
Abstract
A new series of thiazolyl-coumarin-drug conjugates were synthesized through the reaction of 3-bromoacetylcoumarin and N-substituted and N, N-di-substituted thioureas. The synthesized compounds were characterized by1H-NMR,13C-NMR and FTIR spectroscopy and thoroughly analyzed through in-vitro and in-silico studies. Among these, derivatives 8b and 8d demonstrated significant inhibitory effects during in vitro analysis. Compound 8b exhibited a notable inhibition potential exhibiting IC50 = 0.32 ± 0.04 µM against CAIX, while compound 8d showed a potent inhibitory effect with IC50 = 0.38 ± 0.02 µM and 0.61 ± 0.05 µM against CAII and CAXII isozymes, respectively. Surpassing its standard inhibitor acetazolamide. Electronic characteristics of all synthesized hybrid compounds were accessed via density functional theory calculations (DFTs). Furthermore, the results were validated via molecular docking studies. Drug likeness properties were predicted at the end via ADMET analysis, to support the investigations. The comprehensive experimental and computational analyses supported the conclusion that the synthesized hybrid compounds possess the potential to act as inhibitors against different types of carbonic anhydrases. Overall, this study will open new avenues for the development of dual inhibitors of CAII and CAXII, displaying versatile therapeutic applications.
Similar content being viewed by others
Introduction
Carbonic anhydrases (CAs) represent a versatile superfamily of zinc (Zn2+) containing metalloenzymes1, that are crucial for maintaining acid-base balance and participating in various physiological processes across different tissues including the interconversion between CO2 and HCO3− in various biological processes i.e. in respiration, and carbon dioxide transport2. CAs are known in nature as genetically distinct families. These families include the α-, β-, γ-, δ-, and ζ-CAs3,4,5. In mammals, sixteen different alpha-CA (α-CA) isoforms have been isolated, and they play crucial physiological roles6.
The cytosolic carbonic anhydrase isoforms include CAI, CAII, CAIII, CAVII, and CAXIII. These enzymes are predominantly located in the cytoplasm and are involved in processes such as respiration, acid-base balance, and ion transport. For instance, CAII is highly abundant in red blood cells and plays a crucial role in CO₂ transport and pH regulation7. Similarly, CAIII is found in skeletal muscles and is implicated in muscle function and metabolism. CAVII and CAXIII are less characterized but are thought to be involved in various cellular processes, including pH regulation and ion transport8. The membrane-bound carbonic anhydrase isoforms, including CAIV, CAIX, CAXII, CAXIV, and CAXV, are anchored to cellular membranes and play key roles in ion transport, acid-base balance, and extracellular pH regulation. CAIX is overexpressed in various cancers, such as clear cell renal cell carcinoma, where it contributes to tumor progression by enhancing acidosis. CAXII is found in tissues like the kidney and pancreas, where it aids in bicarbonate transport and fluid secretion. While CAXIV and CAXV are less studied, they are believed to participate in similar functions9.
Recent studies have highlighted the therapeutic potential of carbonic anhydrase (CA) inhibitors across various medical conditions10. Notably, methazolamide, a glaucoma medication, has demonstrated efficacy in reducing tau protein accumulation, offering promise for treating tau-related dementias. Additionally, dual-target inhibitors that simultaneously modulate CA activity and other molecular targets are under investigation, aiming to enhance treatment efficacy for complex diseases11. Furthermore, the FDA-approved CA inhibitor acetazolamide is being explored for its potential in managing bacterial infections, suggesting a novel application for existing therapies12.
Clinically, CAIX and CAXII are overexpressed in various types of cancer cells, including breast cancer. Inhibition of these isoforms has shown promise in cancer therapy by influencing intracellular and extracellular pH13, which can affect clonogenic survival, apoptosis, migration, and radiosensitivity of cancer cells14. Several studies have evaluated the efficacy of CAII, CAIX, and CAXII inhibitors in preclinical and clinical settings15.
The primary goal of this study is to synthesize new thiazolyl coumarin-based conjugates to explore therapeutically effective Carbonic anhydrase inhibitors as potential anticancer agents. Coumarins are a class of widely studied compounds containing the 1-benzopyran-2-one core. The structural architect of coumarin consists of a pyrone ring fused with benzene. Coumarin and its derivatives are of great importance in natural and synthetic products16. Coumarins are widely distributed in the plant kingdom and substructure of various natural and pharmaceutical compounds17. The lactone ring attached to the conjugated system shows interaction with various enzymes and receptors in organisms18. Coumarin and its derivatives comprise more than 40 drugs, which are extensively prescribed as medicine19. Furthermore, coumarin derivatives possessing several substituted thiazole rings at carbon-3 display high potency against various diseases. The coumarin nucleus attracts the remarkable attention of synthetic chemists and pharmacologists due to its broad range of biological applications20,21,22.
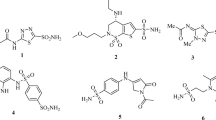
Coumarin derivatives exhibit numerous biological activities such as anticoagulant (e.g., warfarin, acenocoumarol)23,24, insecticide (e.g., coumaphos)25 antibacterial (e.g., novobiocin, clorobiocin)26and anticoagulant rodenticide (e.g., brodifacoum, bromadiolone)27 as shown in Fig. 1. Coumarin and its derivative 7-hydroxycoumarin were tested for their cytotoxic behaviour in several human tumor cell lines. Both compounds retard cell proliferation of a gastric carcinoma cell line28.
Thiazole ring-containing compounds are also a highly active group of heterocyclic pharmacophores. Additionally, all available Penicillins possess a thiazole ring, which improves potency against huge scans of bacteria29. Thiazoles have drawn continued interest due to their use in various medical treatments such as bacterial infections. Moreover, they have found their applications in material science as parent starting material. In addition, 2-aminothiazole derivatives have been reported to show significant pharmaceutical applications30,31.

Previously reported Coumarin containing medicinally active drugs.
Coumarin thiazole analogues show various biological activities such as antibacterial32,33, antiviral34,35,36, anti-inflammatory37,38, anticancer39,40, anticoagulant41, antioxidant42,43,44, anticonvulsant45, antimicrobial46,47,48, antifungal49,50, acetylcholinesterase inhibitors51,52 alkaline phosphatase and ecto-5′-nucleotidase inhibitors53, Anti-Tubercular54, carbonic anhydrase inhibitors55.
Recently, various classes of compounds have been screened to find suitable inhibitor of Carbonic anhydrase isozymes such as azomethine-tethered sulfonamides56, Acyl-3-(Ciprofloxacinyl) Thioureas57, pyrazole-based benzene sulfonamides58, diarylsulfonamides and their bioisosteres59 and 2, 4-thiazolidinedione-tethered coumarins60, Thiazolyl-pyrazoline derivatives61. The practice of combining two or more pharmacophores into a single hybrid skeleton is a common approach in drug designing. Based on these findings the current study aimed to synthesize hybrid pharmacophores containing coumarin with different commercial drugs through thiazole moieties. The molecular hybridization of coumarin and drugs was adopted to improve biological activity62,63.
Experimental
Materials and methods
All solvents and reagents were commercially purchased. Melting points were observed Gallenkamp melting point instrument (MP-D). IR spectra were obtained with a Bruker-alpha infrared spectrometer device. The NMR spectra were recorded using two different instruments operating at 300 MHz and 400 MHz for 1H-NMR, and at 75 MHz and 101 MHz for 13C-NMR, respectively, using DMSO-d6 and CDCl3 (in 8b) solvents.
Synthesis of 3-acetylcoumarin (3)
In a two-neck round bottom flask, a mixture of substituted salicylaldehyde (0.1 mol) and methyl/ethyl acetoacetate in excess was stirred in the presence of the catalytic amount of pyridine for 30–50 min. The yellow colour solid product was formed in a reaction mixture, filtered washed with cold ethanol, and recrystallized in ethanol. The obtained product Rf and melting point values were matched with those given in the literature64.
