
Abstract
Amaranth, a gluten-free pseudo-cereal, is grown, cultivated, and adapted in diverse ecological zones all over the world. In recent years, increased attention has been paid on its nutritional eminence, particularly in terms of its seed protein, which specifically contains high levels of the essential amino acids. This important food and nutritional security crop’s gene pool, which is diverse, needs to be characterized and genotyped in order to broaden its genetic base and to boost production. Genotyping by sequencing data of diverse germplasm serve useful tool for identification of important candidate genes associated with important traits of plants. In the present study, genotyping-by-sequencing (GBS) approach was used to characterize a genetically diverse collection of 192 Amaranth accessions. This resulted in an estimated 41,931 single-nucleotide polymorphisms (SNPs) segregating across the entire collection and several thousand SNPs segregating within every accession. A model-based population structure analysis reveals the presence of three subpopulations among the Amaranth accessions, which are in parallel with the results of phylogenetic analysis. A total of 1796 gene ontology (GO) terms were assigned to SNP-carrying genes for three main categories: biological process, cellular component, and molecular function. High-throughput genotyping and sequencing data generated, will be very useful not only for breeders for further enhancement of Amaranth but also for molecular biologists for isolation and identification of nutritionally important genes from Amaranth, which can be used for biofortification programs.
Similar content being viewed by others

Introduction
Out of the several thousand known edible plant species, just over 30 crops provide 95% of the world food supplies and four major crops viz.; rice, maize, wheat and potato provide 60–70% global dietary requirements1. The shrinking portfolio of world food basket raises serious concerns on how a few major crops can ascertain the food and nutritional security of the ever-increasing population. Further, these major crops (rice, wheat maize and potato) are the reservoir of carbohydrates, providing high amount of calories but lacking the essential amino acids and minerals for a balanced nutrition2. Therefore, diversifying the food basket by incorporation of long-forgotten ancient pseudocereals with unparalleled nutritional profile is eventually a leading strategy to address the global nutritional security and combat the health infirmities and prevalent hidden hunger3.
Grain amaranth, Amaranthus hypochondriacus, is one such pseudocereal with tremendous but under-explored source of nutraceutical properties and has the potential to diversify the cropping systems around the world. Amaranth grains are known for high concentration of essential amino acids, gluten-free protein, low glycemic index and exceptional mineral profile compared to the major crops4. Additionally, amaranth is the rich source of several functional compounds and bioactive flavonoids, including squalene, tocopherols, tocotrienols, sterols and other lipophilic constituents5. Several therapeutic studies have confirmed the role of these bioactive compounds in reducing the risk of cardiovascular disease, cancer, obesity and type 2 diabetes6,7,8. Furthermore, the C4 photosynthetic pathway and ability to withstand harsh environmental conditions and fragile ecosystems makes this crop an ideal candidate for climate resilient agriculture and an alternative food grain for the development of sustainable agriculture systems.
Despite its immense potential, efforts in order to bred highly nutritive and productive cultivars for the amaranth have been limited. Critical impediments hampering the progress of breeding for agronomic and nutraceutical traits in grain amaranth are the poorly characterized germplasm collections at the genome level and the undeciphered genetic mechanisms governing these traits. Only scattered germplasm holdings of grain amaranth are maintained in in situ conditions across the world1. To some extent, these collections have been assessed for morphological and agronomic traits in different agro-ecological conditions and utilized in breeding programs. Genetic diversity has been investigated in these collections with a variety of genetic markers; however, most studies are small scale, utilizing only a few number of PCR based markers9,10,11,12.
High-throughput genomic technologies can now be applied to crops such as Amaranth13,14, which are still considered subsistence crops, to analyze molecular markers, transcript sequences, gene space, genome structural variations, and genome sequences. Low sequencing costs have facilitated the development of GBS, a high-density marker genotyping technology that is robust, cost-effective, and highly multiplexed. Studies of genetic variation in a wide variety of species are possible using GBS by analyzing a large number of SNPs. There are several advantages to using SNPs in plant genetic analysis, including their widespread genomic distribution, co-dominant inheritance, reproducibility, and location on particular chromosomes15,16,17,18,19, and20. High throughput technologies of sequencing and genotyping are expected to improve breeding progress in developing countries in several ways. For instance, these technologies facilitate selection of parents for new crosses and enable introduction of novel alleles from exotic germplasm through high-resolution germplasm fingerprinting with low coverage sequencing or SNP genotyping. Because of lack of characterization of the amaranth gene pool at genome level, it is difficult to find genetically diverse accessions that have beneficial traits to be used in breeding programs. Furthermore, the crop has not been extensively investigated for understanding its diversity, taxonomy, and evolution at genome level. To date, there are only few reports on utilization of next generation SNP markers for determining genetic structure of 21 amaranth diversity collections21,22,23. It has been realized that a detailed genome level characterization of genetic diversity and population structure analysis of the diverse amaranth germplasm collections would provide insights on genetic relationship between the different accessions.
In the present study, we used GBS technology to genotype a diverse amaranth germplasm collection. The objectives of this study were to: (1) generate SNP markers by GBS technology and evaluate their characteristics; (2) determine the population structure and genetic diversity of the amaranth diversity panel and (3) Functional annotation of the generated SNPs and assigning the gene ontology (GO) terms to SNP-carrying genes in amaranth. Such studies would provide comprehensive knowledge on the value of locally adapted and exotic germplasm and allow for its effective utilization in breeding programs to develop nutritionally rich and stress tolerant cultivars.
Results
High-Throughput Genotyping of Amaranthus hypochondriacus accessions was performed by Genotyping-by-sequencing analysis using 192 genotypes, which comprised of a set of 120 (IC) accessions from India, 69 (EC) accessions and 3 NAIP accessions from different regions of the world (Fig. 1). ApeKI restriction enzyme was used for preparing the library because it is methylation sensitive. Methylation-sensitive restriction enzymes have been shown to be effective in selecting genomic DNA for exonic gene-containing regions and reducing genomic regions with repeat elements24. Single-nucleotide polymorphisms were called using the TASSEL GBS pipeline25.

