
Abstract
Hydroxyapatite (HAp, Ca10(PO4)6(OH)2) is the major inorganic component of bones, with high bioactivity and biocompatibility, and pores in the 50–200 μm range can facilitate cell anchorage and proliferation. HAp was synthesised through a rapid sol-gel method, avoiding the usual long aging process typically required for sol-gel HAp. Acetate and nitrate precursor salts were compared, to produce bioceramics having different porosities induced via the addition of hydrogen peroxide (H2O2) pore-forming agent. 3–10 wt% H2O2 was added, and the resulting bioceramics calcined at 400 and 700 °C. Microstructure, composition, specific surface area and macro/mesoporosity were analysed, and bioactivity and cytotoxicity/biocompatibility evaluated by immersion in simulated body fluid (SBF) and MTT assays on MG63 osteoblast cell lines. When heated to 400 °C HAp was the only calcium phosphate phase present, but after heating to 700 °C they were a mixture of HAp and β-tricalcium phosphate (β-TCP, Ca3(PO4)2). The bioceramics exhibit high bioactivity, crystallising HAp from SBF, and most were biocompatible, with cell viabilities of 110–139% for samples with 3 wt% H2O2 derived from nitrates, or from acetates heated to 700 °C. This is the first time that HAp-based bioceramics derived from a rapid sol-gel process have been produced with such induced porosity.
Similar content being viewed by others

Introduction
Bone defects caused by severe internal or superficial traumas, congenital deformities and different bone diseases, are very common and can cause serious changes in the quality of life of the patients1. These conditions can limit the ability to accomplish basic tasks, such as walking, and may also cause social and psychological problems. The current solutions available for these problems are essentially the use of bone graft transplants, or implants based on different types of materials. These types of intervention are needed to ensure the regeneration of the bone tissue and the complete restoration of its functions2. However, when bones are not able to restore themselves, bone tissue engineering shows great therapeutic potential, since the challenges associated with current orthopaedic solutions can be solved by the development of suitable porous and resorbable scaffolds, made from different types of osteobiological materials3.
Within this class of materials, there is the group of bioactive ceramics that possess specific biological or physiological functions, which allows them to be used directly in the human body. In particular, calcium phosphate (CaP) ceramics are the most widely used due to the fact that they are minerals composed of calcium cations and phosphate anions, which are known to be the major inorganic material in all native human bones (approximately 60 wt%)4. Besides that, calcium phosphate ceramics are known to be biocompatible, osteo-conductive and osteo-inductive, which means that they are able to form direct bonds with the bone tissue and the surrounding soft tissues, making them an important asset for bone regeneration5. Furthermore, they have a high affinity for the adhesion of proteins and growth factors, which stimulates the proliferation and differentiation of osteoprogenitor cells, without the modification of their chemical surface6. However, the use of additives or surface coatings in CaP materials and implants can change their properties in order to enhance the processes of angiogenesis and osteogenesis. It is important to choose the type of calcium phosphate that best fits the desired application, since the osteo-conductivity and osteo-inductivity of CaP depends on its physical and chemical characteristics. Depending on the ratio of Ca/P, the temperature, or the presence of water and impurities, calcium phosphate usually crystallises in two forms: calcium hydroxyphosphate, also known as hydroxyapatite (Ca10(PO4)6(OH)2, HAp), and β-tricalcium phosphate (Ca3(PO4)2, β-TCP)7. Of these two forms, HAp is thermodynamically the most stable calcium phosphate ceramic compound in solution, when the pH, temperature and composition are closest to those of physiological body fluids. In particular, hydroxyapatite is highly biocompatible, which enables the cell growth, cell attachment to the surface and cell proliferation, and it is able to form direct bonds with the host tissues in a short period of time, resulting in osteo-conduction and osteo-induction8.
Another important feature that must be considered is the porosity, in particular the porous structure and pore size, for cell migration and penetration, tissue ingrowth and vascularisation9,10 - pores sizes within the range of 1–50 μm are noted to facilitate cell anchorage and proliferation11,12,13 and protein adsorption14. For all these reasons, hydroxyapatite is widely used as a starting material for endo-osseous implants and coating materials for metal or polymeric prostheses. Moreover, HAp crystallises in the hexagonal system and, given the complexity of its lattice, it can often contain variant ions such as carbonate (CO32−), reticular defects and vacancies, which give origin to the class of non-stoichiometric or calcium-deficient hydroxyapatite14,15. For this reason, some forms of non-stoichiometric HAp can show a photocatalytic behaviour in the presence of oxygen vacancies16. Porous HAp ceramics have been produced in the past, via the burning out of sacrificial organic pore formers (e.g., polyurethane sponges) or foaming with the use of a hydrogen peroxide (H2O2) pore-forming agent17,18,19,20,21,22,23, although never with HAp derived from a rapid sol-gel process.
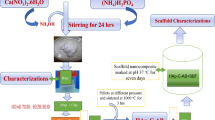
This work focuses on the synthesis of HAp powder through a rapid sol-gel method24 and subsequently the production of several ceramics having different induced porosities for biomedical applications, by the addition of 3–10 wt% H2O2. For the first time, nitrate salts were also used to make HAp via this rapid sol-gel process. The characteristics of the hydroxyapatite porous ceramics and scaffolds produced via rapid sol-gel synthesis were assessed by means of scanning electron microscopy (SEM), X-ray diffraction (XRD) and specific surface area analysis. The in-vitro biocompatibility properties were assessed by performing MTT assays on the MG63 cell line, and the bioactivity was evaluated with a bio-mineralisation assay using a simulated body fluid (SBF). This is the first time that such induced porosity studies have been carried out on HAp bioceramics derived from a rapid sol-gel process, which can be used to produce HAp in only 2 h with no ageing required.