Synthesis of 3-(2-bromoacetyl)-2H-chromene-2-one (4)
The substituted 3-acetyl coumarin 3 was dissolved in chloroform and bromine solution in chloroform was added dropwise using a dropping funnel at 0 °C for 15 min. After the addition of bromine, the temperature of the system increased to 30 °C with strong stirring for 3 h. The solvent evaporated at reduced pressure and the obtained solid was collected and recrystallized in ethanol64.
Synthesis of thiourea derivatives (7a–j)
Acyl thiourea derivatives (6a–j) were synthesized according to the reported method. The acyl part was removed by hydrolysis with KOH in the ethanol-water mixture to obtain thiourea derivatives65.
General procedure for thiazolyl-coumarin (8a–j)
An equimolar mixture of 3-bromoacetyl coumarin 4 (4.0 mmol) and drug-substituted thiourea 7a–j in absolute dry ethanol was refluxed for 3 h. During the reaction yellow precipitate appears. After completion of the reaction precipitate was filtered and washed with ethanol as given in Fig. 2.

Synthesis of thiazolyl coumarin conjugates (8a–j).
In-vitro enzyme Inhibition assay
Carbonic anhydrase inhibition activity was conducted using a previously established method with slight modifications66. In this approach, hCAII, hCAIX, and hCAXII were employed to hydrolyze p-nitrophenyl acetate into p-nitrophenol, and the resulting product was quantified spectrophotometrically. The reaction mixture comprised 60 µL of 50 mM Tris-sulfate buffer (pH 7.6, containing 0.1 mM ZnCl2), 10 µL (0.5 mM) of the test compound in 1% DMSO, and 10 µL of carbonic anhydrase. After thorough blending, the components were preincubated for 10 min at 25 °C. The plate reader at 348 nm was used to measure the plate. For the preparation of p-nitrophenyl acetate, a 6 mM stock solution was created using < 5% acetonitrile in the buffer and used immediately. To ensure a 0.6 mM concentration, 20 µL of the substrate solution was added to each well. The total assay volume was adjusted to 100 µL. After a 30-minute incubation at 37 ℃, the ingredients were thoroughly mixed, and a reading was taken at 348 nm. Acetazolamide served as the positive control, while 1% DMSO served as the negative control. The reported results represent the mean of three independent experiments (± SEM) and are expressed as percent inhibitions, calculated using the following formula:
\({\text{Inhibition }}\left( \% \right){\text{ }}={\text{ }}\left[ {{\text{1}}00{\text{ }} - \left( {{\text{Abs of test comp}}/{\text{Abs of control}}} \right){\text{ x1}}00} \right]\)
IC50 values of selected compounds exhibiting > 50% inhibition activity at 1.0 mM were further evaluated to measure IC50 value through serial dilution. The IC50 values of the synthesized compounds were calculated by using the non-linear curve fitting tool PRISM 5.0 (GraphPad, San Diego, CA, USA), as reported earlier67.
Density functional theory
The study employed Density Functional Theory (DFT) with the B3LYP method to meticulously assess the electronic characteristics of the molecules under investigation68. The widely used B3LYP method was chosen for these calculations, allowing predictions of molecular geometries, vibrational frequencies, and other properties. The optimization of compound geometries was carried out comprehensively using Gaussian 09, Revision D.01 (https://gaussian.com/gaussian09/) software69 Additionally, frequency calculations, Frontier Molecular Orbital (FMO) analysis, and the determination of global reactivity descriptors were conducted using the 6-31G basis set70. This basis set incorporates both primitive and Gaussian-type orbitals, offering the advantage of facilitating precise FMO analysis. Additionally, the energy gap between the HOMO and LUMO was computed to determine chemical hardness and softness, offering valuable information about the stability and potential reactivity of synthesized compounds69,71. For the visualization of generated files, GaussView 06 ( https://gaussian.com/gaussview6/) software is utilized72.
Molecular Docking studies
Molecular docking analysis, a computational technique used in drug discovery, predicts the binding interactions between a protein receptor and a small molecule ligand59. A newly synthesized series of compounds 8a–j was subjected to docking analysis to gain insights into their binding relationship with CA-II (carbonic anhydrase II), CA-IX (carbonic anhydrase IX) and CA-XII (carbonic anhydrase XII). The 3D crystal structures of human carbonic anhydrase II (PDB ID: 3V7X, resolution: 1.03 Å), human carbonic anhydrase IX (PDB ID:5FL4, resolution: 1.82 Å) and human carbonic anhydrase XII (PDB ID:5MSA, resolution: 1.20 Å) were retrieved from the protein data bank (http://www.rcsb.com/ )73,74.
To prepare the target protein structure, MGL tools version 1.5.7 (http://mgltools.scripps.edu/) were utilized, involving the removal of heteroatoms and water molecules, along with the addition of polar hydrogen atoms and Kollman charges. The protein underwent filling for any missing amino acid residues75. The optimized 3D structures of all newly synthesized compounds were generated using ChemDraw 3D ultra 20.0 (https://perkinelmerinformatics.com/products/research/chemdraw/)76.
The AutoDock version 4.2 (http://autodock.scripps.edu/) software’s default genetic algorithm served as the scoring function, and specific grid box dimensions were set for each target: CAII (x = 12.242480, y = 0.126193, z = 16.248152), CAIX (x = 24.497735, y=-19.652808, z = 59.811285), and CAXII (x = 14.024518, y = 0.706553, z=-12.901974). The procedure was validated by re-docking the co-crystal ligand followed by the validation of docking of prepared protein with positive control. Approximately 100 different configurations of docking complexes were generated for each compound (8a–j) with the active pockets of CAII, CAIX, and CAXII. The most stable configuration from the ensemble of configurations was selected for further analysis and data collection. To better understand the binding interactions, including hydrogen bonding, van der Waals interactions, pi-pi stacking, hydrophobic interactions, and covalent bonding, within the active sites of CAII, CAIX, and CAXII, 2D and 3D models were developed77.
ADMET analysis
The detailed assessment of the physicochemical and pharmacokinetic features of the synthesized derivatives was carried out using the SwissADME platform, available at (http://www.swissadme.ch/). This advanced computational tool provides a comprehensive analysis of various pharmacokinetic parameters, covering aspects of absorption, distribution, metabolism, and excretion (ADME)78. SwissADME employs advanced predictive algorithms to evaluate the drug-likeness and potential toxicity of compounds79. The bioavailability radar chart, driven by cutting-edge machine learning and statistical techniques, was finely calibrated using extensive molecular datasets with precisely defined properties. The BOILED-Egg model within this framework conclusively determines critical ADME properties, including blood-brain barrier (BBB) permeation, passive human gastrointestinal absorption (HIA), and the classification as a substrate or non-substrate for permeability glycoprotein80.
Results and discussion
Chemistry
Our approach was to develop new thiazolyl coumarin conjugate drug derivatives and explore their biological activities. Already reported inhibitors of Carbonic anhydrase isozymes containing 2, 4-thiazolidinedione-tethered coumarins60, and thiazolyl-pyrazoline derivatives61 possessed potent inhibitory activity.