Global geographical distribution of 192 accessions of Amaranthus hypochondriacus used in the present study using GBS.
The present study utilized version 2.1 of the Amaranthus hypochondriacus reference genome, sourced from (https://phytozome-next.jgi.doe.gov/info/Ahypochondriacus_v2_1). UNEAK commands are run as TASSEL plugins via the command line (Linux or Mac operating systems). Around 400 million raw reads were generated and filtered. A detailed final SNP statistic, showing number of heterozygous, homozygous and total single-nucleotide polymorphisms (SNPs) per accession is given in Supplementary Table S1. 6,588,854 SNPs were identified among the accessions, of which 5,477,353 were homozygous and 1,111,501 were heterozygous.
The SNP having maximum frequency of allele occurrence is G/C with an overall allele occurrence of 10,041, with a frequency of about 23.95 per cent. The SNP having least frequency of occurrence is T/G with an overall occurrence of 1017, with a 2.42 per cent frequency. Single-nucleotide polymorphism statistics in the entire dataset showing number and percentage frequency of allele occurrence is detailed in Table 1.
In plant genomes, due to biased mutational processes (e.g. cytosine deamination), the ratio of transitions to transversions for bona fide polymorphisms is expected to be much greater than one. In the present study, total number of transitions were 14,488 as compared to 27,327 number of transversions resulting in the transition/transversion ratio of 0.54 for Amaranth which represents a bias towards transversion type of base substitutions. In previous studies, this ratio has been reported as high as 2.5 in maize26 and 2.4 in Arabidopsis sp27 which is due to higher rate of transition than that of transversion. Hoshikawa et al. (2023) have reported this ratio as 1.28 for SNP data of Amaranth tricolor accessions reflecting higher rate of transition than transversion23. The data regarding the rates of transitions and transversions for SNP data of grain Amaranth accessions used in the present study are graphically represented in Fig. 2.

The rates of various substitution mutations grouped into transitions and transversions.
Chromosome 1 contained the highest number of SNPs, totaling 4361, whereas chromosome 16 had the lowest count, with only 1315 SNPs. 363 SNPs were present within the unplaced contigs of the Amaranthus hypochondriacus. The total number of SNPs present in each of the chromosomes is given in Fig. 3a. A heat map portraying SNP density per chromosome at one Mb window size is shown in Fig. 3b.

(a) The total number of SNPs in each chromosome of A. hypochondriacus. (b) Heatmap portraying the densities of SNPs on individual chromosomes within one Mb window size.
A triangle plot for pair wise LD between marker sites was created. To visualize LD throughout the genome, heat maps were produced based on pair wise R2 estimates, and corresponding p-values were calculated using permutations for all marker pairs (Fig. 4a). The R2 values for each marker pair are presented in the bottom half of the heat map and are represented by shades of red, increasing in intensity in equal increments of 0.1 from 0.0 (white) to 1.0 (red). Haplotype block analysis was performed using LD values calculated for each marker pair in TASSEL 5.0, followed by analysis using R Studio. The average size of haplotype blocks was estimated at 295,883 bp (Fig. 4.b). This estimation was based on the LD decay threshold of r² = 0.1, which is widely used in population genomics studies as it represents a balance between detecting meaningful associations and accounting for background LD28,29.
The linkage disequilibrium decay was calculated using the linkage disequilibrium values as revealed by TASSEL 5.0. The average size of haplotype blocks was estimated at 295,883 bp with the help of the LD values per marker pair obtained from TASSEL 5.0 followed by analysis using R Studio (Fig. 4b).

(a) Linkage Disequilibrium Plot. Markers were ordered on the x- and y-axes based on genomic location; hence, every cell of the heat map represents a single marker pair. (b) Linkage Disequilibrium (LD) decay plot across the genome. The x-axis represents the physical distance between SNP pairs in base pairs, while the y-axis shows the LD measure (r²). Each point represents a pair wise SNP comparison. The dark blue line indicates the LD decay threshold of r² = 0.1, which was used to estimate the average haplotype block size of 295,883 bp. The vertical (green) line signifies the average LD decay distance.
