
Abstract
At present, there are currently no molecular biomarkers for the early diagnosis of sepsis cardiomyopathy (SCM) in clinical practice. This study focuses on an in-depth examination of the DNA hydroxymethylation profiles within plasma extracellular vesicles and explores potential molecular biomarkers during the process of SCM. The 5hmC-Seal sequencing technology was utilized to examine the hydroxymethylation modifications of extracellular vesicles DNAs in 13 patients with septic cardiomyopathy, 18 patients with sepsis without cardiomyopathy, and 8 patients without sepsis. Additionally, a diagnostic model was constructed using machine learning methods based on the differential hydroxymethylation modifications to screen for candidate biomarkers. The accuracy of the diagnostic model was 0.962, with a sensitivity and specificity of 92.3% and 88.89%, respectively. Furthermore, the diagnostic accuracy was validated using the GEO dataset, with an accuracy rate reaching 1 (GSE79962 and GSE66890), and the differential diagnostic accuracy rates also reached 0.959 and 0.944 (GSE79962). Together, the results suggest that extracellular vesicles DNAs hydroxymethylation markers can be used for diagnosis of septic cardiomyopathy.
Similar content being viewed by others

Introduction
Septic cardiomyopathy (SCM) is defined as a sepsis (SI) induced acute syndrome of non-ischemic cardiac dysfunction, manifested in systolic and/or diastolic left ventricular (LV) dysfunction and/or right ventricular dysfunction1. SCM is gaining worldwide attention in the field of critical care due to its high morbidity and mortality. The incidence fluctuates between 20-50%2 and the mortality rate is between 16.5-66%3, depending on previously different definitional criteria. Currently, the precise diagnosis of septic myocardiopathy remains a worldwide challenge.
At present, the diagnosis of SCM mainly relies on bedside echocardiography4,5,6. A recent database study suggested that echocardiographic assessment of myocardial function influenced hemodynamic management of patients with sepsis, which resulted in earlier discontinuation of vasopressor administration, and decreased 28-day mortality7. But due to the timing of initiating ultrasonography and the difference in the level of bedside physicians, there is maybe a delay in the diagnosis of septic cardiomyopathy which may influence hemodynamic management and increase cardiac preload resulting in deterioration of heart function, ultimately exacerbating patients’ death, so the exploration of specific markers for the early diagnosis of septic cardiomyopathy is of great significance. Although Troponin8 and Natriuretic peptides4,9 as cardiac biomarkers have prognostic value for patients with sepsis, they are not specific to diagnosis of SCM. Clinically, there is an urgent need for effective liquid biopsy technology for molecular diagnosis of septic myocardiopathy.
Extracellular Vesicles (EVs) are small membrane vesicles released by cells into the extracellular environment, playing a crucial role in cell-to-cell communication by carrying biological molecules such as proteins, lipids, and nucleic acids to modulate the function of recipient cells10,11,12. In recent years, the role of EVs in cardiovascular diseases has gained increasing attention, including their involvement in SCM13. Research has found that resident macrophages in the heart, characterized by high expression of TREM2, maintain mitochondrial metabolic homeostasis in cardiomyocytes by taking up EVs containing damaged mitochondria from cardiomyocytes, thereby exerting a protective effect against SCM14. This indicates that EVs play a key role in the communication between cardiomyocytes and immune cells and may be involved in regulating the stress response and repair processes of cardiomyocytes. Furthermore, The miRNA and DNA in these EVs can also mirror gene - expression changes in myocardial tissue. Research has shown that EVs released by ischemic cardiomyocytes are rich in mtDNA. This mtDNA can activate inflammatory responses via TLR9 and inhibit the expression of myocardial repair genes like VEGF and HIF − 1α15. Moreover, extracellular vesicle DNAs (evDNAs) can act as a signaling molecule to trigger intracellular DNA - sensing pathways or, through gene transfer, supply extra genetic information to recipient cells, influencing their phenotype16. In summary, EVs and their carried molecules might indicate the pathological state of cardiomyocytes, offering a new angle for the early diagnosis and treatment of SCM.
5 - hydroxymethylcytosine (5hmC), as the “sixth base” in epigenetic modifications, plays crucial roles in gene expression regulation, cell differentiation, embryonic development, and disease occurrence. As an intermediate product of DNA demethylation, the distribution of 5hmC in gene bodies and promoter regions is closely related to the activation of gene expression17. 5hmC can regulate gene transcriptional activity by increasing chromatin accessibility and promoting transcription factor binding. In some cases, it can also inhibit gene expression, showing a dual regulatory effect. Moreover, the dynamic changes of 5hmC during embryonic development are essential for cell fate determination and tissue differentiation18. During the differentiation of mouse embryonic stem cells, the dynamic changes in 5hmC levels are closely related to the expression regulation of pluripotency - related genes. In recent years, 5hmC has been widely used to explore disease biomarkers19. Our laboratory have demonstrated that the 5hmC signatures from cell-free DNA (cfDNA) could serve as early warning biomarkers for myocardial Injury in COVID-19 patients20 indicating that 5hmC may have potential value in SCM warning and discovery of target therapy. So we conducted a prospective study to explore the value of 5hmC for early warning biomarkers of SCM patients.
In this study, we utilized the 5hmC-Seal technique to obtain the genome-wide 5hmC profiles from plasma evDNAs of 13 SCM patients, 18 sepsis patients without cardiomyopathy, and 8 non-sepsis individuals. Our results indicate that SCM patients, sepsis without cardiomyopathy, and non-sepsis exhibit distinct 5hmC signatures, and 5hmC markers selected by machine learning algorithms can be employed for the diagnosis of septic cardiomyopathy.
Materials and methods
The study was approved by The Ethical Review Board of the Peking University Third Hospital (No. 202205802).