Experimental
Rapid sol-gel synthesis of hydroxyapatite powders
The synthesis of pure hydroxyapatite (HAp) powder was achieved by performing two rapid sol-gel procedures, differing in the use of calcium acetate hydrate, Ca(CH3CO2)2·H2O (94%, Supelco, Sigma-Aldrich), or calcium nitrate tetrahydrate, Ca(NO3)2·4H2O (≥ 99.0%, Fluka Analytical), as precursors for calcium ions24. This method is based on the rapid sol-gel technique developed by Ben-Arfa and Pullar, which can produce either bioactive glass25 or HAp24 in only 2 h, without any need for the usual long ageing processes required of several days to weeks for sol-gel. This approach is 50–200 times quicker than previous aqueous sol-gel routes for HAp production.
One of these two salts (each 0.0995 mol) was dissolved in 100 mL of distilled water in a 250 mL beaker, to produce a yield of 10 g of HAp. Subsequently, orthophosphoric acid (H3PO4, ≈ 85% w/w, AnalaR, BDH Laboratory Supplies, 0.0597 mol)) was added dropwise (volH3PO4 = 4 mL) with a Pasteur pipet, and the solution was heated to 45 °C while stirring at 500 rpm. Then the necessary amount of ammonium hydroxide (NH4OH, 30% w/w NH3 solution, AnalaR, BDH Laboratory Supplies) was added to adjust the pH to 7–8, which was kept constant during the synthesis (around 15 mL required). After 30 min, the synthesis was completed, and the final precipitate was immediately dried rapidly using a rotary evaporator for 1 h, with a water bath temperature of 60 °C and under a constant pressure of 60 millibar, in a 500 mL pear-shaped flask to hold the slurry. This resulted in the production of a HAp powder that was left overnight at room temperature in the rotary evaporator, under a constant pressure of 60 millibar, to ensure complete drying. The resulting material was a pure white HAp powder. Powders obtained from calcium acetate and calcium nitrate are named Ac and Nit, respectively.
Preparation of porous HAp ceramics
Hydrogen peroxide (H2O2, 30% w/w, GPR, BDH Laboratory Supplies) was added to a mixture of 10 g of the sol-gel HAp powders in distilled water (between approximately 8–20 ml), to act as a pore-forming agent. 3 wt%, 5 wt% and 10 wt% hydrogen peroxide (as a 30% w/w solution) was added to the HAp powder and diluted with the minimum amount of distilled water to obtain pastes with the desired viscosity. The resulting samples were named as Nit3, Nit5, Nit10 and Ac3, Ac5 and Ac10, respectively, for the calcium nitrate and calcium acetate precursors. The viscous white pastes were then slowly poured into hemispherical glazed ceramic moulds to give them the shape of hemispheres of about 1 cm in diameter.
The drying process was carried out slowly, placing the samples in a desiccator with silica gel for some hours, followed by drying in an oven at 120 °C for one day. Subsequently, the samples were removed from the ceramic moulds and sintered in air on alumina sheets, at either 400 °C for 2 h (heating rate of 200 °C/h) or 700 °C for 2 h (heating rate of 100 °C/h). The 400 and 700 °C temperatures were chosen in order to observe the behaviour of the different calcium precursors and to evaluate the phase composition obtained, since pure hydroxyapatite had formed at 400 °C and by 700 °C a mixed β-TCP/HAp phase had formed.
Characterisation
The phase composition of the powders was assessed by X-ray powder diffraction (XRD) conducted on a PANalytical X’Pert Pro3 diffractometer (Malvern, The Netherlands), equipped with a fast RTMS detector (PANalytical PIXcel 1D, Malvern, The Netherlands), using Cu Kα radiation (45 kV and 40 mA, 10–80° 2θ range, with a virtual step scan of 0.020° and virtual time per step of 2 s at room temperature). For this analysis, the samples were ground in an agate mortar, until the whole sample passed through a 63 μm sieve. The phase identification was executed using the PANalytical X’Pert High Score Plus PRO3 software. Phase ratios were estimated from the relative peak intensities of the strongest (100%) peaks.
The morphology of the powders was first evaluated by optical microscope (Leica EZ4HD, Germany) and then observed by scanning electron microscopy (SEM, Hitachi SU-70, Tokyo, Japan) with an accelerating voltage of 15 kV. For SEM examination, the samples were placed on aluminium specimen stubs with conducting carbon cement (Leit-C, Conducting Carbon Cement, AGG3300, Germany), dried in the oven at 60 °C and then coated with a thin carbon film by sputtering (Emitech, K950, USA) to provide a conducting layer.
The specific surface area (SSA) of the ceramic samples synthesised with 10 wt% hydrogen peroxide was measured via physical nitrogen adsorption isotherms (outgas temperature: 150 °C) using the Brunauer-Emmett-Teller (BET) method (Micromeritrics Gemini 2370 V5.00, Norcross, GA, USA). The Barrett-Joyner-Halenda (BJH) method was used for mesopore and macropore sizes and distribution analysis, and Density Functional Theory (DFT) for mesopore sizes and distribution.
In-vitro tests of biomedical properties
Simulated body fluid Preparation
In order to prepare 1 L of simulated body fluid (SBF)26, 700 mL of deionised water was placed in a 1 L beaker and set at 37 ± 1.0 °C in a water bath. The reagents were slowly added sequentially, as listed in Table 1, with an accuracy of ± 0.1 mg, under continuous stirring. The pH was monitored throughout the process, maintaining it at 7.4 to avoid any rapid increase, which could cause precipitation. Once all the reagents were mixed, the SBF solution was transferred to a 1 L volumetric flask and filled to the 1 L mark with deionised water. The solution was allowed to cool down to room temperature before being stored in a refrigerator at 5 °C.
Biomineralisation assay with simulated body fluid
The bioactivity of the bioceramics was evaluated by immersing 0.22 mg of each sample in 14.5 mL of SBF in an airtight polyethylene container, previously sterilised for 10 min under a UV lamp (STERIL BOX germicidal UV lamp). The dissolution vessels were incubated in an oven at 37 °C for 1 day, 3 days and 7 days. At the end of each period, the samples were removed from the incubator and the solids were collected by filtration (particle retention 1–2 μm). The collected solids were immediately washed three times with deionised water and the filtered SBF solution was measured to obtain the final pH and temperature. The washed samples were dried in an oven at 37 °C overnight, and then prepared for SEM analysis as described above.