Phylogenetic analysis
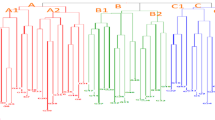
In the present study, the genetic diversity analysis was done among the global collection of 192 Amaranth genotypes. The phylogenetic tree was created through neighbor-joining method of TASSEL software. In the resulting dendrogram, the genotypes segregated into three major clusters A, B and C (Fig. 5). Cluster C consisted of only the Indian genotypes, Clusters B also consisted of Indian genotypes except one exotic genotype whereas Cluster A was comprised of exotic and Indigenous genotypes. Cluster A, the largest cluster, contained 159 accessions; 68 out of the total 69 exotic accessions were present in this cluster along with 90 indigenous genotypes. Cluster A can be broadly categorized into 11 sub clusters, namely A1, A2, A3, A4, A5, A6, A7, A8, A9, A10, and A11. A1 consisted of eight genotypes, all of which were indigenous accessions: 1 from Gujarat, 5 from Maharashtra, and 2 from Himachal Pradesh. A2 consisted of 7 accessions: 3 exotic accessions (1 from China, 1 from Germany, and 1 from France) and 4 indigenous accessions (2 from Maharashtra and 2 from Himachal Pradesh). A3 consisted of 13 accessions, of which 6 were exotic: one each from Russia and Germany, and 2 each from China and U.S.A. The remaining 7 indigenous genotypes were: 4 from Maharashtra, and one each from Meghalaya, Himachal Pradesh, and Uttarakhand. A4 consisted of 8 accessions, of which 3 were exotic accessions (one each from U.S.A., Germany, and Poland) and 5 indigenous accessions (one each from Uttarakhand and Himachal Pradesh, and 3 from Maharashtra). A5 cluster contained 2 indigenous accessions from unknown locations. A6 consisted of 24 accessions, of which the majority were exotic (17) – 6 from Germany, 5 from U.S.A., 4 from Russia, and one each from France and Latvia. It also contained 7 indigenous accessions: 2 each from Himachal Pradesh and Jammu & Kashmir, and one each from Madhya Pradesh, Uttarakhand, and Meghalaya. A7 consisted of 12 accessions; of which five were exotic (4 from the U.S.A., 1 from Germany) and 7 were indigenous (1 each from Uttarakhand, Himachal Pradesh, Sikkim, Madhya Pradesh, Meghalaya, and 2 from Maharashtra). A8 consisted of 2 accessions: one exotic genotype (Germany) and one indigenous genotype (Himachal Pradesh). Sub-cluster A9 consisted of 5 accessions, of which 2 were exotic (one each from Kenya and Germany) and 3 were indigenous (2 from Himachal Pradesh and 1 from Madhya Pradesh). Sub-cluster A10 consisted of 21 accessions, of which 5 were exotic: 3 from the U.S.A., and 1 each from Latvia and Benin. Among the 16 indigenous accessions, 5 were from Maharashtra, 4 from Madhya Pradesh, 3 from Himachal Pradesh, and one each from Sikkim, Gujarat, Uttarakhand, and Karnataka. Sub-cluster A11 contained the highest number of accessions among the subclusters of cluster A, with 57 accessions, including the highest number of exotic genotypes (26): 8 from U.S.A., 5 from Russia, 3 each from China and Netherlands, 2 from Germany, 2 from Latvia, and one each from South Africa, Nepal, and Poland. Among the indigenous accessions, there were 3 from Uttarakhand, 12 from Himachal Pradesh, 9 from Maharashtra, 2 from Sikkim, 3 from Gujarat, and one each from Rajasthan and Madhya Pradesh.
Cluster B contained 26 genotypes, which are all indigenous. This cluster included 10 genotypes from Maharashtra, 4 from Sikkim, 2 each from Uttarakhand, Himachal Pradesh, Madhya Pradesh, and Gujarat, and one each from Karnataka, Goa, and Chhattisgarh. One accession’s location was unknown.
Cluster C comprised seven A. hypochondriacus genotypes; all except one were indigenous genotypes. The exception was an exotic genotype, EC-120,051, from China. The indigenous genotypes were from Maharashtra, Uttarakhand, and Madhya Pradesh.

Phylogenetic tree of 192 A. hypochondriacus accessions of this present study generated through neighbor-joining method of TASSEL software.
Population structure analysis
To infer the right number of subpopulations (K), k was set from 1 to 10 and 3 runs for each k value were performed. In each run, a burn-in of 5000 iterations was followed with an extra 50,000 replications. To choose the best k value, an impromptu statistic ΔK based on the rate of change in the log probability of data between successive K values was used. The maximum ΔK value was observed at K = 3. The inferred ancestry at K = 3 suggested that the Amaranthus genotypes were grouped into three subpopulations (Fig. 6). As expected, there was a strong correlation between the geographic pattern observed in the phylogenetic analysis and the population structure identified using STRUCTURE software with some prominent exceptions. Since the ancestries of most of the genotypes were not known earlier, an admixture model was used to infer their ancestry. A model, which allows for hybrids is more flexible and often provides a better fit for complicated population structure. Figure 7 displays the proportional allocations of each of 192 A. hypochondriacus accessions to K ancestral gene pools inferred using ADMIXTURE30, under K = 3, shown as a bar plot.

Identification of the appropriate subpopulation number (K): Subpopulation number (K) against delta K and the maximum K value observed at K = 3.

Three subpopulations of 192 accessions based on ADMIXTURE (Alexander et al., 2015) each rectangular bar represents one copy of the accession, and its probability of being classified into one of the three subpopulations can be determined by the length of the differently colored (red, yellow, green, blue) lines. Y-axes, Q value; X-axes, the ID of each accession.
The discriminant analysis of principal components (DAPC)
Two-dimensional DAPC of 192 accessions has further confirmed the separation of different accessions of A. hypochondriacus into three major clusters. The cluster 1, remained a distant isolated group, whereas, some amount of overlapping or admixture was observed between clusters 2 and 3 as per their phylogenetic relatedness (Fig. 8.).

DAPC of 192 Amarathus hypochondriacus accession based on 41,931 filtered single nucleotide polymorphisms. The axes represent the first two principal components (PCs).
The multidimensional scaling analysis conducted was conducted on the SNP data by grouping the accessions from five different countries and two continents and labeling them in separate colors (Fig. 9). The plot revealed again that the entire population separated into three separate groups, with some admixtures. All the members from the African continent were present within cluster 1. The remaining accessions did not form any differentiation based on their geographical location and were found to be present in all the three clusters.

Multidimensional scaling analysis to study population structure of A. hypochondriacus accessions from five countries and two continents.
Genetic diversity and population differentiation analysis
Various indices of genetic diversity within accessions among the five countries (China, Germany, India, Russia and the United States of America) and two continents (Africa & Rest of Europe) analyzed are given in Table 2. India displayed the highest segregation sites (35556), contrasting with Russia, which exhibited the lowest (31103). Nucleotide diversity (π) ranged from 0.183 (Africa) to 0.309 (Rest of Europe). The Watterson estimator values spanned from 0.232 (India) to 0.413 (United States of America). Analysis of expected (He) and observed (Ho) heterozygosity revealed discrepancies within populations, the variations ranging from 0.022 (China) 0.104 (Russia). Tajima’s D values were consistently negative, ranging from − 0.008 in India to −0.185 in China. Systematic inbreeding coefficient (FIS) ranged from 0.056 in Africa to 0.412 in Russia. Similarly, within India (Table 3), the Himalayan Region displayed the most segregation sites (39343), while the North-East Region displayed the fewest (31924). Nucleotide diversity (π) ranged from 0.1677 in the North-East Region to 0.2455 in Central India. Watterson estimator values varied from 0.258 in the North-East Region to 0.294 in Central India. The difference between expected and observed heterozygosity varied from 0.026 (Himalayan region) and 0.081 (Central India). Tajima’s D values were consistently negative, with the highest observed in Central India (−0.048) and the lowest in the North Eastern Region (−0.090). The FIS was lowest in the Himalayan Region (0.169) and highest in Central India and the Southern and Western Region (0.344).