Study population
We carried out a prospective cohort study in the department of Critical Care Medicine in Peking University Third Hospital. From November 2020 to April 2022, we consecutively enrolled 39 patients aged at least 18 years. Patients were classified into three groups: non-sepsis group, sepsis group, SCM group which was defined as having sepsis, and elevated troponin and/or natriuretic peptides values, and one of the following four items appeared, respectively LV ejection fraction less than 50%, lateral E’ wave < 10 cm/sec, lateral E/e’ ratio > 14 cm/sec21, RV dysfunction with Tricuspid annular plane systolic excursion < 16 mm22. Sepsis group was defined with sepsis-3.0 criteria but without SCM, but patients with the history of chronic heart failure and chronic renal failure were excluded. Non - sepsis was defined as patients aged > 18 years, with no evidence of infection, coronary heart disease, chronic kidney disease, and a post - operative state. The following data were recorded: demographic characteristics; infection focus induced sepsis; Sequential Organ Failure Assessment (SOFA) score at admission of ICU; death; hemofiltration.
Study design
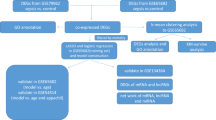
The study aimed to discover 5hmC biomarkers using the efficient 5hmC-Seal technology, in order to identify potential diagnostic biomarkers for SCM. A total of 39 patients were included in the cohort, consisting of SCM (n = 13), sepsis (n = 18), and non-sepsis (n = 8). The workflow diagram of plasma evDNAs sequencing is shown in Fig. 1.

Flow chart of the experiment. It includes sample collection, EVs extraction, and DNA sequencing analysis of EVs from plasma vesicles of study cohort.
Plasma separation
Blood samples (8 ml) were collected in blood collection tubes containing EDTA, then centrifuged sequentially at 1350×g and 3000×g for 12 min at 4 °C, and the supernatant was taken after each centrifugation, and finally centrifuged at 10,000×g for 5 min at 4 °C, and the supernatant was taken and stored at -80 °C until further analysis.
Isolation of extracellular vesicles
EVs were isolated from plasma samples using an exosome is olation kit (Wayen, China). Add 20 ul of Reagent C to 1 ml of plasma and mix well, incubate at 37 °C for 15 min. After shaking, the sample was centrifuged at 10,000 × g for 10 min at room temperature, the supernatant was removed, 250 µL of Reagent A was added and mixed, and the sample was incubated at 4 °C for 30 min. The supernatant was removed by centrifugation at 3000 × g for 10 min and the sample was centrifuged again to remove the residual supernatant. The EV pellet was resuspended in 1 ml of phosphate-buffered saline, Reagent B was added and treated as Reagent A. Finally, the EV pellet was resuspended in 200ul of phosphate-buffered saline, and the precipitated EVs were used immediately.
Isolation of total DNA
Total DNA was extracted from plasma-isolated exosomes using the Quick-DNA™ Miniprep Plus Kit (ZYMO, USA) according to the manufacturer’s instructions. 200ul of BioFluid & Cell Buffer and 20ul of Prteinase k were added to the isolated exosomes, mixed, and then digested by incubation at 55º C for 10 min of incubation for digestion. The mixture was transferred to a Zymo-Spin™ IIC-XLR column packed in a 2 mL collection tube, and after centrifugation the DNA adhered to the column membrane, which was rinsed with DNA Pre-Wash Buffer and DNA Wash Buffer; DNase/RNase-Free water was then added and centrifuged at 13,500 × g for 1 min, and the eluted evDNAs was added back to the rotating column by aspiration and centrifugation. The effluent collected was the total DNA of exosomes. The DNA concentration was detected by Qubit 3.0 and the data were recorded. The amount of DNA obtained after total DNA isolation is shown in Supplementary Table 1.
5hmC library construction and high-throughput sequencing
With the application of high-efficiency 5hmC-Seal technology, we constructed 5hmC libraries for all samples. Given the highly sensitive nature of the chemical labeling method, we assigned low values of 1–10 ng for the input evDNAs. And the 5hmC-modified DNA was extracted with KAPA HyperPlus Library Preparation Kit (KAPA Biosystems, USA), Invitrogen Dynabeads® M-270 Streptavidin Kit (Thermo Fisher Scientific, USA), and Beckman Binding Magnetic Bead Kit (Beckman Coulter, USA). First, fragment DNA using KAPA FCS, Frag Buffer, and Frag Enzyme interrupter. In accordance with the next-generation sequencing demands, the plasma-derived evDNAs was subjected to end-repairing, followed by the use of Hyper Prep Kit (KAPA Biosystems, USA) for 3′-adenylation and the use of compatible adapters (Illumina, USA) for ligation. Then, we supplemented the ligated evDNAs to a 25 µL glycosylation reaction solution including 25 mM MgCl2, 50 mM pH-8.0 HEPES buffer, 100 µM UDP-6-N3-Glc and 1 µM β-glucosyltransferase and incubated at the temperature of 37 °C for 2 h. Afterwards, the purification of evDNAs was performed via the DNA Clean & Concentrator Kit (ZYMO, USA). Incubation of the purified DNA proceeded using DBCO-PEG4-biotin (1 µL; Click Chemistry Tools; 4.5 mM stock in dimethyl sulfoxide) under the temperature of 37 °C for 2 h. Likewise, by adopting the foregoing ZYMO kit, purification of DNA was again performed. Subsequently, streptavidin beads (2.5 µL; Life Technologies) in 1× buffer (1 M NaCl, 5 mM pH-7.5 Tris, 0.5 mM EDTA, and 0.2% Tween 20) was directly supplemented to the reaction solution, and incubated for half an hour under the condition of room temperature. In the end, buffers 1–4 were utilized to accomplish each 5-min rinsing of the beads 8 times. In addition, the entire procedures of binding and washing were conducted with mild rotation at ambient temperature. Afterwards, RNase-free water was used to resuspend the beads, followed by amplification of beads through a polymerase chain reaction (PCR; 14–16 cycles). In line with the instruction of the manufacturer, AMPure XP beads (Beckman, USA) were adopted for purifying the PCR products. We measured the concentration of the libraries with the aid of Qubit 3.0 fluorometer (Life Technologies, USA). In addition, high-throughput sequencing of a paired-end 39-bp sequence was carried out on the Next Seq 500 platform.