For this designed strategy different thiazolyl coumarin conjugate drug derivatives (8a–j) were prepared in multiple steps as outlined in Fig. 1. All the synthesized compounds were yellow to light yellow amorphous solids having melting point ranges from 188 to 330 °C and their structure elucidation was done by analyzing 1H and 13C NMR and IR spectra. In IR spectra, differentiating peaks appeared in the range of 3389 − 3332 (N-H), 1720 − 1686 (C = O lactone), and 1610 − 1600 (C = N) which indicate synthesis of thiazolyl coumarin. In proton NMR spectra signal of amine was detected at about 10.75 to 5.23ppm. The single proton of the coumarin ring was observed at about 8.79-8.15ppm as a singlet. The single thiazole ring proton was embedded in the aromatic region. In addition, signals of aliphatic protons appeared in their respective ranges. From 13C NMR spectra, the signals of the 159.2 to 157.2 ppm can be for the carbonyl of the lactone ring. The signals of aliphatic and aromatic carbons were observed at 53 − 29 ppm and 156 − 108 ppm. The characteristic signal of thiazole ring carbon attached to two nitrogen and one Sulphur appeared at 162 ppm. The 1H-NMR,13C-NMR and IR of some selected compounds are given in supplementary data (Figure S1-S28).
Characterization data of Thiazolyl coumarins hybrids (8a–j)
3-(2-(((3s,5s,7s)-Adamantan-1-yl) amino) thiazol-4-yl)-2 H-chromen-2-one (8a)
Light yellow solid; yield 72%; Rf = 0.50; hexane/EtOAc 40%; mp 188–189 °C. FTIR (νmax/cm− 1): 3361.69 (N-H), 2941.16 (C-H aliphatic, asymmetric, sp3, 2835.11 (C-H aliphatic, symmetric, sp3, 1699.10 (C = O lactone), 1604.54 (C = N); 1H NMR (300 MHz, DMSO-d6): δ8.46 (s, pyranone, C-H, 1 H), 7.83 (d, 1 H, J = 7.5 Hz, Ar-H), 7.59 (t, 1 H, J = 7.5 Hz, Ar-H), 7.5 (s, 1 H, Ar-H), 7.48 (s, 1 H, Ar-H), 7.41 (d, 1 H, J = 8.1 Hz, Ar-H), 7.59 (t, 1 H, J = 7.5 Hz, Ar-H) 2.1–1.74 (m, 15 H, adamantyl); 13C NMR: (75 MHz, DMSO-d6): δ 165, 159.2, 152.5, 143.7, 138.3, 131.8, 129.2, 125.06, 121.1, 119.7,116.2,108.4,53.4,41.4,36.5,29.4. Anal. calcd. For C22H22N2O2S (378.14): C, 69.81; H, 5.86; N, 7.40; O, 8.45; S, 8.47 Found: C, 69.75; H, 5.80; N, 7.35. MS: m/z: 378.02 (96%).
3-(2-(((3s,5s,7s)-Adamantan-1-yl) amino) thiazol-4-yl)-6-bromo-2 H-chromen-2-one (8b)
Yellow solid; yield 80%; Rf = 0.45; hexane/EtOAc 40%; mp 264–266 °C. FTIR (νmax/cm− 1): 3389.46 (N-H), 2971.66 (C-H aliphatic, asymmetric, sp3, 2845.21 (C-H aliphatic, symmetric, sp3, 1720.08 (C = O lactone), 1600.34 (C = N); 1H NMR (300 MHz, CDCl3): δ8.42 (s, pyranone, C-H, 1 H), 7.74 (s 1 H, Ar-H), 7.71 (d, 1 H, J = 1.5 Hz, Ar-H), 7.56–7.60 (dd, 1 H, J = 1.8hz, J = 8.7 Hz, Ar-H ), 7.23 (d, 1 H, J = 9 Hz, Ar-H), 5.33 (s, 1 H, NH), 2.19–1.74 (m, 15 H, adamantyl); 13C NMR: (75 MHz, CDCl3): δ 164.9, 159, 151.5, 136.8, 133.7, 130.3, 121.6, 121.2, 118, 117, 110.3, 41.7, 36.2, 29.5. Anal. calcd. For C22H21BrN2O2S (456.05): C, 57.77; H, 4.63; Br, 17.47; N, 6.12; O, 7.00; S, 7.01 Found: C, 57.55; H, 4.38; N, 6.03. MS: m/z: 456.00 (93%).
N-(4-((4-(2-Oxo-2 H-chromen-3-yl)thiazol-2-yl)amino)-2 phenoxyphenyl)methanesulfonamide (8c)
Yellow solid; yield 76%; Rf = 0.51; hexane/EtOAc 40%; mp 181–182 °C. FTIR (νmax/cm− 1); 3334.58 (N-H), 3255.47 (N-H), 2924.33 (C-H aliphatic, asymmetric, sp3, 2852.99 (C-H aliphatic, symmetric, sp3, 1719.69 (C = O lactone), 1610.02 (C = N); 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1 H, NH), 9.22 (s, 1 H, NH), 8.31 (s, 1 H, CH), 7.77 (s, 1 H CH), 7.74–7.67 (m, 1 H, Ar-H), 7.65 (ddd, J = 8.5 Hz, 7.2, 1.6 Hz, 1 H, Ar-H), 7.50–7.38 (m, 7 H, Ar-H), 7.29–7.18 (m, 4 H, Ar-H), 7.17–7.03 (m, 2 H, Ar-H), 2.98 (s, 3 H, CH3) 13C NMR (101 MHz, DMSO-d6) δ 162.0, 158.6, 156.0, 152.2, 151.6, 143.4, 140.0, 138.2, 131.7, 130.0, 129.0, 128.8, 124.6, 123.9, 120.7, 120.1, 119.3, 119.0, 115.8, 112.0, 110.3, 106.4, 40.2. Anal. calcd. For C25H19N3O5S2 (505.07): C, 59.39; H, 3.79; N, 8.31; O, 15.82; S, 12.68 Found: C, 59.32; H, 3.68; N, 8.25. MS: m/z: 505.01 (97%).
N-(4-((4-(6-Bromo-2-oxo-2H-chromen-3-yl) thiazol-2-yl) amino)-2-phenoxyphenyl)methanesulfonamide (8d)
Yellow solid; yield 83%; Rf = 0.55; hexane/EtOAc 40%; mp 247–249 °C. FTIR (νmax/cm− 1): 3287.87 (N-H), 3255.47 (N-H), 3109.13 (C-H aliphatic, asymmetric, sp3, 3086.43 (C-H aliphatic, symmetric, sp3, 1699.76 (C = O lactone), 1601.02 (C = N); 1H NMR (300 MHz, DMSO-d6): δ 10.46 (s, 1 H, NH), 9.23 (s, 1 H, NH), 8.15 (s, 1 H) 7.80 (d, 1 H, J = 2.1 Hz, Ar-H ), 7.76 (s, 1 H, Ar-H) 7.75–7.71 (dd, 1 H, J = 2.1 Hz, J = 8.7 Hz, Ar-H ), 7.48–7.36 (m, 6 H, Ar-H) 7.22–7.13 (m, 3 H, Ar-H, Ar-H) 2.97 (s, 1 H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 162.4, 158.6, 156.5, 151.7, 151.5, 143.5, 140.4, 137.1, 134.3, 130.9, 130.4, 129.5, 124.2, 121.5, 121.3, 121.2, 119.4, 118.5, 116.7, 112.7, 111,6, 107.0, 38.9. Anal. calcd. For C25H18BrN3O5S2 (582.98): C, 51.38; H, 3.10; Br, 13.67; N, 7.19; O, 13.69; S, 10.97 Found: C, 51.32; H, 3.04; N, 7.15. MS: m/z: 505.01 (97%).
