
Abstract
Metagenomic next-generation sequencing (mNGS) is widely used to diagnose complex infections in hospitalized patients, particularly those associated with COVID-19 which has garnered significant concern over the past five years. To investigate the molecular epidemic of the viral variant and the potential co-infection pathogens, we conducted retrospective mNGS analysis of 254 SARS-CoV-2-positive specimens collected from 200 hospitalized patients between March and September 2023. Phylogenetic analysis of the identified Omicron subvariants showed minimal evolutionary divergence, with no association between sub-lineages and pneumonia severity. Notably, mNGS demonstrated enhanced detection of polymicrobial coinfections, identifying bacterial, fungal, and viral co-pathogens in 92.5% (185/200) of cases. Pneumonia severity was associated with advanced age (proportion of elderly patients: 61.1 vs 78.3%; p = 0.032) and comorbid conditions, particularly diabetes mellitus (OR 2.03, 95% CI 1.03–4.02, p = 0.041), but showed no correlation with SARS-CoV-2 sub-lineages or coinfecting pathogens. While mNGS enhances coinfection diagnosis, COVID-19 outcomes are predominantly driven by host factors rather than Omicron subvariant evolution. Prioritized monitoring of elderly and comorbid individuals remained critical for severe pneumonia management.
Similar content being viewed by others


Introduction
Metagenomic next-generation sequencing (mNGS), an efficient, high-throughput approach for pathogen detection, is increasingly used in clinical laboratories1,2. It is particularly effective in identifying rare, novel, and hard-to-detect pathogens3. A prominent example is the rapid identification of SARS-CoV-2 within one week in the early breakout of coronavirus disease 2019 (COVID-19) pandemic using mNGS4. In addition to diagnosing infectious diseases, mNGS also performed well in phylogenetic analysis of SARS-CoV-2 compared to whole-genome sequencing5. Since its initial emergence, SARS-CoV-2 has evolved from the ancestral strain through multiple generations of Omicron subvariants6. As the persistent emergence of SARS-CoV-2 variants in recurrent waves, particularly frequent sequential reinfections with Omicron subvariants, necessitates updated vaccination strategies7, the evolution of virus and its clinical significance should be monitored8,9.
SARS-CoV-2 often leads to co-infections secondary infections with bacteria, fungi, and other viruses10. Compared with traditional diagnostic methods such as real-time PCR, microbial culture, and antigen or antibody detection, mNGS can simultaneously detect all microorganisms present in a specimen11,12. Combined diagnostic approaches with mNGS have resulted in a five-fold increase in the detection rate of the pathogens in adult patients with severe community-acquired pneumonia (SCAP), compared with only routine cultures13. Metagenomic next-generation sequencing has demonstrated established clinical utility in infection diagnosis and therapeutic regimen adjustment, particularly through its capability to comprehensively characterize polymicrobial infections14,15.
In this study, we conducted a retrospective analysis to gain detailed insights into SARS-CoV-2 variants and potential co-infections in COVID-19 cases using metagenomic next-generation sequencing (mNGS) data collected over six months.
Material and methods
Study design and SARS-CoV-2 positive samples collection
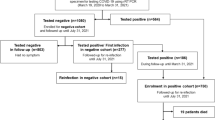
A total of 5,686 samples were collected retrospectively from March to September 2023 in Ruijin Hospital Affiliated to Shanghai Jiaotong University School of Medicine, of which 254 (4.47%) were positive for SARS-CoV-2. Only one result was included for each patient. Fifty-four samples were excluded due to repeated patient sources (Fig. 1). Most samples were from respiratory tract specimens, including 116 sputum (58%), 42 bronchoalveolar lavage fluid (BALF) (21%), and 38 nasopharyngeal swabs (19%). Four patients tested positive for SARS-CoV-2 in blood, with no other types of samples collected from them.

The flowchart of sample and clinical data collection of COVID-19 patients. COVID-19 corona virus disease 2019. *Only one sample of each patient was enrolled.