Read mapping
Sequence quality was assessed using FastQC (version 0.11.5). In addition, raw reads were aligned with the human genome (hg19 version) using Bowtie2 (version 2.2.9)23. Then, SAM tools (version 1.3.1; parameters adopted: SAMtools view -f 2 -F 1548 -q 30 and samtoolsrmdup) were applied for further read filtration24, so that the nonduplicate unique matches with the genome could be reserved. After extension, paired-end reads were converted into BedGraph format and then normalized to the total number of paired reads using bedtools (version 2.19.1)25. Meanwhile, with the aid of UCSC bedGraphToBigWig, we also converted the paired-end reads into the BigWig format, so that the Integrated Genomics Viewer-assisted visualization could be achieved.
5hmC data processing and analysis
Firstly, all 5hmC peaks from 39 samples were merged using “Bedtools merge” (Bedtools, version 2.27.1), resulting in a total number of peaks. Subsequently, “multicov” (bedtools 2.27.1) was exploited to quantify the read counts in every merged peak for the entire samples, followed by count normalization using the aforementioned method. For signal enhancement, the entire biological replicates from identical samples were integrated. The p-value evaluation was performed on every peak in a sample based on the Poisson distribution. Using one-tailed Poisson distribution, the probability of read counts per peak for a single sample was evaluated, with λ being the mean value for other samples. Finally, the DESeq2 package 1.30.0 in R version 3.5.0 was adopted to identify differentially hydroxymethylated regions (DhMRs), where the filtering cutoffs were set at P < 0.05 & |log2FoldChange| ≥0.5. To estimate the intragenic 5hmC content, reads belonging to UCSC genes were counted, which were identified by the corresponding annotated transcription start sites (TSS) and transcription termination sites (TTS). According to the read counts overlapping the hydroxymethylated regions (hMRs) computation of reads per kilobase of transcript per million reads mapped (RPKM) was accomplished.
Mapping of 5hmC-modified genes
Distribution mapping of SCM, sepsis, and non-sepsis 5hmC-modified genes using the ggplot2 package in R26. Use of the ggpubr package in R27 to map the upsets of SCM, sepsis, and non-sepsis 5hmC-modified genes in the Promoter, Intron, Exon, and Donstream regions.
Difference analysis
Gene expression profiles were constructed for each sample of SCM patients, sepsis patients and non-sepsis. Differential analysis between sepsis patients and non-sepsis was performed after using the limma package in R28. A screening threshold of P < 0.05 and |log2FoldChange| ≥0.5 were set to obtain the differentially hydroxymethylated modified genes (DhMGs) between sepsis patients and non-sepsis. Similarly, patients with SCM and patients with sepsis were differentially analyzed to obtain differentially expressed genes for SCM and sepsis.
Enrichment analysis
Functional enrichment was performed using the Gene Ontology (GO) database (http://www.geneontology.org/). The Database for Annotation, Visualization and Integrated Discovery (Metascape) Bioinformatics resource was used to conduct functional enrichment analysis and delineate the underlying biological processes and pathways of aberrantly expressed intersecting genes based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database29.
Gene clustering trend analysis
Gene expression matrices of SCM patients, sepsis patients and non-sepsis individuals were plotted as gene clustering trends using the Mfuzz package in R30. Clusters of genes that were consistently up-regulated and consistently down-regulated from the control, sepsis to SCM groups were screened, and functional enrichment of genes within gene clusters.
Feature selection and classifier construction
For the genes within the screened gene clusters, the train_test_split function from the Scikit-Learn 0.22.1 module in Python 3.6.10 was used, and a predictive model was built with the Least Absolute Shrinkage and Selection Operator (LASSO). In the study cohort, DhMRs were detected using DESeq2 1.30.0 in R 3.5.0, with a threshold of P < 0.05 and |log2FoldChange| ≥ 0.5. To prevent overfitting, we conducted 5 rounds of 10 - fold cross - validation, with 100 runs per round. Finally, markers observed in at least 3 rounds were used to construct the final predictive model in the training set. The parameter α was selected to maximize the accuracy in the validation set across a grid of values ranging from 0.1 to 0.9. Furthermore, the area under the curve (AUC) and 95% CIs were generated to evaluate the model performance31.
External data set collection
The GSE66890 and GSE79962 datasets were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) using the GEOquery package. Files were read in, and the oligo package was used for background correction, normalization, and expression quantification. Expression matrices were then extracted from the processed data, merged for the two datasets, and samples were grouped based on their information. The GSE79962 dataset includes array-based gene expression profiles of myocardial tissue from 20 patients with SCM, 11 patients with ischemic cardiomyopathy (ICM), and 9 patients with nonischemic dilated cardiomyopathy (DCM). The GSE66890 dataset includes array-based gene expression profiles of whole blood from 28 sepsis patients.
Statistical analysis
Statistical analysis was performed using IBM SPSS 27.0.1. For patient baseline data, three - group comparisons used one - way ANOVA (mean ± SD) for normally distributed continuous variables and Kruskal - Wallis H test (median [IQR]) for non - normally distributed ones. For two - group comparisons, non - paired t - tests and Mann - Whitney U tests were used for normally and non - normally distributed continuous variables respectively.Categorical variable comparisons used χ² tests when theoretical frequency was > 5, otherwise Fisher’s exact test. P < 0.05 was statistically significant.