Biocompatibility of the samples: MTT assay
The cytocompatibility of the porous bioceramics was determined using the thiazolyl blue tetrazolium bromide (MTT) assay, using a MG63 osteosarcoma cell line (osteoblasts). Cells were seeded in two different 96-well plates in the presence of the samples, at a density of 5.0 × 103 and 1.0 × 104 per well, and incubated at 37 °C in 5% CO2 for 24 h. The medium was then removed and replaced within a serum-free medium containing 2 mg/mL MTT and incubated for 2 h at 37 °C. The MTT reagent was then removed, and the formazan crystals were solubilised using dimethyl sulfoxide (DMSO). The absorbance of the solution at 570 nm was measured on a microplate reader (CLARIOstar Plus, BMG Labtech). The absorbance of the controls was subtracted, and the percentage control was calculated as the absorbance of the treated cells/control cells. Cell viability was assessed using the equation:
All tests were conducted in triplicate to ensure reproducibility.
Results and discussion
Phase composition of porous bioceramics
The XRD patterns of the samples calcined at 400 °C are shown in Fig. 1a. The samples synthesised using calcium acetate (Ac3-Ac10) are less crystalline, with broader peaks and lower intensities compared to those synthesised from calcium nitrate (Nit3-Nit10), which display more well-defined peaks. The lower crystallinity of Ac3-Ac10 can be attributed to the incomplete decomposition of the acetate, leaving some carbon in the sample, as evidenced by the grey colour observed in Sect. 3.2. However, no carbon peaks were observed in the XRD patterns. The main XRD peak for hexagonal crystalline α-graphite occurs at 26.4° 2θ (JCPDS 75–2078), and a very broad peak would be expected for poorly crystalline graphite. Instead, a sharp peak at around 26° 2θ corresponds to the 002 peak of HAp, consistent with the fourth most intense peak in the HAp standard pattern (JCPDS 04–010-6314) and representing a relative intensity of 35%. Indeed, despite this low calcination temperature, all samples appear to have only one phase (HAp). The XRD patterns obtained for Ac3-Ac10 resemble those previously reported for HAp ceramics produced via this rapid sol-gel route24 when calcined at 400 °C, made with calcium acetate without the addition of pore-forming agents.

(a) The XRD patterns of Ac3-10 and Nit3-10 after heating to 400 °C. (b) shows the patterns of Nit3-10 between 10–55°, with HAp peaks marked and the CaCO3 peak indicated.
The XRD patterns for Nit3-Nit10 powders heated to 400 °C show that they are much more crystalline (Fig. 1a), possibly due to the exothermic combustion of the nitrate salts aiding crystallisation even at only 400 °C. These three samples have very similar XRD patterns, and the only calcium phosphate crystalline phase present is HAp. However, all show a peak, that is not HAp or β-TCP at 29.4° 2θ, which is the major 104 peak of calcite (CaCO3)27. This can be seen in Fig. 1b, showing the XRD patterns of these samples between 10 and 55° 2θ with peaks identified. The strongest CaCO3 peak is observed for Nit10 sample, made using the greatest quantity of H2O2, and is weakest for Nit3. CaCO3 was not detected previously in the HAp made by this rapid sol-gel method using calcium acetate as a precursor. Therefore, it seems to be a feature of using either calcium nitrate (with exothermic decomposition) or the H2O2 additive. The fact that the quantity of CaCO3 appears to be lower in the sample prepared with less H2O2, and the absence of CaCO3 when acetates are used, suggests that a combination of the two factors is the cause, resulting in the formation and precipitation of calcium carbonate in these samples.
Carbonate is present is water in low levels as carbonic acid (H2CO3) from CO2 absorbed from the atmosphere, but the levels are probably too low (~ 10 ppm) to account for a quantity of CaCO3 detectable by XRD. However, calcium nitrate is a strong oxidiser, and when it is combusted in air it forms CaO28, which is itself unstable, and will react with carbon dioxide in the air to form CaCO3. This will be further exacerbated by the presence of H2O2 that is also a strong oxidiser – hence the effect is greater with more H2O2 present, and more CaCO3 is observed as a secondary phase. Therefore, we suggest that this process is responsible for the formation of CaCO3 as a minor secondary phase, while the bulk of the Ca reacts to form HAp at 400 °C. At a higher temperature of 700 °C, all of the Ca reacts to form HAp or β-TCP, hence the absence of CaCO3 at this temperature.
The XRD patterns of the samples calcined at 700 °C are shown in Fig. 2a, and indicate the coexistence of two crystalline phases: HAp and β-TCP (JCPDS 01–073-4869) in all samples. β-TCP can form from non-stoichiometric HAp (Ca: P ratio = 1.67 in HAp and 1.5 in β-TCP) or at higher temperatures, but this is still a relatively low temperature for the formation of β-TCP. However, this seems to be a feature of this rapid sol-gel method. The co-existence of β-TCP was reported before in the literature for HAp made by this method without adding H2O2, and a ratio of β-TCP: HAp of 63:3724. The lower crystallinity of the HAp originally formed favours its transformation into β-TCP at only 700 °C. Similar results have been reported for HAp produced by a different sol-gel method29, and in other HAp ceramics made with the addition of pore-forming agents17.

(a) The XRD patterns of Ac3-10 and Nit3-10 after heating to 700 °C. (b) Shows the patterns of Nit3-10 between 10–55°, with significant (> 5%) HAp and β-TCP peaks marked.