Wet-lab pipelines for mNGS
We employed metagenome, meta-transcriptome and targeted metagenome to detect pathogen as previously described2. All samples were centrifuged at 12,000 × g for 5 min to collect the pathogens and human cells. Next, 50 μL of precipitate was taken to deplete of host nucleic acid using 1 U of Benzonase (Sigma) and 0.5% Tween 20 (Sigma), incubated at 37 °C for 5 min. The reaction was stopped with 400 μL of a terminal buffer16. The unique molecular sample index (UMSI), composed of quantified DNA fragments, was spiked into each sample as a maker and internal control. The mixture (600μL) was triturated in new tubes with 500 μL of ceramic beads utilizing a Minilys personal TGrinder H24 homogenizer (Tiangen, China). Then, nucleic acid was extracted and eluted in 65 μL of elution buffer by QIAamp UCP pathogen minikit (Qiagen, Germany)2. For RNA extraction and reverse transcription, 200 μL of the samples was extracted following manufacturer’s instructions with QIAamp Viral RNA Mini Kit (Qiagen, Germany). cDNAs were generated using the QIAseq FastSelect -rRNA HMR Kit (Qiagen, Germany); Both DNA and cDNA were quantified using a Qubit double-stranded DNA (dsDNA) high-sensitivity (HS) assay kit (Invitrogen, USA). Indexed DNA libraries were constructed with a TruePrep DNA Library Prep Kit (Vazyme Biotech, China)17,18. 4) An aliquot of 750 ng library from each sample was used for hybrid capture-based microbial probe19 enrichment through one round of hybridization (SeqCap EZ Library, Roche, USA). Library was quantified with a Qubit dsDNA HS Assay Kit and assessed using an Agilent 2100 Bioanalyzer and a High Sensitivity DNA Kit (Agilent Technologies, USA). Sequencing was performed with an Illumina NextSeq 550 sequencer (Illumina, USA) with 75-cycle single-end sequencing.
Bioinformatics analyses for microorganism
Bioinformatics analyses were performed as previously described with adjusted details20. Trimmomatic21 and K-complexity22 were employed to remove low quality and low complexity reads, adapter contamination, and duplicate reads, as well as those shorter than 40 bp. Human sequence data were discarded by mapping to a human reference genome (hg38) using Burrows-Wheeler Aligner software23. The final database of representative assemblies of microorganisms and pathogen lists consisted of approximately 20,000 genomes24. SNAP v1.0beta.18 (https://github.com/amplab/snap) was used to align the microbial reads. DNA or RNA viruses reported as positive required coverage of three or more non-overlapping regions on the genome. A species/genus-positive detection was defined when the reads per million (RPM) ratio, or RPM-r, was more than 5, where RPM-r was defined as the RPM in the clinical sample divided by the RPM in the negative control25. Each finalized report was reviewed by two qualified medical technicians. The presence of co-infections was determined through a) potential pathogenicity assessment of detected pathogens at the sampling site; b) relevance to conventional laboratory results; c) final clinical assessment by clinicians.
SARS-CoV-2 whole genome and phylogenetic analysis
Adapter sequences and reads of low-quality bases(Q20 < 10) and shorter than 40 bp were removed by fastp26. High-quality reads were aligned to the reference genome of SARS-CoV-2 (GenBank: MN908947.3) utilizing the Burrows-Wheeler Aligner (BWA)27. Reads with high mapping quality over 25 were reserved using SAMtools28. A mutant allele was designated if its frequency was ≥ 0.7, while a degenerate nucleotide was assigned if the mutant allele frequency ranging 0.3 to 0.7; otherwise, the reference allele was assigned. Variants with a minor allele frequency (MAF) > 0.3 and their impacts on genes, transcripts, protein sequences, and regulatory regions were annotated using the ensemble variant effect predictor (VEP)29,30. Sequencing depth and genome coverage were calculated based on high-quality mapped (HQM) reads without duplications. SARS-CoV-2 genomes were analyzed using the Nextclade tool (https://clades.nextstrain.org) for multiple alignment and phylogenetic tree construction31. SARS-CoV-2 lineage was followed Pongo lineage with PUSHER-v 1.19 database32. Tree visualization was carried out using iTOL33.