3-((4-(6-Bromo-2-oxo-2H-chromen-3-yl) thiazol-2-yl)amino)benzenesulfonamide (8e)
Yellow solid; yield 85%; Rf = 0.29; hexane/EtOAc 40%; mp 311–314 °C. FTIR (νmax/cm− 1): 3344.58 and 3314.23 (NH2 stretche ), 1705.38 (C = O lactone), 1601.58 (C = N), 1544.78 (N-H bending); 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1 H, NH), 8.73 (s, 1 H, CH), 8.71 (t, 1 H, J = 4 Hz, Ar-H), 8.05 (d, J = 2.4 Hz, 1 H), 7.86 (s, 1 H CH), 7.82–7.79 (m, 2 H, Ar-H), 7.57 (t, J = 7.9 Hz, 1 H), 7.48, 7.47 7.47 (d, J = 8.6 Hz, 1 H), 7.46 (s, 2 H, SO2NH2) 13C NMR: (101 MHz, DMSO-d6): δ 161.6, 158.1, 152.8, 144.1, 142.8, 137.2, 138.1, 133.1, 132.3, 130.4, 126.5, 121.5, 121.1, 119.8, 118.5, 115.7, 113.4, 112.4. Anal. calcd. For C25H18BrN3O5S2 (476.94): C, 45.20; H, 2.53; Br, 16.70; N, 8.78; O, 13.38; S, 13.40 Found: C, 45.18; H, 2.58; N, 8.72. MS: m/z: 476.89 (98%).
4-((4-(6-Bromo-2-oxo-2H-chromen-3-yl) thiazol-2-yl) amino)benzoic acid (8f)
Yellow solid; yield 79%; Rf = 0.32; hexane/EtOAc 40%; mp 326–328 °C. FTIR (νmax/cm− 1): 3332.61 (N-H), 3290.17 -2665.91 (broad OH carboxylic acid), 1708.10 (C = O carboxylic acid), 1686.71 (C = O lactone), 1600.23 (C = N); 1H NMR (300 MHz, DMSO-d6): δ 12.50(s, 1 H, COOH), 10.75 (s, 1 H, NH), 8.69 (s, 1 H), 8.3 (d, 1 H, J = 2.1 Hz, Ar-H ), 7.99 (d, 2 H, J = 8.7 Hz, Ar-H), 7.88 (d, 2 H, J = 8.7 Hz, Ar-H) 7.87 (s,1 H), 7.77–7.73 (dd, 1 H, J = 2.4 Hz, J = 8.7 Hz, Ar-H ), 7.41 (d, 1 H, J = 8.7 Hz, Ar-H); 13C NMR: (75 MHz, DMSO-d6): δ 167.5, 162.2, 158.8, 151.8, 145.0, 143.9, 137.9, 134.3, 131.4, 123.4, 121.7, 121.5, 121.1, 118.5, 116.7, 112.1. Anal. calcd. For C19H11BrN2O4S (441.96): C, 51.48; H, 2.50; Br, 18.03; N, 6.32; O, 14.44; S, 7.23 Found: C, 51.28; H, 2.43; N, 6.25. MS: m/z: 440.97 (99%).
6-Bromo-3-(2-(4-methylpiperazin-1-yl) thiazol-4-yl)-2H-chromen-2-one (8g)
Yellow solid; yield 78%; Rf = 0.31; hexane/EtOAc 40%; mp 318–320 °C. FTIR (νmax/cm− 1): 2959.16 (C-H aliphatic, asymmetric, sp3, 2877.21 (C-H aliphatic, symmetric, sp3, 1694.08 (C = O lactone), 1606.34 (C = N); 1H NMR (400 MHz, DMSO-d6) δ 8.67 (s, 1 H, CH), δ 8.11 (d, J = 2.3 Hz, 1 H), 7.84 (s, 1 H, CH), 7.77 (dd, J = 8.7 Hz, 2.5 Hz, 1 H), 7.43 (d, J = 8.8hz, 1 H), 4.03–3.78 (bs, 6 H, CH2), 2.86 (s, 1 H, CH3) 13C NMR (101 MHz, DMSO-d6) δ 168.7, 158.2, 151.3, 143.6, 137.6, 133.9, 130.6, 121.1, 121.0, 118.1, 116.3, 111.5, 51.4, 45.0, 42.2. Anal. calcd. For C17H16BrN3O2S (405.01): C, 50.26; H, 3.97; Br, 19.67; N, 10.34; O, 7.88; S, 7.89 Found: C, 50.21; H, 3.90; N, 7.79. MS: m/z: 404.97 (99%).
3-Bromo-4-((4-(6-bromo-2-oxo-2H-chromen-3-yl) thiazol-2-yl)amino)benzenesulfonamide (8h)
Yellow solid; yield 82%; Rf = 0.30; hexane/EtOAc 40%; mp 330–331 °C 1H NMR (300 MHz, DMSO-d6): δ 9.97 (s, 1 H, NH), 8.79 (d, 1 H, J = 9 Hz, Ar-H ), 8.6 (s, 1 H) 8.25 (d, 1 H, J = 2.4 Hz, Ar-H ),8.06 (d, 1 H, J = 2.4 Hz, Ar-H) 7.94 (s,1 H), 7.96–7.92 (dd, 1 H, J = 2.1 Hz, J = 8.7 Hz, Ar-H ) 7.78–7.74 (dd, 1 H, J = 2.1 Hz, J = 8.7 Hz, Ar-H ) 7.43 (d, 1 H, J = 9 Hz, Ar-H) 7.40 (s, 2 H, NH2) 13C NMR: (75 MHz, DMSO-d6): δ 162.6, 158.8, 151.8, 143.2, 141.8, 138.5, 137.9, 134.3, 131.4, 130.7, 126.9, 121.7, 121.5, 120.4, 118.5, 116.7, 113.4, 111.4. Anal. calcd. For C18H11Br2N3O4S2 (554.85): C, 38.80; H, 1.99; Br, 28.68; N, 7.54; O, 11.48; S, 11.51 Found: C, 38.75; H, 1.95; N, 7.54. MS: m/z: 554.80 (96%).
3-(2-(((1s,3s)-Adamantan-1-yl)amino)thiazol-4-yl)-6-methoxy-2H-chromen-2-one (8i)
Yellow solid; yield 80%; Rf = 0.45; hexane/EtOAc 40%; mp 264–266 °C. FTIR (νmax/cm− 1): 3389.46 (N-H), 2971.66 (C-H aliphatic, asymmetric, sp3, 2845.21 (C-H aliphatic, symmetric, sp3, 1720.08 (C = O lactone), 1600.34 (C = N); 1H NMR (400 MHz, DMSO-d6) δ 8.45 (s, 1 H), 7.53 (s, 1 H), 7.41–7.33 (m, 2 H), 7.18 (dd, J = 9.1, 3.0 Hz, 1 H), 4.48 (s, 1 H, NH) 3.84 (s, 3 H, OMe), 2.54–2.49 (m, 2 H), 2.10 (s, 8h), 1.71 (q, J = 12.3 Hz, 7 H).; 13C NMR (101 MHz, DMSO-d6) δ 164.6, 158.9, 155.7, 146.5, 143.1, 137.7, 120.6, 119.7, 119.2, 116.8, 110.5, 108.1, 55.8, 52.9, 40.9, 36.0, 28.9. Anal. calcd. For C23H24N2O3S (408.15): C, 67.62; H, 5.92; N, 6.86; O, 11.75; S, 7.85; Found: C, 67.53; H, 5.90; N, 6.80. MS: m/z: 408.09 (95%).
4-((4-(6-Methoxy-2-oxo-2H-chromen-3-yl)thiazol-2-yl)amino)benzoic acid (8j)
Yellow solid; yield 79%; Rf = 0.32; hexane/EtOAc 40%; mp 326–328 °C. FTIR (νmax/cm− 1): 3332.61 (N-H), 3290.17 -2665.91 (broad OH carboxylic acid), 1708.10 (C = O carboxylic acid), 1686.71 (C = O lactone), 1600.23 (C = N); 1H NMR (400 MHz, DMSO-d6) δ 12.58 (s, 1 H, COOH), 10.75 (s, 1 H, NH), 8.73 (s, 1 H, CH), 8.05–7.97 (m, 2 H), 7.93–7.86 (m, 3 H), 7.57 (d, J = 3.0 Hz, 1 H), 7.40 (d, J = 9.0 Hz, 1 H), 7.22 (dd, J = 9.0, 3.0 Hz, 1 H), 3.87 (s, 3 H, OMe).; 13C NMR (101 MHz, DMSO-d6) δ 167.0, 161.7, 158.9, 155.8, 146.7, 144.6, 143.7, 138.8, 130.9, 122.9, 120.2, 119.7, 119.5, 116.9, 116.2, 110.9, 110.8, 55.7. Anal. calcd. For C20H14N2O5S2 (384.06): C, 60.91; H, 3.58; N, 7.10; O, 20.28; S, 8.13 Found: C, 60.88; H, 3.46; N, 7.08. MS: m/z: 384.01 (98%).