The pairwise genetic distances (above diagonal) and fixation indices FST (below diagonal) between five countries and 2 continents were calculated between the five countries and two continents and are presented in Table 4. Pairwise FST values ranged from 0.005 (Germany-India) to 0.076 (Africa-Russia). Nei’s genetic distances (D) varied from 0.015 (China-India & India - USA) to 0.094 (Africa-Rest of Europe& Africa - Russia). The Pair wise FST and Nei’s genetic distance among four regions within India were also calculated (Table 5). Pair wise FST values ranged from 0.007 (Central Region–Southern and Western region) to 0.028 (Himalayan region–North Eastern Region). Nei’s genetic distance (D) ranged from 0.004 (Central Region–Southern and Western Region) to 0.019 (Central Region–North Eastern Region).
Functional annotation of Single-Nucleotide polymorphisms
GO terms were assigned to A. hypochondriacus SNP-carrying genes, with proteins annotated in GO terms. A total of 2,815 GO terms were collected with the most frequent terms related to biological processes (1107) followed by molecular function (984) and cellular components (724). The gene ontology categorization of SNP-carrying genes into three main categories is shown in Fig. 10.

Gene ontology annotation categorization.
Among the biological processes, leaving out the unknown and uncharacterized genes, maximum number of genes corresponded to metabolic process (39.65%) and cellular process (30.8%), followed by localization (8.03%). In case of molecular function, binding and catalytic activity proteins were found to have the highest share with 43.5% and 43.2%. In cellular component category, it’s in the proportion of cell (19.06), cell part (19.06), membrane (17.54), organelle (13.25), protein contain complex (12.84), Gene ontology (GO) term assignment for different categories of A. hypochondriacus single-nucleotide polymorphism carrying genes is shown in Fig. 11.

Gene ontology (GO) term assignment of A. hypochondriachus single-nucleotide polymorphism carrying genes classified into three main categories: biological process, cellular component, and molecular function.
Discussion
A breeder’s ability to manage crop genetic resources efficiently depends greatly on having adequate knowledge of existing genetic diversity. Molecular, biochemical, and morphological analysis are all possible methods for analyzing genetic diversity. Since the above methods have limitations, next-generation sequencing technologies (such as Genotyping by sequencing or GBS) are being applied to a wide range of crop species to study diversity among population in precise manner. In addition to providing a powerful and rapid way to identify the genetic basis of agriculturally important traits, precise phenotyping of agronomically significant traits in combination with genotyping help in predicting the breeding value of individuals in breeding populations. GBS has now made it possible for underutilized pseudocereal crop like Amaranth to conduct a variety of genetic analyses that were previously too expensive and unaffordable. The GBS based approach generate data which are very useful in conducting a number of analyses including Genome wide association mapping (GWAS), marker based identification of QTL, Germplasm diversity analysis, germplasm fingerprinting. Analysis SNPs across the genome revealed an unexpected bias towards transversions. A transition to transversion (Ts: Tv) ratio of 0.5302 was observed in our dataset. Our observation of a bias towards transversions, while unusual, is not unprecedented in genomic research. Several studies have reported similar patterns across diverse organisms. In cotton (Gossypium sp.) mitochondrial DNA, Zhang et al. (2020) found an extremely low Ts/Tv ratio of 0.2831. Similarly, Zhang et al. (2012) observed a Ts/Tv ratio approaching 1 in certain Hassawi rice (Oryza sativa L.) genomes, some genotypes had a ratio less than 132. In grasshoppers, Keller et al. (2007) reported a transversion-biased ratio of 0.48 after accounting for potentially methylated cytosines33. These findings collectively suggest that departure from the typically observed transition bias may be more common than previously thought, occurring across various taxonomic groups and genomic contexts. Our results contribute to this growing body of evidence, highlighting the complexity and diversity of mutational patterns in genomes. There were three major clusters identified based on the cluster analysis of genotypes based on geographical distribution. However, the dendrogram did not classify genotypes unequivocally based on country of origin. The main reason for this could be attributed to broad diversity of accessions of A. hypochondriacus used in the present study. Another reason is that different genotypes or accessions of A. hypochondriacus have spread to different geographical locations from its single center of origin i.e., Mexico, but some of them retained the same genetic background. The domestication of Amaranth took place in USA around 6,000 years ago34. The observation that the majority of Asian and European Amaranth accessions clustered with accessions from the U.S.A. suggests that amaranth spread to Europe and Asia after its domestication in the U.S.A. The latter is supported by a report, which states that Amaranth was introduced and grown in Germany in the sixteenth century and spread throughout Europe by seventeenth century5. It then spread to the Himalayan region and interior China in 19 th century. Its cultivation started in China, India, Africa, Europe, North and South America in 20 th century. Cluster A comprises indigenous accessions largely clustered with accessions from Germany, China, and the USA. It is therefore obvious that most of accessions from India have evolved from the accessions of these countries. In addition, it could also be interpreted that the different accessions within the subclusters of cluster A may have same genetic and biochemical makeup, which in turn have similarity in terms of nutrients or antinutrients composition. However, the same needs verification through future experimentation. Cluster B contains one accession from China, EC-120,051, clustered with accessions from India. Remaining six accessions of China clustered with other Indian accessions along with exotic accessions from Germany, Russia and USA. It suggests the spread of Amaranth from Europe to China from which it spread to India due to close geographical proximity between two countries and possible gene flow. Cluster C contains only indigenous accessions, which may have evolved separately due to divergence from ancestral accession of China or Europe. Since indigenous accessions are grouped together with accessions from different countries, therefore it suggests that allelic reshuffling has occurred due to exchange of Indian and exotic genetic resources followed by hybridization and selection. The division of a plant population into genetically pure and admixed lines is an important feature of population structure analysis. In the present investigation, the population diversity analysis was done based upon the geographical origins of different accessions, which led to division of entire collection of accessions in three subpopulations. The accessions in cluster A have shown admixture of 54% with most of these accessions originating from the USA. The presence of only one Chinese accession along with Indian accessions in cluster B and six Chinese accessions in cluster C indicates some level of admixture with accessions from China. In order to improve Amaranth breeding program, there were made crosses between well adapted US accessions and European or Asian accessions. The presence of different alleles in accessions of Cluster A and Cluster B, as observed in the present study might result from recombination and reshuffling of alleles due to hybridization between accessions of diverse geographical origins. The admixture between US and Indian accessions suggests that many accessions of Amaranth may have resulted from intercrossing between Indian and US accessions as a part of breeding strategy. However, it also suggests that some accessions have also been derived from some European ancestors. The presence of a Chinese accession in Cluster B also suggests the possible gene flow from the Chinese accession to Indian accessions as a result of breeding activities. However, Cluster C contains only Indian accessions, which are grouped together but fall in a separate cluster. This indicates that these indigenous accessions might have evolved from either Chinese or U.S. accessions followed by their complete divergence to form a separate population. The DAPC and the MDS further support the above findings. From the MDS, it was found that except for accessions from Africa, accessions from all other locations were dispersed across all three clusters. A lack of clear differentiation among accessions from different geographical regions into separate clusters indicates substantial gene flow and genetic admixture, potentially facilitated by human-mediated dispersal, breeding activities, and natural migration patterns. This also proves that the accessions around the world are from a common origin, namely the USA.