Clinical data collection
Clinical characteristics, physical examination data, medications and treatments of COVID-19 patients were extracted from admission and discharged records. Severe pneumonia was assessed by clinical physicians according to multiple scoring systems, including CURB-6534, Pneumonia Severity lndex (PSI)35, CURXO36 and SMART-COP37. Patients were divided into two groups according to the presence or absence of severe pneumonia. There were 69 cases in the severe pneumonia group and 131 cases in the non-severe pneumonia group. Radiological images were reviewed by qualified physicians. White blood cell (WBC), neutrophil and lymphocyte counts were collected from the reports closest to the sampling time of the mNGS specimen involved and no more than a week. The study was approved by the Ethics Committee of Ruijin Hospital in accordance with the Helsinki Declaration.
Statistics
Continuous variables not normally distributed are presented as medians (interquartile range [IQR]), while categorical variables are described as counts (%). Blood cell counts were analysed using the mean and corresponding 95% confidence interval (95% CI). The Wilcoxon signed-rank test and the Mann–Whitney U test were used for comparative analysis, as appropriate. The chi-square test (χ2) was employed to assess distributions across different groups. The Hosmer–Lemeshow test were performed in the unconditional logistic regression analysis, where the variables were selected by back-ward procedure. Graphs were created using GraphPad Prism version 8.4.0 software. Venn diagrams were generated with jvenn, an interactive Venn diagram viewer. Statistical analyses were conducted using SPSS version 27.0 software, and a two-sided p-value of < 0.05 was considered statistically significant.
Results
Pango lineage of SARS-CoV-2 variants
Reliable SARS-CoV-2 Pango lineages were available from mNGS data for 122 (61%) patients, with sufficient reads of SARS-CoV-2 covering 96.30 to 99.89% of the genome (median reads: 29,420 [13,973–47,422] vs. 612.5 [119.3–2,300], p < 0.0001) (Fig. S1). The most prevalent sub-lineages were FU.1 (20.5%), EG.5.1.1 (17.2%), FY.3 (11.5%), and XBB.1.16.1 (11.5%), accounting for more than half of the cases (Fig. 2a). Over time, XBB.1.16 was the most frequently detected sub-lineage from May to June, but then gradually decreased, while FU.1 was consistently detected during this period, and HK.3 began to emerge in August and increased steadily (Fig. 2b). When we compared the frequency of these sub-lineages in the severe group and the non-severe group (Fig. 2c), we found no significant differences in the distribution of lineages between the two groups (Table 1). Eleven patients exhibited blood viremia (Supplementary Table 1). Subvariants EG.5.1, FR.1, FL.2, and FU.1 were identified in severe cases, while XBB.1.16 and XBB.2.3 were detected in non-severe cases. Since phylogenetic analysis indicated minimal variation in the evolutionary branches of Omicron variants from April to September 2023 (Fig. 3), no significant association was observed between SARS-CoV-2 sub-lineages and either viremia occurrence or disease severity (Supplementary Table 1 and Supplementary Table 2). Although no specific lineage predominated in COVID-19 patients, the proportions of different sub-lineages fluctuated monthly.

Distribution and relative frequencies of SARS-CoV-2 sub-lineages from March to September, 2023. (a) The proportion of different Pango lineages available in 122 patients. (b) the weekly relative frequencies of SARS-CoV-2 sub-lineages over time. The week numbers started from the first week of 2023. (c) The proportion of different Pango lineages available in severe and non-severe pneumonia patients.

Phylogenetic analysis of SARS-CoV-2 lineages and clinical correlates. The amino acid sequences were converted from nucleotide sequences using MEGA-X (10.1.8). Phylogenetic construction was performed by IQ-TREE (1.6.12). The GTR + F model was used for nucleotide sequences, while the Blosum62 model was used for amino acid sequences. Maximum-likelihood phylogenetic tree (scale bar: 10 nucleotide substitutions per site) reconstructed from SARS-CoV-2 genomic sequences, highlighting lineages circulating between April and September 2023. Red stars (*) denote six viral isolates from patients with confirmed viremia, mapped to specific branches of the Omicron subvariants. Lineage nomenclature follows PANGO classification, with temporal distribution of dominant variants annotated monthly. The detection of viremia-associated strains across phylogenetically distinct clusters underscores the need for genotype-agnostic monitoring of systemic infection risks.