Results
Clinical characteristics of SCM patients
The clinical summary, including baseline characteristics and laboratory data, of all 39 patients was shown in Table 1. Of the 39 patients, the median age of all the patients was 66 years. The cohort of patients who met the diagnostic criteria is as follows: SCM in 13 patients (33.33%), sepsis in 18 patients (46.15%), non-sepsis in 8 patients (20.51%). Among them, 19 individuals have Hypertension, 8 have Diabetes, and 9 have Coronary Artery Disease. Finally, the mean of SOFA, N-terminal pro-B-type natriuretic peptide (NT-proBNP), and Troponin I (TNI) for all patients was 7 scores, 1720.0 pg/ml and 0.02 ng/ml, respectively.
Landscape of 5hmC profiles of EvDNAs in patients with SCM
To explore whether there are differences in the 5hmC signals in the whole-genome 5hmC profiles of DNA derived from EVs among patients with SCM, sepsis, and non-sepsis, the genomic distribution of 5hmC in patients with SCM, patients with sepsis, and non-sepsis was determined using chemical labeling of hydroxymethylated DNA and high-throughput sequencing technology.
Firstly, we compared the distribution of 5hmC along the genome among the SCM group, sepsis group, and the non-sepsis group, and we found that SCM had the highest level of hydroxymethylation, followed by septic patients, while control patients had the lowest level of hydroxymethylation (Fig. 2A). In addition, hydroxymethylated regions (hMRs) were found to be predominantly distributed in intron and intergenic regions (Fig. 2B). The venn diagram results show that there are both common and distinct hMRs on different positions of the genome among the SCM, sepsis, and non-sepsis groups (Fig. 2C-D, Fig. S1A-C). Thus, loss of 5hmC during disease progression occurs mainly in the intergenic region and tends to accumulate slowly on the GB. These findings indicate that 5-hydroxymethylcytosine (5hmC) markers derived from evDNAs could serve as potent epigenetic biomarkers for the diagnosis of SCM through liquid biopsy, potentially enhancing the diagnostic process of SCM.

Characteristics of 5hmC distribution in the evDNAs of SCM patients. (A) Genome-wide distribution of 5hmC in plasma samples of SCM, sepsis patients, and non-sepsis individuals. (B) Presence of 5hmC peaks that overlapped within biological replicates on different genomic elements. (C) Venn diagram showing gene number associated with peaks observed of those three groups; peaks that overlapped with promoter were employed to perform the analysis. (D) Venn diagram showing gene number associated with peaks observed of those three groups; peaks that overlapped with introns were employed to perform the analysis. SCM septic cardiomyopathy, SI sepsis, GB gene body, 5hmC 5-hydroxymethylcytosine, UTR untranslated region.
5hmC profiles differ among sepsis and non-sepsis group
Differential analysis between sepsis and non-sepsis revealed 331 hyper hydroxymethylated modified genes and 578 hypo-hydroxymethylated modified genes (|log2FoldChange| ≥ 0.5, P < 0.05) in sepsis (Fig. S2A). KEGG network analysis and GO analysis (Fig. S2B, Fig. S3A, B) revealed that genes with high hydroxymethylation were enriched in genomes associated with the cancer pathway, the MAPK signaling pathway, and the JAK-STAT signaling pathway, and hyper hydroxymethylated genes were enriched in the Rap1 signaling pathway, T-cell receptor signaling pathway, NF-kappa B signaling pathway, and PI3K-Akt signaling pathway. Meanwhile, the hubs of the KEGG functional interaction networks (Fig. S4) showed that these genes, including BCL2 Apoptosis Regulator (BCL2)、Spleen Associated Tyrosine Kinase (SYK)、Mitogen-Activated Protein Kinase Kinase Kinase 7 (MAP3K7)、Janus Kinase 2 (JAK2), participated in the regulation of inflammatory response pathway.
5hmC profiles differ among SCM and sepsis
Differential analysis between SCM and control revealed 469 hyperhydroxymethylated modified genes and 145 hypo-hydroxymethylated modified genes (|log2FoldChange| ≥0.5, P < 0.05) in SCM (Fig. S2C). KEGG network analysis and GO analysis (Fig. S2C, Fig. S3C, D) revealed that genes with high hydroxymethylation were enriched in genomes associated with Calcium signaling pathway, MAPK signaling pathway and Apoptosis, and hyperhydroxymethylated modified genes were enriched in inflammatory and immune pathways including the TNF signaling pathway, T cell receptor signaling pathway and B cell receptor signaling pathway. Meanwhile, the hubs of the KEGG functional interaction networks (Fig. S5) showed that these genes, including NFKB Inhibitor Alpha (NFKBIA), TNF Alpha Induced Protein 3 (TNFAIP3), Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha (PIK3CA) and Jagged Canonical Notch Ligand 1 (JAG1), participated in the regulation of inflammatory response pathway.
5hmC markers reflect the progression of septic cardiomyopathy
Then, we investigated how DNA hydroxymethylation is coordinated during SCM. Gene clusters were calculated for three specific stages, each containing genes with similar GB hydroxymethylation patterns. K-means analysis was used to classify these differentially regulated hMRs into nine clusters based on the different expression patterns of the unique biological features (Fig. 3, Fig. S6) and selected three clusters in which cluster 4 and 5 of hMRs progressively increased and cluster 7 decreased with the disease progressed (Fig. 3A, C, E). Next, GO network analyses were performed on the genes involved in clusters 4, 5 and 7 (Fig. 3B, D, F). DhMGs in cluster 4 are mainly enriched in cellular response to growth factor stimulation, positive regulation of JNK cascade, DNA repair, positive regulation of stress-activated protein kinase signaling cascade, and positive regulation of stress-activated MAPK cascade signal pathways (Fig. 3B). DhMGs in cluster 5 are mainly enriched in the process of glycerolipid metabolism, negative regulation of gene expression, epigenetic regulation of gene expression, calcium ion transmembrane transport, vascular morphogenesis, cell-cell adhesion (Fig. 3D), and DhMGs in cluster 7 are predominantly enriched in activation of GTPase activity, muscular system processes, negative regulation of cytosolic processes, epigenetic regulation of gene expression, response to growth factors signal pathways (Fig. 3F).