However, both Ac3-10 and Nit3-10 display near-identical XRD patterns when heated to 700 °C, with similar variations in the ratio of HAp to β-TCP with quantity of H2O2 added, and similarly high crystallinity. The peaks with > 5% intensity for HAp and β-TCP have been identified in Fig. 2b – this has only been shown for Nit3-10, as the Ac3-10 patterns are virtually identical. It can be observed that the quantity of β-TCP present varies with the amount of H2O2 added, but not in a linear manner. Nit3 and Ac3 have the lowest amount of β-TCP, with the 50% intensity 214 peak at 28.0° 2θ and the 100% intensity 217 peak at 31.2° 2θ being the clearest markers for β-TCP. However, in Nit5 and Ac5 β-TCP appears to be the major crystalline phase present, as reported for the acetate-derived ceramics without H2O2 addition, with an approximate ratio of 60:40 β-TCP: HAp. For Nit10 and Ac10 there is a considerable amount of β-TCP, but HAp is the major phase with an approximately inverse ratio of 40:60.
This variation is repeated in both series of samples, and so it is unconnected to the calcium salt used for synthesis, and would seem to be due the amount of H2O2 added. It would appear that β-TCP is more soluble in H2O2 (log Ksp value = −28 at 21 °C) than HAp (log Ksp = −117)30. A study of β-TCP for dental implants found that β-TCP could dissolve in H2O2 to recrystallise as needle-like HAp31, whereas HAp was unaffected by H2O2. This study found that a concentrated 30% solution of H2O2 heated to 90 °C was sufficient to completely recrystallise all β-TCP as HAp. Therefore, the results observed in our work suggest that while a 5% addition of H2O2 was not enough to significantly reduce the quantity of β-TCP in Nit5, 10% H2O2 was able to begin partially dissolving β-TCP and recrystallising it as HAp, leading to a lower amount of β-TCP being present in Nit10.
It should also be noted that no traces of remaining CaCO3 were detected in Nit3-Nit10 after heating to 700 °C. This is positive because, while a mixture of β-TCP and HAp can improve bioactivity and resorbability, the presence of CaCO3 or CaO can be detrimental.
Morphology and microstructure of porous bioceramics
Optical microscopy showed that the surface of all the samples was very irregular with pores and cavities of different sizes (Fig. 3). The samples synthesised from calcium nitrate (Nit3-Nit10) were white after heating to 400 °C and 700 °C, while those produced from calcium acetate (Ac3-Ac10) exhibit a brownish-grey colour when heated to 400 °C, becoming white after heating at 700 °C. This colour change is due to the presence of a small quantity of residual carbon from the acetate salt, which remained after heating to 400 °C, but was completely burned out by 700 °C. Larger pores, several hundred microns in diameter, were observed in all samples. The number of pores, induced by the pore-forming agent (H2O2), increased with the quantity of H2O2 added. These pores are of a suitable size range to facilitate cell anchorage and proliferation11,12.

Optical microscope photos of (a) Ac10 after heating 400 °C, (b) Ac5 after heating to 700 °C, (c) Nit5 after heating to 400 °C and (d) Nit5 after heating to 700 °C (scale bar = 500 μm). Inserts have scale bar of 100 μm.
The microstructure of the ceramics heated to 400 °C and 700 °C can be seen in the SEM images presented in Figs. 4 and 5. Ac3-10, after heating to 400 °C, exhibited a network of flat, plate-like particles with poor crystallinity (as also observed in the XRD patterns) covered by much smaller fine plates or needles with nanoscale thickness, which are typical of HAp growth at lower temperatures or initial crystallisation from solution (Fig. 4a, c & e). This explains the highly amorphous appearance of the XRD pattern of Ac3-10 calcined at 400 °C. After heating to 700 °C, both these fine needles and amorphous looking plates transformed into larger, more rounded looking particles or elongated assemblies of these, but still with sub-micron diameters (Fig. 4b), d) & f)). On the macroscale, the appearance of the materials had not significantly changed, and there was no evidence of any sintering (densification) occurring, only grain growth (Ostwald ripening) was observed. No significant differences in grain structure were observed between the ceramics with different quantities of H2O2 added, despite differences in their β-TCP amount. However, Ac5 and Ac10 appeared to contain fewer elongated assemblies.

SEM images of Ac3-10 at 1 K and 20 K magnification: (a) Ac3 400 °C, (b) Ac3 700 °C, (c) Ac5 400 °C, (d) Ac5 700 °C, (e) Ac10 400 °C, (f) Ac10 700 °C.

SEM images of Nit3-10 at 1 K and 20 K magnification: (a) Nit3 400 °C, (b) Nit3 700 °C, (c) Nit5 400 °C, (d) Nit5 700 °C, (e) Nit10 400 °C, (f) Nit10 700 °C.
In contrast, Nit3-10 were already much more crystalline after heating to 400 °C (Fig. 5a, c & e), as also reflected by their XRD patterns. Their overall microstructure appears denser and more compact, but they still consist of nanoscale particles, with a less platy appearance. Upon heating to 700 °C, there was a clear increase in particle size, but still little sintering or further densification (Fig. 5b, d & f). Nit5, with the greatest proportion of the β-TCP phase, appears to have the largest grain size after heating to 700 °C (Fig. 4d), although all are still submicrometric.
This is a preliminary study, and 3%, 5% and 10% H2O2 were chosen arbitrarily as initial levels to be studied. Further addition would be expected to increase porosity, but also further weaken the bioceramics. We will report mechanical tests of actual scaffolds with H2O2 in a follow-up paper dedicated to scaffolds.
The nitrogen adsorption isotherms for the powders with the greatest amount of H2O2 added, Ac10 and Nit10, after heating to 400 °C and 700 °C, are shown in Fig. 6. The N2 adsorption isotherms were generally Type V, with no inflection to indicate monolayer coverage, and a hysteresis which began almost immediately. This suggests that adsorbent-adsorbate interaction was weak, with capillary condensation occurring in mesopores to form the hysteresis loop. All samples displayed a mix of type H1 and H4 hysteresis, typical of mesoporous materials. H1 (vertical parallel loop sides) indicates a narrow range of uniform mesopores with cylindrical pore geometry and good pore connectivity, while H4 (horizontal parallel sides) indicates narrow slit like mesopores32,33. It should be noted that none of the N2 adsorption methods discussed here are capable of measuring the micron-scale pores expected from the addition of H2O2, but mesoporosity and macroporosity will still aid bioactivity and cell adhesion.