In-vitro enzyme Inhibition assay
In the current investigation of various synthesized hybrid compounds 8a–j, it was found that derivatives 8b and 8d exhibited the most pronounced inhibitory effects. Specifically, compound 8b demonstrated a noteworthy IC50 value of 0.32 ± 0.04 µM against CAIX, while compound 8d displayed inhibitory activity with IC50 values of 0.38 ± 0.02 µM and 0.61 ± 0.05 µM against CAII and CAXII isozymes, respectively. Upon a detailed examination of derivative 8b, it was deduced that its substantial inhibitory effect could be attributed to the presence of 6-bromo-2 H-chromen-2-one. The involvement of the oxygen atom and terminal benzene ring in hydrogen bonding and pi-pi stacked interactions, respectively, contributed significantly to its inhibitory activity. Similarly, the investigation of compound 8d revealed potential interactions facilitated by the thiazole moiety, amino group, and terminal phenyl ring. The nitrogen atom of the thiazole moiety and the amino group formed hydrogen bonds within the active site of CAIX. Additionally, the sulphonamide moiety of compound 8d exhibited potent interactions with the active site amino acids of CAXII.
Remarkably, all synthesized hybrid compounds displayed greater inhibitory potential compared to acetazolamide, the reference inhibitor. The results were also compared with the previously reported compounds Notably, compound 8d emerged as a promising dual inhibitor of CAIX and CAXII, suggesting its potential therapeutic utility in addressing chronic diseases such as hypertension and cancer. The IC50 values for all synthesized hybrid compounds 8a–j are presented in Table 1.
Structurally, thiazolyl and coumarin ring in the parent structure are crucial for biological activity of these pharmacophore hybrid derivatives as described in Fig. 3. Furthermore, the substitution on these moieties facilitates the interaction of these compounds with the targeted proteins. The presence of adamantane ring on thiazolyl moiety on R2 position helps in the formation of hydrogen bond within the active pocket of CAIX. Similarly, phenoxyphenyl substitution at R2 position facilitate the interaction of 8c and 8d within the active site of CAXII.

Illustration of R groups on newly synthesized compounds 8a–8e.
In addition, substitution of 2-Br-SO2NH2Ph and 4-COOHPh at thiazolyl ring at R2 position renders 8h and 8j derivative as a good inhibitor of CAIX respectively. It was observed that the presence of electron withdrawing group i.e., phenyl group at terminal position facilitate the bonding interactions of these derivatives as shown in Fig. 4.

Illustration of R groups on newly synthesized compounds 8f–8j.
The coumarin moiety significantly enhances the activity of thiazolyl-coumarin hybrids by contributing to their binding affinity, selectivity, and pharmacological efficacy81. Coumarin derivatives have been identified as promising non-classical inhibitors of carbonic anhydrase (CA) isoforms, particularly CAIX and CAXII, which are overexpressed in various cancers. The hybridization of coumarin with thiazole units has been shown to yield compounds with potent inhibitory activity against these tumor-associated CA isoforms, with potent inhibitory potential. These hybrids leverage the structural advantages of both scaffolds to achieve enhanced potency and selectivity, making them promising candidates for targeted cancer therapy82..
Density functional theory
In the current study, the electronic properties of synthesized hybrids were predicted through density functional theory (DFT) calculations. 631-G’, the widely accepted basis set in quantum chemistry, was employed for detailed analysis of the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO), as given in Fig. 2. These orbitals provided a comprehensive detail regarding the electronic structures and reactivity of synthesized hybrid compounds, as given in Table 2.
The HOMO-LUMO energy difference profoundly influences a molecule’s reactivity. A larger gap indicates a “hard” molecule with heightened kinetic stability and reduced chemical reactivity. This significant gap creates a substantial energy barrier, impeding electron transfer and reactions, and results in a more even electron density distribution. Conversely, a “soft” molecule has a smaller HOMO-LUMO gap, making it less stable and more reactive. The smaller gap allows for easier electron transfer, facilitating chemical reactions and resulting in a greater electron density imbalance. Table 3 presents computed parameters based on HOMO and LUMO energy values to comprehensively assess reactivity and stability.
The synthesized hybrid compound 8e possesses the highest energy gap (0.137 eV) exhibiting remarkable stability and hardness (0.069 eV). While compounds 8d and 8g exhibit the lowest energy gap (0.120 eV) mitigating the more reactivity. The detailed geometry of HOMO and HUMO is shown in Fig. 5.

HOMO and LUMO geometries of synthesized hybrid compounds 8a–j.
DFT is primarily concerned with the ground-state electronic structure of an electronic system. Optimized structures predict molecular geometry, providing insights into the spatial arrangement of atoms. Various chrematistics including dipole moments and magnetic properties are derived from the accurate representation of the optimized structure as given in Fig. 6.

Energy-minimized 3D structures of synthesized hybrid compounds 8a–j obtained through molecular mechanics optimization.
The structures were geometry-optimized using DFT/B3LYP/6-31G in Gaussian, ensuring conformational stability for further in silico studies. Conclusively, the possible reaction sites of the synthetized compound were predicted by DFT analysis, where, the HOMO-LUMO analysis confirmed the electronic transitions among orbitals. The molecular docking approach was used to explore the interactions of the synthetized compound with the targeted enzymes.
Molecular Docking studies
In current study, the newly synthesized hybrid compounds 8a–j were docked within the active sites of target proteins, namely CAII, CAIX and CAXII. The most stable configuration with the least binding energy was retrieved and analyzed in 2D and 3D positions to elaborate the interactions formed between synthesized compounds 8a–j and targeted proteins.
The interactions of compound 8b within the active site of CAIX and compound 8d within the active site of CAII and CAXII demonstrated notable binding affinities as shown in Table 4. The key interactions between the compound 8b and the active site of Carbonic Anhydrase IX (CAIX) are crucial for its binding and inhibitory action as shown in Fig. 7. The amino group, attached to the thiazole ring, forms hydrogen bonds with polar residues in the enzyme’s active site, stabilizing the compound in place. These interactions are enhanced by the thiazole ring, which may also coordinate with the enzyme’s zinc ion, an essential component for CAIX’s catalytic activity, further supporting the inhibition process. The adamantane ring provides a rigid, hydrophobic structure that fits well into the enzyme’s hydrophobic pocket, contributing to the stability and specificity of the binding. Additionally, the chromone scaffold of the compound, with its aromatic nature, engages in π-π stacking interactions with aromatic residues like PHE243 and TRP17, adding further strength to the binding. The bromine substitution at position 6 of the chromone ring plays a role in modifying the electronic properties of the molecule, likely enhancing its interaction with nonpolar regions of the enzyme’s active site through hydrophobic interactions. Together, these interactions ensure that the compound binds efficiently to CAIX, potentially inhibiting its function and offering therapeutic benefits, particularly in cancer therapy.

The Most probable 3D(A) and 2D(B) binding pose of synthesized hybrid compound 8b within the active pocket of CAIX.