Identification of persistently dysregulated DhMRs during progression from sepsis to SCM. (A, C, E) K-means were clustered for DhMRs with P < 0.05 between SCM and sepsis groups. (B, D, F) GO (BP) analysis of up-regulated persistently dysregulated genes and down-regulated persistently dysregulated genes.
Diagnostic model based on 5hmC markers can effectively diagnose SCM
The clear separation of the SCM group from the sepsis group on the PCA plot indicates significant differences between the two groups (Fig. 4A, B).We constructed a classification model based on the 5hmC markers from cluster 4, cluster 5, and cluster 8 obtained through temporal clustering analysis, using the Least Absolute Shrinkage and Selection Operator (LASSO) algorithm. To assess whether the sample size was sufficient for model training, we reduced the sample size and evaluated the performance of the classifier, finding that the classification ability gradually declined when the sample size was reduced to 6. To confirm the effectiveness of the selected markers, we reduced the number of markers used for screening and determined that six markers were sufficient for robust classification.

5hmC markers’ diagnosis for SCM in the study cohort. (A) Principle component analysis (PCA) plot of 5hmC markers in evDNAs from SCM and sepsis samples. (B) Heatmaps of 6 5hmC markers with treatment response, batch and sex information labeled in the study cohorts. Unsupervised hierarchical clustering was performed across genes and samples. (C) ROC curves showing the results of the prediction model based on 5hmC markers differentiating patients with SCM from sepsis. (D) Confusion matrix showing model prediction ability for validation cohort. (E) ROC curves showing the results of the prediction model based on 5hmC markers differentiating patients with SCM from sepsis. (F) Confusion matrix showing model prediction ability for validation cohort.
We applied a recursive feature elimination algorithm based on LASSO to optimize the selection of 5hmC markers, achieving the best cross-validation score (Fig. S7A). This approach ultimately identified six key 5hmC-based markers (Fig. S7B). Using these markers, we developed an SCM classification model (Supplementary Table 2) that achieved high classification accuracy during screening (AUC = 0.962, Fig. 4C), achieving 92.3% sensitivity and 88.89% specificity (Fig. 4D). The model performance evaluation metrics are shown in Supplementary Table 3. Compared to other SCM risk factors like NT-proBNP, LVEF, WBC, PCT, TnI, and some combined indices, which have AUCs of 0.904, 0.851, 0.524, 0.750, 0.840, and 0.833 respectively, this approach’s advantage is evident. (Fig. S8, 9) respectively. By utilizing the GSE79962 and GSE66890 datasets for further evaluation of the diagnostic model, the verification results indicated that the model’s AUC could reach 1 (Fig. 4E), achieving 100% sensitivity and 100% specificity (Fig. 4F). The PCA plots from the external validation cohort demonstrate good separation between SCM and Sepsis (Fig. S10A), further validating the reliability of the results.
In conclusion, the findings suggest that in addition to echocardiographic and serum cardiac biomarkers, markers such as MAP2K5, KCP, BAHD1, PLCL2, ANKRD17, and UBE2V2 in EVs DNA may be of value in effectively differentiating between SCM and sepsis.
Diagnostic model can distinguish SCM from ICM and DCM
To further verify the ability of the diagnostic model to distinguish between SCM and other types of cardiomyopathy. ROC analyses were performed on samples from patients with SCM, patients with ICM, and patients with DCM from the publicly available dataset of the GEO database (GSE79962). The AUCs of ICM and DCM versus SCM were 0.959 (Fig. 5A), achieving 100% sensitivity and 90.9% specificity (Fig. 5B), and 0.944 (Fig. 5C), achieving 80% sensitivity and 100% specificity (Fig. 5D), respectively, and the analysis showed that these genes better distinguished SCM from ICM and DCM. The PCA plots from the external validation cohort demonstrate good separation between SCM and ICM, as well as between SCM and DCM (Fig. S10B, C).

ROC curves for SCM, ICM and DCM. (A) ROC curves showing the results of the prediction model based on GSE79962 differentiating patients with SCM from ICM. (B) Confusion matrix showing model prediction ability for validation cohort. (C) ROC curves showing the results of the prediction model based on GSE79962 differentiating patients with SCM from DCM. (D) Confusion matrix showing model prediction ability for validation cohort.
By searching the GeneCards database, the string database, and the PathCards database, these genes, MAP2K5, KCP, BAHD1, PLCL2, ANKRD17, UBE2V2, were found to have potentially related pathways, including Jak-Stat signaling pathway, MAPK signaling pathway, IL17 signaling pathway, to sepsis and SCM (Fig. S11). Through the pathway analysis of these target genes, a series of standardized biological pathways were identified, which are responsible for cell differentiation, regulation of cell component biogenesis, gene expression and apoptosis processes.
Discussion
EvDNAs essentially represents fragments of cellular DNA derived from the nucleus or mitochondria, which are the result of regular DNA metabolism and/or the accumulation of damaged-induced DNA32. An increase in the secretion of evDNAs has been shown to serve as a defense mechanism against oxidative stress in disease-damaged or aging cells33. Additionally, studies have indicated that an increase in evDNAs levels after neoadjuvant therapy is significantly associated with disease progression and EVs have the capability to provide clinically relevant information about the growth and progression of diseases34. Therefore, liquid biopsy based on evDNAs have the potential to become a rapid and precise method for disease diagnosis. Numerous studies have shown that 5hmC is associated with the epigenetic activation of various diseases, serves as a marker of disease progression, and has the potential to act as a biomarker for various diseases35. In summary, the signal characteristics of 5hmC in blood evDNAs may be a reliable biomarker for different diseases. At present, there are no molecular markers available for the diagnosis of SCM in clinical practice. In this study, we conducted an in-depth study of the hydroxymethylation profiles of DNA in plasma EVs using 5hmC-Seal sequencing technology, in an attempt to discover reliable biomarkers for SCM.