Nitrogen adsorption/desorption isotherms and BET specific surface area values for Ac10 and Nit10 heated to 400 °C (a-b) and 700 °C (c-d). The insets show the single point BET isotherms.
We previously observed that Ac10, after heating to 400 °C, still contains residual carbon from the acetate precursor. This is reflected in its high specific surface area (SSA) of 232.33 m²/g, as determined by BET (Fig. 6a), which is attributed to the presence of mesoporous carbon that is known to have very high surface area. Pullar has previously published papers on mesoporous Tuna Bone Char (TBC) from pyrolysed tuna bones, composed of calcium phosphate (HAp) and graphitic carbon. The TBC was a nanostructured material (particle size 30–60 nm), with a surface area of 100.67 m2/g. Hence, even a small amount of mesoporous carbon present would greatly enhance the SSA of the bioceramic samples34.
The BJH SSA was found to be similar, at 194.533 m²/g for adsorption and 210.455 m²/g for desorption, indicating that most of this porosity was indeed from mesopores or smaller macropores up to around 200 nm (the approximate limit of this technique). The t-plot also did not detect any microporosity. The BJH pore volume was 0.558 cm3/g for pores up to 324 nm in diameter, with around 50% of that being in pores under 20 nm in diameter. DFT gave the mesopore volume as 0.390 cm3/g for pores between 2 and 26 nm in diameter, with around 2/3 of that being in pores under 14 nm. Smaller pores have a relatively greater SSA and so contribute more to the total pore area. BJH showed that pores under 20 nm in diameter contributed to approximately 90% of the pore area, while DFT suggested that pores under 25 nm accounted for a pore area of 138 cm2/g, with approximately 90% of that coming from pores under 16 nm in diameter. The hysteresis of the isotherm for Ac10 also more closely resembled the H4 type, which is typical of narrow mesopores. This relatively high SSA and degree of mesoporosity could make Ac3-10 suitable for applications as adsorbents to combat pollutants when heated to 400 °C, retaining a portion of mesoporous carbon along with the well-known cation exchange capabilities of HAp35.
Nit10 contained no source of carbon and, thus, had a considerably lower SSA of 49.78 m2/g (Fig. 6b). The BJH SSA was also very similar at 50.12 m²/g, indicating that this porosity was due to mesopores or smaller macropores. The t-plot also did not detect microporosity. The BJH pore volume was 0.345 cm3/g from pores under 200 nm in diameter, mostly from pores between 20 and 200 nm, which are considerably larger than those in Ac10. DFT gave a mesopore volume of 0.232 cm3/g for pores between 2 and 25 nm in diameter, mostly from pores > 15 nm. However, approximately 80% of the BJH surface area was from pores < 40 nm in diameter, and DFT gave a pore area of around 47 m2/g, with approximately 80% of that deriving from pores < 20 nm in diameter. Therefore, although it lacked the contribution of residual carbon to its SSA, the relatively high SSA of Nit10 was also from mesoporosity.
As can be seen in Fig. 6c), Ac10 heated to 700 °C proved to be difficult to characterise by these techniques, also indicated by the non-linear single point BET plot in the inset of Fig. 6c). Nevertheless, a BET SSA of 21.80 m2/g was obtained, lower than all others and an order of magnitude lower than when heated to only 400 °C. After heating to 700 °C, the sample was white and no longer contained any residual carbon. The BJH pore volume was 0.132 cm3/g for pores under 300 nm diameter, mostly from pores < 20 nm, and DFT gave a mesopore volume of 0.098 cm3/g, also from pores < 20 nm. These pore volume results are less than half those measured for Nit10, suggesting that the low BET SSA value may indeed be accurate. It also matches well the values previously reported for the acetate-derived bioceramics produced by this rapid sol-gel method after heating to 700 °C, without added H2O2, which was 19.1 m2/g24. The pore surface area was almost entirely derived from pores smaller than 15 nm according to BJH, and less than 8 nm from DFT, with no microporosity detected by t-plot. The DFT pore surface area was 36.72 m2/g, greater than the BET value, but still lower than all other ceramics reported here. Although the SSA was lower, the size of the mesopores providing most of the surface area for Ac10 was smaller than for Nit10, and the upper limits (around 15–20 nm diameter) for ~ 90% of the pore area were similar to those measured for Ac10 heated to 400 °C, indicating that the pore structure of the actual ceramic had remained unchanged with an increase in temperature.
This observation is further supported by the results for Nit10 when heated to 700 °C (Fig. 6d), which had a lower BET SSA of 46.03 m2/g, compared to when heated at 400 °C. However, the BJH pore volume was considerably lower, at 0.111 cm3/g from pores under 250 nm diameter, but still mostly from pores between 20 and 200 nm, and DFT gave a mesopore volume of only 0.0605 cm3/g for pores between 2 and 25 nm in diameter, but still mostly from pores > 15 nm. Despite the much lower pore volume values, the size range of the pores contributing to this was similar to that for Nit10 heated to 400 °C, and larger than in Ac10. Similarly, although the values were lower for mesopore area (BJH = 29.86 m²/g, DFT = 16.4295 m²/g), the dimensions of the pores contributing to this had remained the same as when heated to only 400 °C, in the mesopore range (< 20 nm diameter).
These results suggest that the measured SSA of these bioceramics is almost entirely due to mesoporosity. Although SSA may decrease (due to fewer pores), the actual pore structure/dimension does not significantly change with an increase in heating temperature from 400 °C to 700 °C. The unusually high SSA observed for Ac10 after heating to 400 °C can be attributed to residual finely mesoporous carbon.
Bioactivity and biomineralisation
The morphology and structure of the sol-gel derived ceramics, before and after their immersion in simulated body fluid (SBF), were evaluated by observing new HAp crystallisation via SEM. The SEM images taken before, and after 1 and 3 days in SBF, are shown in Figs. 7 and 8. All samples showed evidence of bioactivity with the formation of new HAp on their surface.