The compound 8d interacts with the active site of Carbonic Anhydrase II (CAII), displaying key structure-activity relationship (SAR) features in Fig. 8. The thiazole ring in the compound 8d plays a crucial role in coordinating with the zinc ion at the enzyme’s active site, which is essential for CAII’s catalytic activity. The amino group attached to the thiazole enhances the binding through hydrogen bonding with residues like ASN A:1011 and GLU A:1239, while the chromone scaffold engages in π-π stacking interactions with PHE A:1231, stabilizing the binding. The bromo-chromone ring provides an electronic modulation of the aromatic system, improving interaction with hydrophobic residues like TRP A:1005. Additionally, the methanesulfonamide group forms hydrogen bonds with HIS A:1010 and HIS A:1064, contributing to the overall binding affinity. The phenoxyphenyl group further stabilizes the complex through hydrophobic interactions, providing additional binding strength. These interactions ensure strong and selective inhibition of CAII, with the potential for therapeutic applications in diseases involving CAII, such as glaucoma and other CA-related disorders.

The Most probable 3D (A) and 2D (B) binding pose of synthesized hybrid compound 8d within the active pocket of CAII.
Similarly, Fig. 9 illustrates a comprehensive analysis of the 3D and 2D binding interactions between compound 8d and CAXII. The bromo-chromone scaffold forms π-π stacking interactions with aromatic residues like PHE A:1231, while the thiazole ring coordinates with the enzyme’s zinc ion and forms hydrogen bonds with SER A:130 and SER A:133, enhancing binding stability. The methanesulfonamide group forms additional hydrogen bonds with residues such as ASN A:64 and GLN A:89, while the phenoxyphenyl group provides hydrophobic interactions with LEU A:197. Overall, these interactions ensure strong binding and inhibition of CAIX, making the compound a promising candidate for cancer therapy targeting this enzyme. 2D and 3D binding modes of all other synthesized compounds are given in the Supplementary file (Figure S29-S55).

The Most probable 3D (A) and 2D (B) binding pose of synthesized hybrid compound 8d within the active pocket of CAXII.
Similarly, Acetazolamide interacts with the active sites of CAII, CAIX, and CAXII through several key interactions given in Fig. 10, contributing to its inhibitory effects. In CAII, the sulfonamide group forms hydrogen bonds with HIS A:1004 and GLY A:1006, while also interacting with HIS A:1008 and TYR A:1007, stabilizing the enzyme-inhibitor complex. In CAIX, the sulfonamide group engages in hydrogen bonding with THR A:200 and ASN A:596, while the bromo group facilitates π-π stacking interactions with PHE A:1231, enhancing binding specificity. For CAXII, the sulfonamide group interacts with GLU A:253 and LYS A:253, while the phenyl group contributes to hydrophobic interactions with LEU A:26 and GLN A:27, further supporting inhibition. These interactions enable acetazolamide to effectively inhibit carbonic anhydrase isoforms, making it useful for treating conditions like glaucoma and metabolic disorders.

The Most probable 3D (A) and 2D (B) binding pose of acetazolamide within the active pocket of CAII, CAIX and CXII.
ADMET analysis
The study involved evaluating the physicochemical properties of synthesized hybrid compounds 8a–j using the SwissADME tool. Notably, compounds 8a, 8b, 8g, and 8j exhibited the highest gastrointestinal (GI) absorption, suggesting their potential as promising candidates for pharmacological effects. In terms of pharmacodynamics, a significant number of the compounds demonstrated inhibition of key enzymes, namely CYP2C19, CYP2C9, and CYP3A4. The detailed physicochemical and pharmacokinetic profiles of these derivatives are presented in Table 5.
The comprehensive depiction of ADMET properties of synthesized hybrid compounds 8a–j is shown via a radar chart in Fig. 11. Each parameter is plotted on a separate axis, and the data points are connected to form a polygon, providing a visual overview of the overall pharmacokinetic profile.

Radar charts describing physicochemical and pharmacokinetic properties of synthesized hybrid compounds 8a–j.
The visual representation, depicted in Fig. 12, the boiled egg diagram, serves as a tool for visualizing drug-likeness properties, particularly concerning blood-brain barrier (BBB) permeation. Among the compounds examined, only compound 8g demonstrated the ability to cross the BBB. The compounds 8c, 8d, 8e, 8f, 8h, and 8i, were found to possess reduced absorption and distribution across the cells or tissues.

BOILED-Egg model illustrating the predicted gastrointestinal absorption and blood-brain barrier (BBB) permeability of synthesized hybrid derivatives 8a–j.
Furthermore, the SwissTarget Prediction tool was employed to predict potential protein targets, with rankings based on probability scores, as illustrated in Fig. 13. A higher score indicates a greater likelihood of the molecule binding to that specific protein target. To establish a conclusive understanding, the results were cross-validated with experimental data. Notably, carbonic anhydrase, classified as a lyase enzyme, emerged as a potential target. Experimental data support the conclusion that nearly all synthesized hybrid derivatives 8a–j may serve as potential inhibitors against carbonic anhydrase isozymes.

Predicted protein targets for compounds 8a–j using the SwissTarget Prediction tool.
Conclusions
A novel series of thiazolyl-coumarin-drug conjugates (8a–j) was synthesized via reflux condensation reaction. The synthesized compounds were comprehensively analyzed through in-vitro and in-silico studies, as potential carbonic anhydrase inhibitors. During in vitro analysis, it was found that derivatives 8b and 8d exhibited the most potential inhibitory effects on the targeted enzymes. Among all the compound 8b demonstrated a notable IC50 value of 0.32 ± 0.04 µM against CAIX, while compound 8d exhibited potent inhibitory activity with an IC50 values of 0.38 ± 0.02 µM and 0.61 ± 0.05 µM against CAII and CAXII isozymes, respectively. The DFT analysis showed that the reduced HOMO/LUMO energy gaps of all the synthesized hybrid compounds and increased dipole moment were responsible for the higher binding affinity, especially 8b and 8d compounds. Moreover, the docking study depicted their strong molecular interactions within the binding pocket of CAII, CAIX and CAXII isozymes. The ADMET analysis of newly synthesized hybrid compounds was accessed for the drug-likeness properties, particularly concerning blood-brain barrier (BBB) permeation. Among the compounds examined, only compound 8g showed the ability to cross the BBB. The results were confirmed by cross-validating with experimental data. This work establishes new possibilities for developing dual inhibitors with pharmacological activity against CAII and CAXII, exhibiting versatile biological applications.
Data availability
Data will be available on request by the corresponding author.
References
Supuran, C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery. 7, 168–181 (2008).
Supuran, C. T. Vol. 8 25 (MDPI, 2018).
Akocak, S. & Supuran, C. T. Activation of α-, β-, γ-δ-, ζ-and η-class of carbonic anhydrases with amines and amino acids: A review. J. Enzyme Inhib. Med. Chem. 34, 1652–1659 (2019).
De Simone, G. & Supuran, C. T. In) organic anions as carbonic anhydrase inhibitors. J. Inorg. Biochem. 111, 117–129 (2012).
Lomelino, C. L., Andring, J. T. & McKenna, R. Crystallography and its impact on carbonic anhydrase research. Int. J. Med. Chem. (2018). (2018).
Supuran, C. T. & Scozzafava, A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg. Med. Chem. 15, 4336–4350 (2007).
Haider, M. B. et al. Design, synthesis, characterization and exploration of biological efficacy of iminothiazoline sulfonamide hybrids as efficient inhibitor of carbonic anhydrase. J. Mol. Struct. 142331 (2025).
Kciuk, M. et al. Targeting carbonic anhydrase IX and XII isoforms with small molecule inhibitors and monoclonal antibodies. J. Enzyme Inhib. Med. Chem. 37, 1278–1298 (2022).
Jaitak, A., Kumari, K., Kounder, S. & Monga, V. Carbonic anhydrases: moiety appended derivatives, medicinal and pharmacological implications. Bioorg. Med. Chem., 117933 (2024).