To summarize above analysis it has been revealed that source of germplasm for Indian accessions of grain Amaranth is intriguing in the sense that Indian accessions initially originated from the germplasm from USA. Later when some of these accessions migrated to Europe and China then the hybridization and selection may have resulted in new combinations of genes, which led to evolution of new germplasm. Finally, some of the new Indian accessions evolved from the germplasm, which was introduced from Europe and China through new rounds of variation and selection in order to get adapted for growing in different regions of India.
Analysis of genetic diversity found that nucleotide diversity (π) differed significantly among populations: Rest of Europe, USA, and Russia had the greatest diversity while Africa, China and Germany had comparatively low levels. Watterson’s estimator (θ) as having larger effective population sizes potentially35 identified India, the USA, and Russia. Tajima’s D statistic showed that all populations have experienced recent population expansions or selective sweeps; therefore, they are not neutral for evolution and suggest presence of rare variants36.
Different degrees of inbreeding were detected between the populations by inbreeding analysis, where, African populations had low FIS and Russia showed the highest. India, Rest of Europe and the USA also showed high FIS. It is also worth noting that the observed level of heterozygosity is lower than expected in all other populations, except Africa, where the FIS was also low. This indicates certain factors may be affecting genetic variation like inbreeding or selective pressures, geographical barriers and environmental pressures37.
Within India, nucleotide diversity and the FIS were high in the Central region and Southern and Western region. The Watterson estimator values were more or less the same for all regions. The observed heterozygosity was again lower than the expected within the regions in India. In our analysis of Amaranth genetics, we consistently observed lower observed heterozygosity compared to expected heterozygosity, a pattern also noted in many other independent Amaranth studies23,38,39. These findings suggest a trend of heterozygote deficiency in Amaranth, being a self-pollinated crop, indicating potential factors influencing genetic diversity and breeding strategies.
The FST values were consistently high between Africa and other countries, suggesting that the populations have experienced population expansions or selective sweeps, resulting in high differentiation of the African population from the rest40. The higher Nei’s genetic distance value between Africa and other countries also supports this finding.
It is likely that accessions derived from the USA were recently spread to Russia, India, China and Rest of Europe via introduction and dispersal events, after which they were cultivated and preserved as local cultivars. This hypothesis is supported by the low FST scores between the USA and Russia, China, India, Germany and rest of Europe (0.013–0.028), at the same time, the Nei’s genetic distances were also considerably low between the USA and the other countries except Africa (0.015–0.029). Our study corroborates previous findings of a pronounced genetic divergence between the African and American lineages22. It is also worth noting that FST values and genetic distances are low, between India and other populations (except Africa), suggesting significant admixture between these populations. In case of regions within India, a clear correlation between genetic distances and geographical distances can be observed. The genetic distances and FST between populations of nearby regions were lower compared to regions, which were geographically distant, indicating possible influence of isolation by distance on population structure. Our FST and genetic distance results suggest that A. hypochondriacus may have been introduced to Central India and subsequently spread from there.
In the present study, most of the SNP carrying genes of Amaranth, which have been functionally annotated, belong to different metabolic and cellular processes. Using functionally annotated SNPs, it may be possible to establish marker–trait associations and identify genes that control agriculturally important traits.
Conclusions
Amaranth accessions have been classified into three subpopulations based on several thousand SNPs segregating within each accession, and these SNPs have also revealed phylogenetic relationships among them and have provided a valuable dataset. Our analysis highlights substantial gene flow and genetic admixture between A. hypochondriacus populations worldwide, with accessions from different regions dispersed across clusters. While nucleotide diversity varied significantly among populations, Tajima’s D statistic suggested recent population expansions or selective sweeps in all the populations. The significant genetic divergence observed between African and American lineages, alongside evidence of admixture in India, highlights the complex evolutionary history and human intervention mediated dispersal of A. hypochondriacus. Furthermore, our findings suggest a central role of Central India in the dispersal and establishment of this crop species, with implication for genetic conservation and breeding strategies. In the future, Amaranth breeding could be guided by these data. With our SNP genotyping, breeders will be able to select parents and introduce alleles from exotic germplasm into new crosses. Amaranth grains, being very rich in proteins and minerals such as Phosphorous, Calcium, Potassium as well as important phytochemicals such as squalene, are becoming very popular among the health conscious people. Having developed a useful set of SNPs, we aim to identify those associated with high grain protein or calcium content in future investigations. As the genome of A. hypochondriacus is already deciphered, such investigations would pave way to identify genes/QTLs linked to high protein, minerals through GWAS. It is therefore anticipated that these high-throughput genotyping and sequencing technologies will contribute to better breeding techniques and enhancing quality and output of these minor crops.