Bioactivity tests: SEM images at 10 K magnification of crystallisation of new HAp on Ac3-10, before and one and three days after immersion in SBF. (a) Ac3 400 °C, (b) Ac5 400 °C, (c) Ac10 400 °C, (d) Ac3 700 °C, (e) Ac5 700 °C, (f) Ac10 700 °C.

Bioactivity tests: SEM images at 10 K magnification of crystallisation of new HAp on Nit3-10, before and one and three days after immersion in SBF. (a) Nit3 400 °C, (b) Nit5 400 °C, (c) Nit10 400 °C, (d) Nit3 700 °C, (e) Nit5 700 °C, (f) Nit10 700 °C.
For Ac3-10 samples heated to 400 °C (Fig. 7a-c), the formation of a poorly crystalline HAp coating was observed after 1 day. By day 3, this coating had become a more crystalline coverage of smaller needles or larger plate-like crystal structures, oriented in different directions. At higher magnification it was seen that the plate-like structures were made up of assemblies of smaller sub-micron needles. No significant differences were observed between the three samples. After heating to 700 °C, Ac3-10 had more crystalline plates or elongated needles, and these were also covered by a coating of fine HAp needles after one day, and forming the “flower-like” formations of needles often observed for the crystallisation of HAP after 3 days in SBF (Fig. 7d-f). These “flower-like” formations appeared much more predominant in Ac5 and Ac10, which consisted of around 60% and 40% β-TCP, respectively, suggesting that the presence of this phase favoured the formation of these structures over the more plate-like form.
As discussed above, Nit3-10 was already much more crystalline after heating to 400 °C, and after immersion in SBF the formation of larger, more crystalline and less needle-like HAp was observed when compared to Ac3-10 (Fig. 8a-c). A greater quantity of these new crystals could be observed after 3 days in SBF for all three Nit3-10 samples. However, they appeared to more plate-like for Nit5 and Nit10, and greater in size for Nit10, being nanoscale in thickness but up to several microns in diameter. XRD showed an increasing presence of CaCO3 in Nit5 and Nit10 when heated to 400 °C, and the calcite phase seems to have encouraged the formation of these larger plates compared to the other samples. After heating to 700 °C, Nit3-10 appeared to favour the growth of new HAp similar that observed for Ac3-10, with the formation of many submicron HAp needles after 1 day, and the “flower-like” structures after 3 days (Fig. 8d-f). Once again, there appeared to be significantly more of these “flower-like” structures for Nit5 and Nit10, which contained a significant amount of β-TCP. The CaCO3 phase was no longer present, and the larger plates were also absent.
Therefore, to summarise these results:
-
All the materials exhibited considerable bioactivity, evidenced by the crystallisation of new HAp on their surfaces after only 1 day of immersion in SBF, forming fine needles or plates.
-
Ceramics containing only HAp as the calcium phosphate phase formed more plate-like crystals after 3 days in SBF (Ac3-10 and Nit 3–10 heated to 400 °C).
-
The presence of CaCO3 appeared to favour the formation of more, and larger, plates (Nit3-10 heated to 400 °C).
-
The coexistence of β-TCP with HAp in significant quantities (40–60%) favoured the formation of the “flower-like” assemblies of submicron needles after 3 days in SBF (Ac5, Ac10, Nit5 and Nit10 heated to 700 °C).
In-vitro biocompatibility/cytotoxicity tests
The cytocompatibility of the ceramics heated to 400 °C and 700 °C was measured by performing the MTT assay on the MG63 osteosarcoma cell line. The MTT assay is a colorimetric test for assessing cell metabolic activity via oxidoreductase enzymes in the cell mitochondria, which reduce the soluble 3-(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide (MTT) into insoluble formazan, detectable by its purple colour. The amount of formazan produced is directly proportional to the number of living cells present in the culture, used to measure cytotoxicity (loss of viable cells) or biocompatibility (proliferation of cells). It is generally accepted that below 70% viable cells remaining after the test indicates a cytotoxic material.
Initial results showed the presence of some interference signals (high absorption) in the samples heated to 400 °C that were due to the presence of chemical residues, such as nitrate ions. Therefore, the samples were washed in deionised water to remove any ions present before the MTT tests. These results are shown in Fig. 9.

MTT biocompatibility/cytotoxicity tests with MG63 osteoblasts after 24 h, for Nit3-10 and Ac3-10 after heating to 700 °C and 400 °C. The dashed line shows the minimum 70% limit for % cell viability to be considered non-cytotoxic.
It can be seen that all samples were biocompatible, with cell viabilities exceeding 70%, except for Ac5 and Ac10 heated to 400 °C. This is probably due to the presence of residual carbon in these samples. However, the cell viability of all acetate-derived samples decreases with increasing quantities of H2O2 added, although the exact reasons for this is unclear. It is possible that the hydrogen peroxide has an activating effect on the carbon, increasing the level of cytotoxicity with the amount added. A similar trend was observed for Ac3-10 heated to 700 °C (although they have greater % cell viabilities), despite the absence of residual carbon. All of the Nit3-10 samples are biocompatible, with those heated to 400 °C (HAp with some CaCO3) showing superior results compared to those heated at 700 °C (mixed HAp and β-TCP). These results suggest that a dominant calcium phosphate HAp phase (Nit3 and Ac3) provides better cytocompatibility than a more evenly mixed HAp/β-TCP material (Nit5, Nit10, Ac5 and Ac10 heated to 700 °C). Indeed, the lowest cell viability for the nitrate-derived ceramics was observed for Nit5 heated to 700 °C, which contains the greatest amount of β-TCP.