There are several potential confounding factors in the search for SCM biomarkers in extracellular vesicle DNA, such as blood draw timing and stress. To address these, the study took specific measures. Firstly, whole - blood samples from all groups were collected immediately upon diagnosis. Drawing blood before diagnosis in sepsis and SCM patients might not accurately reflect the levels of 5hmC - modified DNA in extracellular vesicles under disease conditions. This is because the disease might be in an early stage, with biomarker levels not yet significantly changed or with unclear trends. As a result, test results might be unrepresentative and fail to accurately show disease characteristics and progression. In contrast, drawing blood after diagnosis can better capture changes in 5hmC - modified DNA within extracellular vesicles during disease36. Therefore, immediate blood sampling after disease diagnosis can more precisely capture 5hmC - modified DNA in disease states. Secondly, sepsis is a systemic inflammatory response syndrome caused by infection. It usually triggers an immune response, leading to increased white blood cell counts, which is the body’s defense against infection. There are significant differences between healthy individuals and sepsis patients in terms of basic health, immune function, and inflammatory status. In this study, the non - sepsis group patients generally underwent other surgeries. Both surgical patients under stress and sepsis patients are in a stress state, though the sources and nature of stress differ36. This similarity makes the two groups comparable in terms of physiological and metabolic changes related to the stress response. This helps more accurately evaluate the impact of sepsis itself on 5hmC - modified DNA in extracellular vesicles.
Firstly, we found significant differences in the enrichment of 5hmC in evDNAs among SCM patients, sepsis individuals, and non-sepsis. Furthermore, this study found that the 5hmC modified genes related to mononucleosis are mainly enriched in calcium signaling pathways and inflammatory signaling pathways, which is consistent with the literature reports. During sepsis, mitochondrial dysfunction, impaired function of SERCA2, and L-type calcium channel dysfunction may disrupt calcium homeostasis within cardiomyocytes, leading to cardiac dysfunction37. During sepsis, a large number of inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), are produced, which can directly damage cardiomyocytes and lead to cardiac dysfunction38. Furthermore, oxidative stress reactions during sepsis lead to increased production of reactive oxygen species (ROS) and reactive nitrogen species (RNS), which can damage proteins, lipids, and DNA in cardiomyocytes, thereby inducing cardiac dysfunction39. Our results show that the signaling pathway involved in 5hmC modified genes is closely related to the occurrence of SCM, which indicates that we use these 5hmC modified genes to construct the SCM diagnostic model is scientific and reliable.
Considering the importance of SCM detection and the deficient detection efficiency between SCM and sepsis samples, we establish an diagnostic classifier model. Firstly, we selected clusters of 5hmC markers that vary with disease progression, including 5hmC clusters that are upregulated (cluster 4 and 5) and downregulated (cluster 7) as the disease advances. Subsequently, using machine learning algorithms to screen for characteristic 5hmC markers from these clusters, we identified six unique 5hmC changes that may serve as promising biomarkers for the diagnosis of SCM. Each marker could accurately distinguish SCM from sepsis. A diagnostic model of SCM constructed by the panel of 6 DhMRs achieved a sensitivity of 92.30% and accuracy of 88.89%. The performance of the diagnostic model was evaluated in GSE79962 and GSE66890 datasets and well discriminated results were also obtained (AUC = 1.000). Importantly, we used the GSE79962 dataset to validate the power of differential diagnosis with the model, and we found that the diagnostic model based on six 5hmC markers can effectively distinguish SCM from different myocardial diseases. Our research results indicate that 5hmC markers extracted from evDNAs can serve as effective epigenetic biomarkers for the minimally invasive diagnosis of SCM.
Of the six identified markers, MAP2K5 (Mitogen - activated protein kinase kinase 5) is a key regulator in the MAPK signaling pathway, involved in cell proliferation, differentiation, apoptosis, and other biological processes. In septic cardiomyopathy, MAP2K5 may contribute to disease progression through inflammation and apoptosis. During sepsis, inflammatory cytokines such as TNF - α and IL - 1β activate the MAPK pathway. As an upstream kinase, MAP2K5 further activates JNK and p38 MAPK, exacerbating myocardial inflammation40. Moreover, activated MAP2K5 upregulates pro - apoptotic proteins like Caspase - 3, inducing cardiomyocyte apoptosis and worsening myocardial injury41. KCP (Potassium channel - interacting protein) is widely expressed in cardiomyocytes, regulating their electrical activity. In septic cardiomyopathy, KCP may be involved in calcium imbalance and cardioprotection. Oxidative stress and inflammation from sepsis can cause calcium overload in cardiomyocytes. KCP may modulate potassium channel activity to regulate intracellular calcium homeostasis, affecting myocardial contractility42. Additionally, KCP may reduce ischemia - reperfusion injury by regulating potassium channel opening, offering protection in septic cardiomyopathy. This is consistent with functional enrichment analysis results, further confirming the reliability of the identified biomarkers.
In summary, our study shows that evDNAs 5hmC features are linked to sepsis and SCM. Especially, SCM patients have unique 5hmC patterns at MAP2K5 and KCP compared to sepsis and non - sepsis patients. The 6 - gene diagnostic model from these features effectively distinguishes SCM patients from the non - sepsis group, highlighting 5hmC modifications’ potential as SCM biomarkers. While our results don’t establish causality, they provide a framework for understanding epigenetic dysregulation’s role in sepsis progression. Future research should explore longitudinal 5hmC dynamics to validate these links and uncover new SCM control mechanisms.