Materials and methods
Plant material
The diversity panel comprised of 192 accessions representing grain amaranth, spanning from countries of origin, landraces, improved cultivars and genetic stocks, were procured from NBPGR, Shimla (Table 6).This collection comprised of accessions which broadly fall in to two categories viz. Exotic collection (EC) and Indigenous collection (IC).The120IC accessions represents the released cultivars, and landraces of grain amaranth from 13 states of India including North western Himalayan region. The IC accessions represent all the amaranth growing agro-ecological conditions (tropical, temperate and semi-arid) prevalent in the country. The remaining 72 EC accessions assembled in the panel belong to USA, Germany, Russian Federation, China, Lativa, Netherlands, France, Poland, Nepal, Benin, Kenya and South Africa. All the genotypes of the diversity panel are derived from self-fertilizing lineages, and as such, heterozygosity is not expected due to the extreme self-pollinating behavior of the crop.
DNA extraction and quantification
DNA was extracted from seeds of 192 accessions of grain Amaranth using CTAB based protocol. After extraction, the quality of extracted DNA was checked on 0.8% agarose gel in which 1 kb DNA ladder was also run as control to check the quality of DNA. In order to verify that restriction enzymes can cut genomic DNA without being inhibited, restriction digestion test using HindIII restriction enzyme was carried out on every genomic DNA sample. This helped in ascertaining the purity and quality of genomic DNA for restriction-site associated DNA sequencing library preparation, hence ensuring better results. DNA was then quantified using the Nanodrop Spectrophotometer 1000 (Thermo Fisher Scientific).
Genotyping by sequencing
The genomic DNA of 192 Amaranthus hypochondriacus accessions were subjected to GBS (Genotyping by Sequencing) analysis. DNA samples were normalized to a final concentration of 5 µg for GBS. Lyophilized DNA samples were sent to NXGenBio Life Sciences, New Delhi, for GBS analysis. GBS libraries were prepared following41 with minimal modification, Libraries were prepared with ApeKI restriction enzyme for genomic DNA digestion followed by ligation with ApekI and common adapters. The purified 192-plex final DNA library was quantified using a Bioanalyzer (Agilent Technologies) and were sequenced on a single lane of Illumina HiSeqTM X10 platform (Illumina® Inc., San Diego, CA, USA) using V4 sequencing chemistry.
The FASTX Toolkit (version 0.0.13) was used to separate samples from combined data based on barcodes. The program fastx_barcode_splitter.pl (part of the FASTX Toolkit) reads FASTA/FASTQ files and splits them into several smaller files based on barcode matching. FastQC (version 0.11.5, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used for quality checking. Universal Illumina Adapters was removed using trim galore (version 0.6.2, https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), a wrapper script to automate quality and adapter trimming as well as quality control. A. hypochondriacus v2.1 downloaded from (https://phytozome-next.jgi.doe.gov/info/A hypochondriacus_v2_1) was used as a reference genome14. Reads were mapped against the reference genome using the MEM algorithm of BWA (version0.7.5) with default parameters.
Single-Nucleotide polymorphism calling and filtering
Variant calling was done using UGBS- GATK pipeline (version 3.6, https://gatk.broadinstitute.org/hc/enus). The GATK is the industry standard for identifying SNP and indels in germline DNA and RNAseq data. In addition to the variant callers themselves, the GATK also includes many utilities to perform related tasks such as processing and quality control of high-throughput sequencing data. Variants were filtered and indels were removed during vcftools (version 0.1.17, http://vcftools.sourceforge.net/). All SNPs were filtered at minor allele frequency of 5% MAF and 20% missing rate (minimum 80% sample have that particular SNPs). A workstation with 250 GB of RAM with 32-core Intel Xeon processor was used for all the bioinformatics work and consequent analyses. The GBS data were obtained in the form of a hapmap file format.
Preparation of phylogenetic tree by neighbour joining (NJ) method
SNP data from 192 accessions of grain Amaranth were loaded into TASSEL 5.0 software in hapmap format. Based on the loaded genotype data, there was computed pair wise genetic distance matrix, which was used for preparation of phylogenetic tree by selecting NJ method under Analyze tab of TASSEL. The resulting phylogenetic relationships were visualized in a dendrogram, with genotypes grouped into distinct clusters for further analysis.
Population structure analysis
In order to undertake research on population structure and evaluate multilocus genotypic data (192 accessions) we have utilized the window-based software package known as Structure, which is openly available. Initially, the hap map file was changed into a variant call format file by TASSEL software and then the PGD Spider V.2.0.8.2 software was used to convert it into the structure format so that it could be used as an input file for the Structure program. To estimate exact population structure, K’s from one to ten (with 10 iterations each) were tested and the LnP (D) value was used to group all the genotypes. For each run, a burn-in of 5000 iterations was followed by an additional 50,000 replications.
Discriminant analysis of principle component
The Discriminant Analysis of Principle Component was carried out to identify genetic clusters within the germplasm using the R package adegenet 2.0.042. The optimal number of clusters, k value was identified to be 3 using Bayesian Information Criterion (BIC) and the DAPC was carried out. In order to examine how the genotypes grouped based on geographical locations, an additional multidimensional scaling analysis was conducted using the SNP data by grouping the accessions from five different countries and two continents and labeling them in distinct colors.