These results suggest that it is the phase composition (presence of carbon or β-TCP), rather than microstructure, that influences the biocompatibility of these ceramics. Nevertheless, all but two were cytocompatible with osteocytes, and Ac3 and Nit3 actively promoted cell proliferation, with cell viabilities above 100% for Nit3 heated to 400 °C (110%) and 700 °C (139%), and Ac3 heated to 700 °C (127%). Despite the presence of induced pores several hundred microns in diameter, a suitable size range to encourage cell anchorage and proliferation, and the number of these increasing with the quantity of H2O2 added, the phase composition appears to be the most important factor for determining cytocompatibility in these bulk ceramics. The next step will be to produce 3D printed (robocast) scaffolds from these ceramic powders, where the effects of the pore-forming agent on the micron-scale porosity can be truly assessed in a rigid scaffold structure.
Conclusions
Bioceramics were produced using a rapid sol-gel process that avoids the long aging times usually required for sol-gel derived HAp, using calcium acetate and, for the first time, calcium nitrate, as precursors. These ceramics also incorporated 3, 5 and 10 wt% H2O2 as a pore-forming agent for the first time, and were formed as bulk ceramic hemispheres and heated to 400 °C and 700 °C. Induced macropores, ranging from 100 to 500 μm, were observed in all samples, increasing in number with the quantity of H2O2 added. This macropore size range is suitable to encourage cell anchorage and proliferation.
When heated to 400 °C, the only calcium phosphate phase present was HAp. Acetate derived samples produced a less crystalline material also containing residual carbon, while nitrate derived ceramics also contained CaCO3 as a secondary phase. Upon heating to 700 °C, all samples exhibited mixed phases of HAp and β-TCP, with HAp being the majority phase with 3% H2O2, and between 60 − 40% β-TCP with 5% and 10% H2O2 added. Carbon and CaCO3 were completely removed after heating to 700 °C, and all samples displayed a similar microstructure characterised by irregular or elongated particles of a few hundred microns, with a high degree of submicron porosity. Nitrogen adsorption showed that mesopores were the main source of porosity below 200 nm, with approximately 90% of the pore area coming from pores between approximately 2–20 nm in diameter. The SSA of the nitrate-derived ceramics ranged from 46 to 49 m2/g, while the acetate-derived samples had a lower SSA of 21 m2/g when heated to 700 °C, but a large area of 232 m2/g when heated to only 400 °C due to residual carbon from the acetate being present.
Bioactivity tests via submersion in SBF for 1 and 3 days demonstrated considerable biomineralisation in all samples. After only 1 day in SBF, new HAp had crystallised on the surface in the form of fine needles or plates. After 3 days, ceramics containing only HAp as the calcium phosphate phase formed more plate-like crystals, and the presence of CaCO3 appeared to favour the formation of more, larger plates. The coexistence of β-TCP with HAp in significant quantities (40–60% β-TCP) favoured the formation of “flower-like” assemblies of submicron needles after 3 days in SBF. All samples were biocompatible, with cell viabilities exceeding 70%, except for the acetate-derived samples with 5–10 wt% H2O2 added and heated to only 400 °C. Cell viabilities for osteoblasts between 110 and 139% were observed for samples with 3 wt% H2O2 added derived from nitrates, or those from acetates heated to 700 °C, without any carbon present. However, although the induced pores were within a suitable size range to encourage cell anchorage and proliferation, and their number increased with the quantity of H2O2 added, the phase composition appears to be the most important factor for determining cytocompatibility in these bulk ceramics. Indeed, the presence of carbon or β-TCP, rather than microstructure, seems to have the greatest influence on their biocompatibility.
The next step will be to 3D print robocast scaffolds from these ceramic powders, to assess the effects of the pore-forming agent on a rigid scaffold structure.
Data availability
Data is available upon request. Please contact the corresponding author, R.C. Pullar (robertcarlyle.pullar@unive.it), for matters regarding this.
References
Ho-Shui-Ling, A. et al. Bone regeneration strategies: engineered scaffolds, bioactive molecules and stem cells current stage and future perspectives. Biomaterials 180, 143–162 (2018).
J Miron, R. Optimized bone grafting. Periodontol 2000. 94, 143–160 (2024).
Hasan, A. et al. Advances in osteobiologic materials for bone substitutes. J. Tissue Eng. Regen Med. 12, 1448–1468 (2018).
Lichte, P. et al. Scaffolds for bone healing: concepts, materials and evidence. Injury 42, 569–573 (2011).
El-Ghannam, A. & Ducheyne, P. Bioactive ceramics in Comprehensive Biomaterials; (ed Ducheyne, P.) 157–179 (Elsevier, (2011).
Jeong, J. et al. Bioactive calcium phosphate materials and applications in bone regeneration. Biomater. Res. 23 https://doi.org/10.1186/s40824-018-0149-3 (2019).
Di Bello, C. & Bagno, A. Biomateriali. Dalla scienza dei materiali alle applicazioni cliniche, second edition, Ch. 4, 149–158 and Ch. 8, 235–253 (Pàtron, Bologna, (2016).
Huang, H. et al. Preparation of multigradient hydroxyapatite scaffolds and evaluation of their osteoinduction properties. Regen Biomater. 9, rbac001. https://doi.org/10.1093/rb/rbac001 (2022).
Tripathi, G. & Basu, B. A. Porous hydroxyapatite scaffold for bone tissue engineering: Physico-Mechanical and biological evaluations. Ceram. Int. 38, 341–349 (2012).
Lee, H. et al. The production of porous hydroxyapatite scaffolds with graded porosity by sequential Freeze-Casting. Materials 10, 367. https://doi.org/10.3390/ma10040367 (2017).
O’Brien, F. J. Biomaterials & scaffolds for tissue engineering. Mater. Today. 14, 88–95 (2011).
Karp, J. M. et al. Scaffolds for tissue engineering. MRS Bull. 28, 301–306. https://doi.org/10.1557/mrs2003.85 (2003).
Lutzweiler, G. et al. The overview of porous, bioactive scaffolds as instructive biomaterials for tissue regeneration and their clinical translation. Pharmaceutics 12, 602. https://doi.org/10.3390/pharmaceutics12070602 (2020).
