-00819-4