Mishra, K. A. & Sethi, K. K. Unveiling tomorrow: carbonic anhydrase activators and inhibitors pioneering new frontiers in Alzheimer’s disease. Arch. Pharm. 358, e2400748 (2025).
D’Ambrosio, K., Di Fiore, A. & Langella, E. Dual targeting carbonic anhydrase inhibitors as promising therapeutic approach: a structural overview. Front. Mol. Biosci. 12, 1511281 (2025).
Nocentini, A., Capasso, C. & Supuran, C. (2023).
Petrenko, M. et al. Combined 3-O-acetylbetulin treatment and carbonic anhydrase IX Inhibition results in additive effects on human breast cancer cells. Chemico-Biol. Interact. 333, 109326 (2021).
Güttler, A. et al. Cellular and Radiobiological effects of carbonic anhydrase IX in human breast cancer cells. Oncol. Rep. 41, 2585–2594 (2019).
Williams, K. J. & Gieling, R. G. Preclinical evaluation of ureidosulfamate carbonic anhydrase IX/XII inhibitors in the treatment of cancers. Int. J. Mol. Sci. 20, 6080 (2019).
Vogel, A. J. A. D. P. Darstellung von benzoesäure Aus D.r Tonka-Bohne und Aus D.n Meliloten‐oder Steinklee‐Blumen. 64, 161–166 (1820).
Guibourt, N. J. B. G. Histoire Abrégée Des Drogues Simples, Vol. 1 (Méquignon-Marvis, 1826).
Peng, X. M., Damu, L. V. & Zhou, H. G.J. C. Current developments of coumarin compounds in medicinal chemistry. 19, 3884–3930 (2013).
Lafitte, D. et al. DNA gyrase interaction with coumarin-based inhibitors: the role of the hydroxybenzoate isopentenyl moiety and the 5 ‘-methyl group of the noviose. 41, 7217–7223 (2002).
Curir, P., Galeotti, F., Dolci, M., Barile, E. & Lanzotti, V. J. Pavietin, a coumarin from Aesculus P.via with antifungal activity. 70, 1668–1671 (2007).
Satyanarayana, V., Sreevani, P., Sivakumar, A. & Vijayakumar, V. J. A. Synthesis and antimicrobial activity of new schiff bases containing coumarin moiety and their spectral characterization. 17, 221–233 (2008).
Arshad, A. et al. Synthesis and antimicrobial properties of some new Thiazolyl coumarin derivatives. 46, 3788–3794 (2011).
Anderson, D. M., Shelley, S., Crick, N. & Buraglio, M. J. T. No effect O. the novel antidiabetic agent nateglinide O. the pharmacokinetics and anticoagulant properties O. warfarin in healthy volunteers. 42, 1358–1365 (2002).
Tàssies, D. et al. Pharmacogenetics of acenocoumarol: cytochrome P450 CYP2C9 polymorphisms influence dose requirements and stability of anticoagulation. 87, 1185–1191 (2002).
Weick, J. & Thorn, R. S. Effects O. acute sublethal E.posure to coumaphos O. Diazinon O. acquisition and discrimination O. O.or stimuli in the honey bee (Hymenoptera: Apidae). 95, 227–236 (2002).
Shia, J. S. et al. Exploring the multifaceted biological activities of 4-((5-Amino-1, 3, 4-Thiadiazol-2-yl) Methoxy) coumarin: antioxidant, antibacterial, and antifungal properties. J. Med. Chem. Sci. 7, 954–968 (2024).
Stone, W. B., Okoniewski, J. C. & Stedelin, J. R. Poisoning of wildlife with anticoagulant rodenticides in New York. 35, 187–193 (1999).
Khan, K. M. et al. Synthesis of coumarin derivatives with cytotoxic, antibacterial and antifungal activity. J. Enzyme Inhib. Med. Chem. 19, 373–379 (2004).
Kashyap, S. J. et al. Thiazoles: Having Diverse Biol. Activities 21, 2123–2132 (2012).
Desai, N., Bhatt, N., Somani, H. & Trivedi, A. J. Synthesis, antimicrobial and C.totoxic activities O. some novel thiazole C.ubbed 1, 3, 4-oxadiazoles. 67, 54–59 (2013).
Bondock, S., Naser, T. & Ammar, Y. A. Synthesis of some new 2-(3-pyridyl)-4, 5-disubstituted thiazoles as potent antimicrobial agents. J. E J. O M C. 62, 270–279 (2013).
Vukovic, N., Sukdolak, S., Solujic, S. & Milosevic, T. J. A. Synthesis and antimicrobial evaluation of some novel 2-Aminothiazole derivatives of 4‐Hydroxy‐chromene‐2‐one. 341, 491–496 (2008).
Chimenti, F. et al. Synthesis and anti-Helicobacter pylori Activity of 4‐(Coumarin‐3‐yl) thiazol‐2‐ylhydrazone Derivatives. 42 (2011).
Hassan, M. Z., Osman, H., Ali, M. A. & Ahsan, M. J. Therapeutic potential of coumarins as antiviral agents. J. E J. O M C. 123, 236–255 (2016).
Olomola, T. O., Klein, R., Lobb, K. A., Sayed, Y. & Kaye, P. T. Towards the synthesis of coumarin derivatives as potential dual-action HIV-1 protease and reverse transcriptase inhibitors. 51, 6325–6328 (2010).
Osman, H. et al. New thiazolyl-coumarin hybrids: design, synthesis, characterization, X-ray crystal structure, antibacterial and antiviral evaluation. 1166, 147–154 (2018).
Ramagiri, R. K. et al. A facile one-step multi-component approach T.ward T.e synthesis of 3-(2-amino-4-thiazolyl) coumarins by using T.imethylsilyl isothiocyanate and T.eir antioxidant and anti-inflammatory activity. 190, 1393–1397 (2015).
Sharma, A. Advances in Structure and Activity Relationship of Coumarin Derivatives, 119–136 (Elsevier, 2016).
Rahman, F. S. A., Yusufzai, S. K., Osman, H. & Mohamad, D. Synthesis, characterisation and cytotoxicity activity of thiazole substitution of coumarin derivatives (characterisation of coumarin derivatives). 27, 77 (2016).
Emami, S. & Dadashpour, S. J. Current developments O. coumarin-based anti-cancer agents in medicinal chemistry. 102, 611–630 (2015).
Satish, G. Advances in Structure and Activity Relationship of Coumarin Derivatives, 151–159 (Elsevier, 2016).
Matos, M. et al. Heterocyclic antioxidants in nature: coumarins 21, 311–324 (2017).
Kostova, I. et al. Coumarins as antioxidants 18, 3929–3951 (2011).
Al-Majedy, Y., Al-Amiery, A., Kadhum, A. A., & BakarMohamad, A. Antioxidant activity of coumarins. Syst. Rev.Pharm. 8 (1), 24 (2017).
Siddiqui, N., Arshad, M. F. & Khan, S. A. Synthesis of some new coumarin incorporated Thiazolyl semicarbazones as anticonvulsants. Acta Pol. Pharm. 66, 161–167 (2009).
Raval, J. P., Desai, J. T., Desai, C. K. & Desai, K. R. J. A. A comparative study of microwave assisted and conventional synthesis of 2, 3-dihydro-2-aryl-4-[4-(2–oxo–2H–chromen–3–yl)–1, 3-thiazol–2–ylamino]-1, 5–benzothiazepines and its antimicrobial activity. 12, 233–244 (2008).
Desai, J., Desai, C. & Desai, K. J. A convenient, rapid and eco-friendly synthesis of isoxazoline heterocyclic moiety containing Bridge at 2-amine as potential Pharmacological agent. J. Iran. Chem. Soc.. 5, 67–73 (2008).
Ansari, M. I., Khan, S. A. Synthesis and antimicrobial activity of some novel quinoline-pyrazoline-based coumarinyl thiazole derivatives. 26, 1481–1496 (2017).