Genetic diversity and population differentiation analysis
To evaluate diverse aspects of genetic diversity within and among populations, various metrics were computed using different software tools. Observed heterozygosity (Heo), segregating sites, nucleotide diversity (π), and Watterson estimator (θ) were determined using SambaR V1.10, while within-population genetic diversity (expected heterozygosity, Hee) was calculated using the R package ‘adegenet’43,44. The Nei’s genetic distance was computed using ‘hierfstat’ and fixation distances (FST) between pairwise populations were assessed using ‘StAMPP’, both within the R environment45,46. Two different analyses were conducted. In one analysis, the population was split into five countries (India, the USA, China, Germany, and Russia) and two continents (Africa and the rest of Europe, which included all european countries except Germany and Russia). In the second analysis, India was divided into four regions: Central India, the Himalayan Region, the North-east region, and the Southern and Western Region.
Linkage disequilibrium and haplotype block
A triangle plot for pairwise LD between marker sites was generated using TASSEL. Heat maps were produced based on pairwise R2 estimates to visualize LD throughout the genome, and corresponding p-values were calculated using permutations for all marker pairs. The R2 values for each marker pair are displayed in the bottom half of the heat map and are represented by shades of red increasing in intensity in equal increments of 0.1 from 0.0 (white) to 1.0 (red).
Haplotype block analysis was performed using LD values calculated for each marker pair in TASSEL 5.0, followed by subsequent analysis in RStudio. An LD decay threshold of r² = 0.1 was applied to estimate the average haplotype block size, consistent with common practices in crop genomics (Hamblin et al., 2011; Flint-Garcia et al., 2003). This threshold balances the detection of meaningful associations with background LD considerations, while providing a conservative estimate of haplotype block size suitable for breeding applications28,29,47.
Phylogenetic analysis
The phylogenetic analysis for the samples was performed with the help of the graphical user interface of TASSEL V. 5.0 standalone using neighbor-joining algorithm formed with node name.
Functional annotation of Single-Nucleotide polymorphisms
The hap map file, which includes flanking sequences for each of the filtered SNPs, was used to infer the possible roles of the SNPs. This most recent iteration of the Plant RefSeq (reference sequence) database is now accessible via a locally hosted BLAST server. Each SNP’s flanking sequence was compared to the Plant RefSeq database using a local BLAST. The server housing the standalone BLAST has 32 cores and 256 GB of RAM configured for multi-threading mode. Data were collected and combined after obtaining the BLAST results, which included the Gene IDs of the corresponding species. Each of these SNPs was assigned a gene ontology identifier (GO ID) based on a search of the GO database, which may have resulted in either single GO ID or several IDs.
Data availability
All sequencing data from the 192 grain amaranth accessions have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject accession PRJNA1208757, comprising 384 SRA records.
References
Joshi, D. C. et al. From zero to hero: the past, present and future of grain Amaranth breeding. Theor. Appl. Genet. 131, 1807–1823 (2018).
Kumar, A., Tripathi, M. K., Joshi, D. & Kumar, V. Millets and Millet Technology (Springer, 2021).
Joshi, D. C. et al. Strategic enhancement of genetic gain for nutraceutical development in buckwheat: a genomics-driven perspective. Biotechnol. Adv. 39, 107479 (2020).
Parihar, M. A. N. O. J. et al. Reviving the forgotten food network of potential crops to strengthen nutritional and livelihood security in North Western Himalayas. Indian J. Agron. 66, S44–S59 (2021).
Rastogi, A. & Shukla, S. Amaranth: a new millennium crop of nutraceutical values. Crit. Rev. Food Sci. Nutr. 53, 109–125 (2013).
Silva-Sánchez, C. et al. Bioactive peptides in Amaranth (Amaranthus hypochondriacus) seed. J. Agric. Food Chem. 56, 1233–1240 (2008).
Khamar, R. & Jasrai, Y. T. Nutraceutical analysis of Amaranth oil, avocado oil, Cumin oil, linseed oil and Neem oil. Int. J. Bioassays. 3, 2090–2095 (2014).
Reyad-ul-Ferdous, M., Shamim, D., Shahjahan, M., Sharif, T. & Mukti, M. Present biological status of potential medicinal plant of Amaranthus viridis: a comprehensive review. Am. J. Clin. Exp. Med. 3, 2–17 (2015).
Chandi, A. et al. Use of AFLP markers to assess genetic diversity in Palmer Amaranth (Amaranthus palmeri) populations from North Carolina and Georgia. Weed Sci. 61, 136–145 (2013).
Khaing, A. A. et al. Genetic diversity and population structure of the selected core set in Amaranthus using SSR markers. Plant. Breed. 132, 165–173 (2013).
Štefúnová, V., Bežo, M., Žiarovská, J. & Ražná, K. Detection of the genetic variability of amaranthus by RAPD and ISSR markers. Pak J. Bot. 47, 1293–1301 (2015).
Gelotar, M. J., Dharajiya, D. T., Solanki, S. D., Prajapati, N. N. & Tiwari, K. K. Genetic diversity analysis and molecular characterization of grain Amaranth genotypes using inter simple sequence repeat (ISSR) markers. Bull. Natl. Res. Cent. 43, 103 (2019).
Clouse, J. W. et al. The Amaranth genome: genome, transcriptome, and physical map assembly. Plant. Genome. 9, 1–14 (2016).
Lightfoot, D. J. et al. Single-molecule sequencing and Hi-C-based proximity-guided assembly of Amaranth (Amaranthus hypochondriacus) chromosomes provide insights into genome evolution. BMC Biol. 15, 74 (2017).
Brookes, A. J. The essence of SNPs. Gene 234, 177–186 (1999).
Cho, R. J. et al. Genome-wide mapping with biallelic markers in Arabidopsis thaliana. Nat. Genet. 23, 203–207 (1999).
Gupta, P. K., Roy, J. K. & Prasad, M. Single nucleotide polymorphisms: a new paradigm for molecular marker technology and DNA polymorphism detection with emphasis on their use in plants. Curr. Sci. 80, 524–535 (2001).
Sachidanandam, R. et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409, 928–933 (2001).
Rafalski, A. Applications of single nucleotide polymorphisms in crop genetics. Curr. Opin. Plant. Biol. 5, 94–100 (2002).