Strengths and limitations
Ideal diagnostic biomarkers are expected to be highly sensitive, specific to SCM, and non-invasively detectable at the early stage. We tested the six biomarkers in a small set of plasma samples, and it showed superior diagnostic performance, indicating that the six DhMRs could be potentially applied as biomarkers in clinical practices. But the sample size was limited in this study and next we are going to recruit more participants to verify the generalization of the model. The development of SCM is a complex process involving multiple genetic, epigenetic, and protein expression alterations. Constructing diagnostic models using hydroxymethylation biomarkers merely to assess the diagnosis of SCM may be inadequate. In the future, we will consider combining multi-omics such as radiomics, DNA fragmentation patterns, and proteomic biomarkers to further improve the diagnostic performance. Furthermore, intensively investigation on the functions of the targeted genes is necessary to clearly elucidate the molecular events occurring in the SCM development and progression.
Conclusion
In summary, the study results indicate that hydroxymethylation marks in evDNAs can reflect the progression of SCM, and these marks can effectively predict and diagnose the progression of SCM. The results of this current work provide a foundation for understanding the functional role of 5hmC modification in the progression of SCM, thus opening up the field to explore new mechanisms that can govern the progression of SCM.
Data availability
The datasets supporting the conclusions of this article are included within the article and its Supplementary files. The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive of the BIG Data Center at the Beijing Institute of Genomics, Chinese Academy of Science, under accession number HRA008552 (accessible at https://ngdc.cncb.ac.cn/gsa-human). Code is available from the corresponding author on reason able request.
References
Boissier, F. & Aissaoui, N. Septic cardiomyopathy: diagnosis and management. J. Intensive Med. 2, 8–16. https://doi.org/10.1016/j.jointm.2021.11.004 (2021).
Parker, M. M. et al. Profound but reversible myocardial depression in patients with septic shock. Ann. Intern. Med. 100, 483–490. https://doi.org/10.7326/0003-4819-100-4-483 (1984).
Sanfilippo, F. et al. Tissue doppler assessment of diastolic function and relationship with mortality in critically ill septic patients: a systematic review and meta-analysis. Br. J. Anaesth. 119, 583–594. https://doi.org/10.1093/bja/aex254 (2017).
Ehrman, R. R. et al. Pathophysiology, echocardiographic evaluation, biomarker findings, and prognostic implications of septic cardiomyopathy: a review of the literature. Crit. Care. 22, 112. https://doi.org/10.1186/s13054-018-2043-8 (2018).
Feng, M. et al. Transthoracic echocardiography and mortality in sepsis: analysis of the MIMIC-III database. Intensive Care Med. 44, 884–892. https://doi.org/10.1007/s00134-018-5208-7 (2018).
Vallabhajosyula, S. et al. Impact of right ventricular dysfunction on Short-term and Long-term mortality in sepsis: A Meta-analysis of 1,373 patients. Chest 159, 2254–2263. https://doi.org/10.1016/j.chest.2020.12.016 (2021).
Beesley, S. J. et al. Septic cardiomyopathy. Crit. Care Med. 46, 625–634. https://doi.org/10.1097/CCM.0000000000002851 (2018).
Bessière, F., Khenifer, S., Dubourg, J., Durieu, I. & Lega, J. C. Prognostic value of troponins in sepsis: a meta-analysis. Intensive Care Med. 39, 1181–1189. https://doi.org/10.1007/s00134-013-2902-3 (2013).
Hollenberg, S. M. & Singer, M. Pathophysiology of sepsis-induced cardiomyopathy. Nat. Rev. Cardiol. 18, 424–434. https://doi.org/10.1038/s41569-020-00492-2 (2021).
Bello-Morales, R., Ripa, I. & López-Guerrero, J. A. Extracellular vesicles in viral spread and antiviral response. Viruses 12 https://doi.org/10.3390/v12060623 (2020).
Bose, S., Aggarwal, S., Singh, D. V. & Acharya, N. Extracellular vesicles: an emerging platform in gram-positive bacteria. Microb. Cell. 7, 312–322. https://doi.org/10.15698/mic2020.12.737 (2020).
Fabbiano, F. et al. RNA packaging into extracellular vesicles: an orchestra of RNA-binding proteins? J. Extracell. Vesicles. 10, e12043. https://doi.org/10.1002/jev2.12043 (2020).
Evans, L. et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 47, 1181–1247. https://doi.org/10.1007/s00134-021-06506-y (2021).
Fajgenbaum, D. C. & June, C. H. Cytokine storm. N Engl. J. Med. 383, 2255–2273. https://doi.org/10.1056/NEJMra2026131 (2020).
Tripathi, A., Bartosh, A., Whitehead, C. & Pillai, A. Activation of cell-free mtDNA-TLR9 signaling mediates chronic stress-induced social behavior deficits. Mol. Psychiatry. 28, 3806–3815. https://doi.org/10.1038/s41380-023-02189-7 (2023).
Ibrahim, A. G., Cheng, K. & Marbán, E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell. Rep. 2, 606–619. https://doi.org/10.1016/j.stemcr.2014.04.006 (2014).
Kriukienė, E., Tomkuvienė, M. & Klimašauskas, S. 5-Hydroxymethylcytosine: the many faces of the sixth base of mammalian DNA. Chem. Soc. Rev. 53, 2264–2283. https://doi.org/10.1039/d3cs00858d (2024).
Shi, D. Q., Ali, I., Tang, J. & Yang, W. C. New insights into 5hmC DNA modification: generation, distribution and function. Front. Genet. 8, 100. https://doi.org/10.3389/fgene.2017.00100 (2017).
Klungland, A. & Robertson, A. B. Oxidized C5-methyl cytosine bases in DNA: 5-Hydroxymethylcytosine; 5-formylcytosine; and 5-carboxycytosine. Free Radic Biol. Med. 107, 62–68. https://doi.org/10.1016/j.freeradbiomed.2016.11.038 (2017).
Chen, H. Y. et al. 5-Hydroxymethylcytosine signatures in Circulating Cell-Free DNA as early warning biomarkers for COVID-19 progression and myocardial injury. Front. Cell. Dev. Biol. 9, 781267. https://doi.org/10.3389/fcell.2021.781267 (2021).