Parveen, S. et al. Computational and biological studies of novel Thiazolyl coumarin derivatives synthesized through Suzuki coupling. 44, 1610–1622 (2020).
Kathiravan, M. K. et al. The biology and chemistry of antifungal agents: A review. 20, 5678–5698 (2012).
Sonmez, F. et al. Design, synthesis and Docking study of novel coumarin ligands as potential selective acetylcholinesterase inhibitors. 32, 285–297 (2017).
Madni, M. et al. Synthesis, quantum chemical, in vitro acetyl cholinesterase Inhibition and molecular Docking studies of four new coumarin based Pyrazolylthiazole nuclei. 1168, 175–186 (2018).
Saeed, A. et al. 3-(5-(Benzylideneamino) thiazol-3-yl)-2 H-chromen-2-ones: A new class of alkaline phosphatase and ecto-5′-nucleotidase inhibitors. 6, 21026–21036 (2016).
Meeran, I. S., Tajudeen, S. S., Dusthakeer, V. A. & Shabeer, T. An Insight into the Anti-Tubercular Potential of Schiff Bases.
Kurt, B. Z. et al. Synthesis, antioxidant and carbonic anhydrase I and II inhibitory activities of novel sulphonamide-substituted Coumarylthiazole derivatives. 31, 991–998 (2016).
Saeed, A., Ejaz, S. A., Ul-Hamid, A., El-Seedi, H. R. & Iqbal, J. Synthesis of and molecular Docking studies of azomethine-tethered sulfonamides as carbonic anhydrase II & 15-lipoxygenase inhibitors. J. Mol. Struct. 1243, 130821 (2021).
Saeed, A. et al. Biochemical characterization, and in-Silico investigations of Acyl-3-(Ciprofloxacinyl) thioureas as inhibitors of carbonic Anhydrase-II. Polycycl. Aromat. Compd. 43, 8946–8964 (2023). Synthesis.
Hussain, T. et al. Synthesis, characterization and biological evaluation of pyrazole-based benzene sulfonamides as inhibitors of human carbonic anhydrase II, IX and XII. RSC Adv. 13, 18461–18479 (2023).
Al-Rashida, M. et al. Diarylsulfonamides and their bioisosteres as dual inhibitors of alkaline phosphatase and carbonic anhydrase: structure activity relationship and molecular modelling studies. Bioorg. Med. Chem. 23, 2435–2444 (2015).
Eldehna, W. M. et al. Discovery of 2, 4-thiazolidinedione-tethered coumarins as novel selective inhibitors for carbonic anhydrase IX and XII isoforms. J. Enzyme Inhib. Med. Chem. 37, 531–541 (2022).
Sever, B., Türkeş, C., Altıntop, M. D., Demir, Y. & Beydemir, Ş. Thiazolyl-pyrazoline derivatives: in vitro and in Silico evaluation as potential acetylcholinesterase and carbonic anhydrase inhibitors. Int. J. Biol. Macromol. 163, 1970–1988 (2020).
Ibrar, A., Shehzadi, S. A., Saeed, F. & Khan, I. Developing hybrid M.lecule therapeutics for diverse enzyme inhibitory action: Active role of coumarin-based structural leads in drug discovery. 26, 3731–3762 (2018).
Zhang, L. & Xu, Z. J. E. Coumarin-containing hybrids and their anticancer activities. 181, 111587 (2019).
Ibrar, A. et al. Facile and expedient access to bis-coumarin–iminothiazole hybrids by molecular hybridization approach: synthesis, molecular modelling and assessment of alkaline phosphatase Inhibition, anticancer and antileishmanial potential. RSC Adv. 5, 89919–89931 (2015).
Woodring, J. L. et al. Disrupting the conserved salt Bridge in the trimerization of influenza A nucleoprotein. 63, 205–215 (2019).
Ali, D. et al. Utilization of transition metal fluoride-based solid support catalysts for the synthesis of sulfonamides: carbonic anhydrase inhibitory activity and in Silico study. RSC Adv. 12, 3165–3179 (2022).
Swift, M. L. GraphPad Prism, data analysis, and scientific graphing. J. Chem. Inf. Comput. Sci. 37, 411–412 (1997).
Hossen, J., Ali, M. A. & Reza, S. Theoretical investigations on the antioxidant potential of a non-phenolic compound thymoquinone: a DFT approach. J. Mol. Model. 27, 173 (2021).
Bhavani, K., Renuga, S. & Muthu, S. Quantum mechanical study and spectroscopic (FT-IR, FT-Raman, 13 C, 1H) study, first order hyperpolarizability, NBO analysis, HOMO and LUMO analysis of 2-acetoxybenzoic acid by density functional methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 136, 1260–1268 (2015).
McGrath, M. P. & Radom, L. Extension of Gaussian-1 (G1) theory to bromine‐containing molecules. J. Chem. Phys. 94, 511–516 (1991).
Vanitha, U., Elancheran, R., Manikandan, V., Kabilan, S. & Krishnasamy, K. Design, synthesis, characterization, molecular Docking and computational studies of 3-phenyl-2-thioxoimidazolidin-4-one derivatives. J. Mol. Struct. 1246, 131212 (2021).
Dennington, R., Keith, T. A. & Millam, J. M. GaussView 6.0.16. (Semichem Inc., 2016).
Burley, S. K. et al. Protein data bank (PDB): The single global macromolecular structure archive. Protein Crystallogr. Methods Protocols, 627–641 (2017).
Prieto-Martínez, F. D., Arciniega, M. & Medina-Franco, J. L. Molecular docking: current advances and challenges. TIP. Rev. Especi. Cienc. Químico-Biol. 21 (2018).
Goodsell, D. S., Sanner, M. F., Olson, A. J. & Forli, S. The AutoDock suite at 30. Protein Sci. 30, 31–43 (2021).
Brown, T. & ChemDraw Sci. Teacher 81, 67 (2014).
Aziz, M. et al. Identification of potent inhibitors of NEK7 protein using a comprehensive computational approach. Sci. Rep. 12, 6404 (2022).
Daina, A. & Zoete, V. Application of the SwissDrugDesign online resources in virtual screening. Int. J. Mol. Sci. 20, 4612 (2019).
Jia, C. Y., Li, J. Y., Hao, G. F. & Yang, G.-F. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov.Today. 25, 248–258 (2020).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23, 3–25 (1997).
Singh, P. et al. Design, synthesis and in vitro evaluation of novel thiazole-coumarin hybrids as selective and potent human carbonic anhydrase IX and XII inhibitors. Int. J. Biol. Macromol. 268, 131548 (2024).
Thacker, P. S. et al. Synthesis and biological evaluation of coumarin-thiazole hybrids as selective carbonic anhydrase IX and XII inhibitors. Arch. Pharm. 355, 2200232 (2022).
Acknowledgements
The authors extend their appreciation to the University Higher Education Fund for funding this research work under the Research Support Program for Central Labs at King Khalid University, Saudi Arabia, through the project number CL/CO/A/8.
Author information
Authors and Affiliations
Contributions
The study plan was designed and supervised by S.A.E., P.A.C., and A.S. The whole experimental work was carried out by M.Y.A. and R.B. Writing of the original draft was carried out by A.A., R.K., and H.M.A.,. Formal analysis and an investigation was carried out by C.L., validated the results of all experimental work. All co-authors read and approved the manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ahmed, A., Kanwal, R., Channar, P.A. et al. Synthesis and evaluation of thiazolyl coumarin drug conjugates as carbonic anhydrase isozyme inhibitors by using integrated invitro and insilico approaches. Sci Rep 15, 22032 (2025). https://doi.org/10.1038/s41598-025-03115-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-03115-3
Keywords
This article is cited by
-
Synthesis, docking, SAR and ADMET evaluation of novel pyrrolo[3,4-c]pyrazole-4,6-dione derivatives
Scientific Reports (2026)