Gupta, P. K. & Rustogi, S. Molecular markers from the transcribed/expressed region of the genome in higher plants. Funct. Integr. Genomics. 4, 139–162 (2004).
Thapa, R., Edwards, M. & Blair, M. W. Relationship of cultivated grain Amaranth species and wild relative accessions. Genes 12, 1849 (2021).
Jamalluddin, N., Massawe, F. J., Mayes, S., Ho, W. K. & Symonds, R. C. Genetic diversity analysis and marker-trait associations in Amaranthus species. PLoS ONE. 17, e0267752 (2022).
Hoshikawa, K. et al. Genetic diversity analysis and core collection construction for Amaranthus tricolor germplasm based on genome-wide single-nucleotide polymorphisms. Sci. Hortic. 307, 111428 (2023).
Fellers, J. P. Genome filtering using methylation-sensitive restriction enzymes with six base pair recognition sites. Plant. Genome. 1, 146–152 (2008).
Glaubitz, J. C. et al. TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS ONE. 9, e90346 (2014).
Jiao, Y. et al. Genome-wide genetic changes during modern breeding of maize. Nat. Genet. 44, 812–815 (2012).
Ossowski, S. et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327, 92–94 (2010).
Vos, P. G. et al. Evaluation of LD decay and various LD-decay estimators in simulated and SNP-array data of tetraploid potato. Theor. Appl. Genet. 130, 123–135 (2017).
Pang, Y. et al. High-resolution genome-wide association study identifies genomic regions and candidate genes for important agronomic traits in wheat. Mol. Plant. 13, 1311–1327 (2020).
Alexander, D. H., Shringarpure, S. S., Novembre, J. & Lange, K. ADMIXTURE 1.3 software manual. (2015). http://software.genetics.ucla.edu/admixture/download.html
Xiong, H. et al. Genetic diversity and population structure of Cowpea (Vigna unguiculata L. Walp). PLoS ONE. 11, e0160941 (2016).
Zhang, T. et al. The organelle genomes of Hassawi rice (Oryza sativa L.) and its hybrid in Saudi Arabia: genome variation, rearrangement, and origins. PLoS ONE. 7, e42041 (2012).
Keller, I., Bensasson, D. & Nichols, R. A. Transition-transversion bias is not universal: a counter example from grasshopper pseudogenes. PLoS Genet. 3, e22 (2007).
He, Q. & Park, Y. J. Evaluation of genetic structure of Amaranth accessions from the united States. Weed Turfgrass Sci. 2, 230–235 (2013).
Ferretti, L. & Ramos-Onsins, S. E. A generalized Watterson estimator for next-generation sequencing: from trios to autopolyploids. Theor. Popul. Biol. 100, 79–87 (2015).
Larsson, H., Källman, T., Gyllenstrand, N. & Lascoux, M. Distribution of long-range linkage disequilibrium and Tajima’s D values in Scandinavian populations of Norway Spruce (Picea abies). G3 (Bethesda). 3, 795–806 (2013).
Zhivotovsky, L. A. Relationships between Wright’s FST and FIS statistics in a context of Wahlund effect. J. Hered. 106, 306–309 (2015).
Delgado, H. & Martín, J. P. Genetic diversity of black Amaranth (Amaranthus Quitensis Kunth) landraces of Ecuadorian Highlands: association genotypes-color morphotypes. Agriculture 13, 34 (2022).
Vats, G. et al. Validation of genome-wide SSR markers developed for genetic diversity and population structure study in grain Amaranth (Amaranthus hypochondriacus). Agriculture 13, 431 (2023).
Meirmans, P. G. & Hedrick, P. W. Assessing population structure: FST and related measures. Mol. Ecol. Resour. 11, 5–18 (2011).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE. 6, e19379 (2011).
Jombart, T., Devillard, S. & Balloux, F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11, 94 (2010).
Jombart, T. & Ahmed, I. Adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071 (2011).
De Jong, M. J., de Jong, J. F., Hoelzel, A. R. & Janke, A. SambaR: an R package for fast, easy and reproducible population-genetic analyses of biallelic SNP data sets. Mol. Ecol. Resour. 21, 1369–1379 (2021).
Goudet, J. & Jombart, T. Hierfstat: Estimation and tests of hierarchical F-statistics. R package version 0.04-22. R core Team, 58. (2015) https://CRAN.R-project.org/package=hierfstat
Pembleton, L. W., Cogan, N. O. & Forster, J. W. StAMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour. 13, 946–952 (2013).
Ersoz, E. S., Yu, J. & Buckler, E. S. Applications of linkage disequilibrium and association mapping in crop plants. in Genomics-Assisted Crop Improvement: Vol. 1: Genomics Approaches and Platforms 97–119Springer Netherlands, Dordrecht, (2007).
Acknowledgements
Authors are thankful to Department of Biotechnology, Government of India, New Delhi for providing financial support through project grants no. BT/BI/01/069/2018.
Funding
The present research work was supported by Department of Biotechnology, Govt. of India in the form of research grants (BT/BI/01/069/2018).
Author information
Authors and Affiliations
Contributions
Conceptualization [D.P] Methodology [R.C., T.J.], Germplasm collection and analysis [D.J., R.C.] Research data analysis and interpretation [S.P. and A.P.] Writing - Original draft preparation [D.P.], Writing- Review and Editing [R.C., S.P., A.T., D.J., R.J.] Funding Acquisition [D.P., A.K.] Resources [J.J., S.S., G.T.]
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All plant experiments were conducted in accordance with relevant institutional, national, and international guidelines and legislation.
Consent for publication
All the authors have consented for publication of content.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chauhan, R., Prabhakaran, S., Tiwari, A. et al. Genetic distances and genome wide population structure analysis of a grain amaranth (Amaranthus hypochondriacus) diversity panel using genotyping by sequencing. Sci Rep 15, 33816 (2025). https://doi.org/10.1038/s41598-025-01626-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-01626-7