Nagueh, S. F. et al. Recommendations for the evaluation of left ventricular diastolic function by echocardiography: an update from the American society of echocardiography and the European association of cardiovascular imaging. J. Am. Soc. Echocardiogr. 29, 277–314. https://doi.org/10.1016/j.echo.2016.01.011 (2016).
Rudski, L. G. et al. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American society of echocardiography endorsed by the European association of echocardiography, a registered branch of the European society of cardiology, and the Canadian society of echocardiography. J. Am. Soc. Echocardiogr. 23, 685–713. https://doi.org/10.1016/j.echo.2010.05.010 (2010).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with bowtie 2. Nat. Methods. 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. https://doi.org/10.1093/bioinformatics/btq033 (2010).
Langmead, B. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics Chap. 11, Unit 11.17 https://doi.org/10.1002/0471250953.bi1107s32 (2010).
Klaus & Galensa. ggplot2: elegant graphics for data analysis (2nd ed.). Computing Reviews (2017).
Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Ready Plots. R package version 0.6.0. https://rpkgs.datanovia.com/ggpubr/ (2023)
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2013).
Kanehisa, M. et al. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
Futschik, M. E. & Carlisle, B. Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol. 3, 965–988. https://doi.org/10.1142/s0219720005001375 (2005).
Abraham, A. et al. Machine learning for neuroimaging with scikit-learn. Front. Neuroinform. 8, 14. https://doi.org/10.3389/fninf.2014.00014 (2014).
Fricke, F. et al. TGFBR2-dependent alterations of Exosomal cargo and functions in DNA mismatch repair-deficient HCT116 colorectal cancer cells. Cell. Commun. Signal. 15, 14. https://doi.org/10.1186/s12964-017-0169-y (2017).
Tian, C. et al. Extracellular vesicles regulate Sympatho-Excitation by Nrf2 in heart failure. Circ. Res. 131, 687–700. https://doi.org/10.1161/CIRCRESAHA.122.320916 (2022).
Bernard, V. et al. Circulating nucleic acids are associated with outcomes of patients with pancreatic Cancer. Gastroenterology 156, 108–118e104. https://doi.org/10.1053/j.gastro.2018.09.022 (2019).
Zeng, C., Stroup, E. K., Zhang, Z., Chiu, B. C. & Zhang, W. Towards precision medicine: advances in 5-hydroxymethylcytosine cancer biomarker discovery in liquid biopsy. Cancer Commun. (Lond). 39, 12. https://doi.org/10.1186/s40880-019-0356-x (2019).
Bi, C. F., Liu, J., Yang, L. S. & Zhang, J. F. Research progress on the mechanism of Sepsis induced myocardial injury. J. Inflamm. Res. 15, 4275–4290. https://doi.org/10.2147/JIR.S374117 (2022).
Rosenberger, P. H. et al. Surgical stress-induced immune cell redistribution profiles predict short-term and long-term postsurgical recovery. A prospective study. J. Bone Joint Surg. Am. 91, 2783–2794. https://doi.org/10.2106/JBJS.H.00989 (2009).
Celes, M. R. et al. Disruption of calcium homeostasis in cardiomyocytes underlies cardiac structural and functional changes in severe sepsis. PLoS One. 8, e68809. https://doi.org/10.1371/journal.pone.0068809 (2013).
Kakihana, Y., Ito, T., Nakahara, M., Yamaguchi, K. & Yasuda, T. Sepsis-induced myocardial dysfunction: pathophysiology and management. J. Intensive Care. 4, 22. https://doi.org/10.1186/s40560-016-0148-1 (2016).
Mager, C. E. et al. p38 MAPK and MKP-1 control the glycolytic program via the bifunctional Glycolysis regulator PFKFB3 during sepsis. J. Biol. Chem. 299, 103043. https://doi.org/10.1016/j.jbc.2023.103043 (2023).
Kim, C. G. et al. Caspase-3/MAPK pathways as main regulators of the apoptotic effect of the phyto-mediated synthesized silver nanoparticle from dried stem of Eleutherococcus senticosus in human cancer cells. Biomed. Pharmacother. 99, 128–133. https://doi.org/10.1016/j.biopha.2018.01.004 (2018).
Morciano, G. et al. Calcium dysregulation in heart diseases: targeting calcium channels to achieve a correct calcium homeostasis. Pharmacol. Res. 177, 106119. https://doi.org/10.1016/j.phrs.2022.106119 (2022).
Funding
This work was supported by Natural Science Foundation of China (82274034), Hygiene and Health Development Scientific Research Fostering Plan of Haidian District Beijing (HP2022-28-501002), Capital Health Research and Development of Special Fund (Grant/Award 20202 − 4094), Peking University Third Hospital Cohort Study Project (BYSYDL2021010) and Beijing Municipal Natural Science Foundation (Grant/Award 7212130).
Author information
Authors and Affiliations
Contributions
Zl-Z and WL were responsible for the sample collection and summary of clinical information. LZ participated in library construct and 5hmC sequence. Bx-Z contributed to bioinformatics analysis. Bx-Z, Hy-C and Zl-Z were responsible for conceptual design, data analysis, manuscript writing, and submission. Hy-C revised the article and gave suggestions. J-L, Q-G, and X-Z provided the funding. In addition, Bx-Z, Zl-Z and Hy-C had equal contributions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declarations
The study was conducted according to the guidelines of the Helsinki Declaration and was approved by the Ethics Committee of Fuwai Hospital. Written informed consent was obtained from all participants.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhen, B., Zhao, Z., Chen, H. et al. 5-Hydroxymethylcytosine signatures as diagnostic biomarkers for septic cardiomyopathy. Sci Rep 15, 23987 (2025). https://doi.org/10.1038/s41598-025-02489-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-02489-8