Orlovskii, V. P. et al. Hydroxyapatite and Hydroxyapatite-Based ceramics. Inorg. Mater. 38, 973–984. https://doi.org/10.1023/A:1020585800572 (2002).
Bhat, S. et al. Functionalized porous hydroxyapatite scaffolds for tissue engineering applications: A focused review. ACS Biomater. Sci. Eng. 8, 4039–4076. https://doi.org/10.1021/acsbiomaterials.1c00438 (2022).
Bystrov, V. S. et al. Oxygen vacancies, the optical band gap (Eg) and photocatalysis of hydroxyapatite: comparing modelling with measured data. Appl. Catal. B Environ. 196, 100–107 (2016).
Uchida, A. et al. Bone ingrowth into three different porous ceramics implanted into the tibia of rats and rabbits. J. Orthop. Res. 3, 65–77. https://doi.org/10.1002/jor.1100030108 (1985).
Uchida, A. et al. Slow release of anticancer drugs from porous calcium hydroxyapatite ceramic. J. Orthop. Res. 10, 440–445. https://doi.org/10.1002/jor.1100100317 (1992).
Slosarzyk, A. et al. Porous hydroxyapatite ceramics. J. Mater. Sci. Lett. 19, 1163–1165 (1999).
Yamasaki, N. et al. Porous hydroxyapatite ceramics prepared by hydrothermal Hot-Pressing. J. Mater. Sci. Lett. 9, 1150–1151 (1990).
Tanner, K. E. Bioactive ceramic-reinforced composites for bone augmentation. J. R Soc. Interface. 7, S541–S557 (2010).
Engin, N. O. & Cüneyt Taş, A. Preparation of porous Ca10(PO4)6(OH)2 and β-Ca3(PO4)2 bioceramics. J. Amer Ceram. Soc. 83, 1581–1584 (2000).
Sepulveda, P. & et at. Properties of highly porous hydroxyapatite obtained by the gelcasting of foams. J. Amer Ceram. Soc. 83, 3021–3024 (2000).
Ben-Arfa, B. A. E. et al. Novel route for rapid Sol-Gel synthesis of hydroxyapatite, avoiding ageing and using fast drying with a 50-Fold to 200-Fold reduction in process time. Mater. Sci. Eng. C. 70, 796–804 (2017).
Ben-Arfa, B. A. E. et al. A hundred times faster: novel, rapid sol-gel synthesis of bio-glass nanopowders (Si-Na-Ca-P system, Ca:P = 1.67) without ageing. Int. J. Appl. Glass Sci. 8, 337–343 (2017).
Maçon, A. L. B. et al. A unified in vitro evaluation for Apatite-Forming ability of bioactive glasses and their variants. J. Mater. Sci. : Mater. Med. 26, 115. https://doi.org/10.1007/s10856-015-5403-9 (2015).
Zhou, G-T. et al. Sonochemical synthesis of Aragonite-Type calcium carbonate with different morphologies. New. J. Chem. 28, 1027–1031 (2004).
Zhao, A. et al. Thermal decomposition paths of calcium nitrate tetrahydrate and calcium nitrite. Thermochim. Acta. 714, 179264. https://doi.org/10.1016/j.tca.2022.179264 (2022).
Feng, W. et al. A simple sol-gel technique for Preparing hydroxyapatite nanopowders. Mater. Lett. 59, 916–919 (2005).
Fernandez, E. et al. Calcium phosphate bone cements for clinical Applications—Part I: solution chemistry. J. Mater. Sci. : Mater. Med. 10, 169–176. https://doi.org/10.1023/A:1008937507714 (1999).
Cuneyt Tas, A. Formation of calcium phosphate whiskers in hydrogen peroxide (H2O2) solutions at 90°C. J. Am. Ceram. Soc. 90, 2358–2362. https://doi.org/10.1111/j.1551-2916.2007.01768.x (2007).
Sing, K. S. W. et al. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 57, 603–619 (1985).
Thommes, M. et al. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC technical Report). Pure Appl. Chem. 87, 1051–1069 (2015).
Miranda, C. et al. Exploring the potential of a Waste-derived bone Char for pharmaceuticals adsorption in Saline-Based wastewater. Sustain. Chem. Pharm. 42, 101761. https://doi.org/10.1016/j.scp.2024.101761 (2024).
Piccirillo, C. et al. Biphasic apatite-carbon materials derived from pyrolysed fish bones for effective adsorption of persistent pollutants and heavy metals. J. Environ. Chem. Eng. 5, 4884–4894. https://doi.org/10.1016/j.jece.2017.09.010 (2017).
Acknowledgements
Anna Bertocco thanks the Cà Foscari University of Venice Erasmus+ scheme for funding a period of research at the University of Aveiro.Marinélia Capela, Ana P. F. Caetano and Maria Paula Seabra are grateful to the project CICECO – Aveiro Institute of Materials, UIDB/50011/2020, UIDP/50011/2020 and LA/P/0006/2020, financed by national funds through the FCT/MEC (PIDDAC).
Author information
Authors and Affiliations
Contributions
AB: Synthesis of samples; acquisition, analysis and interpretation of data; revision of manuscriptMC: Acquisition, analysis and interpretation of dataAPFC: Acquisition, analysis and interpretation of dataAN: Acquisition, analysis and interpretation of dataAQ: Supervision; analysis and interpretation of dataMPS: Supervision; acquisition, analysis and interpretation of dataRP: Conception; supervision; analysis and interpretation of data; revision of manuscriptAB and RP wrote the main manuscript text, AB and RP prepared Figs. 1, 2, 3, 4, 5, 6, 7 and 8, RP and AN prepared Figure 9. All authors reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bertocco, A., Capela, M., Caetano, A.P.F. et al. Porous hydroxyapatite – β-tricalcium phosphate ceramics produced from a rapid sol-gel process. Sci Rep 15, 16422 (2025). https://doi.org/10.1038/s41598-025-01253-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-01253-2