Co-detected pathogens by mNGS
Co-infection patterns were analysed by categorizing pathogens into bacteria, fungi, and viruses, excluding torque teno virus (TTV). A total of 143, 117 and 132 samples of patients tested positive for bacteria, fungi and viruses alongside SARS-CoV-2 (Fig. 4a). A total of 185 patients (92.50%) developed co-infections of varying patterns, as shown in (Fig. 4b). The bacteria-fungi-virus co-infection was the most common combination in the non-severe pneumonia group (42.7%), while bacteria-fungi (21.7%) and bacteria-fungi-virus (21.7%) co-infections had similar proportions in the severe pneumonia group. There was no significant difference in the proportion of co-infection pattern between the two groups. We further analysed the pathogens that were most likely to be detected alongside SARS-CoV-2. The top five bacteria were Enterococcus faecalis (32/200, 16.00%), Acinetobacter baumannii (29/200, 14.50%), Klebsiella pneumoniae (24/200, 12.00%), Stenotrophomonas maltophilia (20/200, 10.00%), and Staphylococcus aureus (20/200, 10.00%) (Fig. 5a). Among fungi, Candida albicans (73/200, 36.50%), Candida glabrata (21/200, 10.50%), Aspergillus fumigatus (20/200, 10.00%), Candida tropicalis (18/200, 9.00%), and Pneumocystis jirovecii (18/200, 9.00%) were the most detected (Fig. 5b). As for virus, Epstein-Barr virus (EBV) (82/200, 41.00%), Human herpesvirus-7 (HHV-7) (63/200, 31.5%), Herpes simplex virus-1 (HSV-1) (31/200, 15.5%), Cytomegalovirus (CMV) (21/200, 10.5%) and Human herpesvirus-6 (HHV-6) (5/200, 2.50%) were widely detected among the patients (Fig. 5c). In addition to these herpesviruses with potential pathogenic significance, the other co-detected virus included Rhinovirus A and C (RV-A, RV-C), Human coronavirus-OC43 (HCoV-OC43), Human metapneumovirus (HMPV), and Human respiratory syncytial virus B (HRSV-B) (Fig. 5d).

Co-infection patterns with SARS-CoV-2. (a) Venn diagrams of pathogens co-detected with SARS-CoV-2. Numbers on the diagram indicate the number of patients who tested positive for bacteria, fungi, or virus. Torque teno virus was excluded. (b) Co-infection patterns of severe and non-severe pneumonia in COVID-19 patients.

Frequency of each pathogen co-detected alongside SARS-CoV-2. (a) Bacteria co-detected with SARS-CoV-2. (b) Fungi co-detected with SARS-CoV-2. (c) Viruses co-detected with SARS-CoV-2 exclude Torque teno virus. (d) Viruses co-detected with SARS-CoV-2 exclude Torque teno virus and herpesviruses.
Clinical characteristics and risk factor analysis of severe-pneumonia in COVID-19 patients
The demographic and clinical characteristics were presented in (Table 2). Severe pneumonia was identified in 69 (34.5%) patients, of whom 55 were male (79.71%), with a median age of 74 years (65.5–79 years). There were more elderly patients (aged > 65) in the severe pneumonia group compared with the non-severe group (p = 0.014), with 80 cases (61.07%) and 54 cases (78.26%), respectively. Common comorbidities included hypertension (102, 51%), malignancies (90, 45%), obesity (BMI > 25, 86, 43%), and diabetes mellitus (62, 31%). Significant differences were observed between the two groups in terms of hypertension (59, 45.04 vs 43, 62.32%), diabetes mellitus (31, 23.66% vs 31, 44.93%), chronic kidney disease (13, 9.92% vs 15, 21.74%), and chemotherapy (40, 30.53 vs 9, 13.04%) (p < 0.05).
Age over 65 years old and comorbidities such as hypertension, diabetes mellitus and chronic kidney disease were correlated with sever-pneumonia in all 200 COVID-19 patients by univariate logistic regression, while gender and the other comorbidities were not associated. In multivariate analysis, only the diabetes mellitus was statistically significant, which could be an independent risk factor of severe pneumonia (Table 3).
Fever and cough were the most frequently reported symptoms, affecting 132 (66%) and 117 (58.5%) patients, respectively. Fever (78, 59.54% vs 54, 78.26%) and shortness of breath (25, 19.08% vs 33, 47.83%) showed significant differences between the two groups (p = 0.0079 and p < 0.0001, respectively). Radiological findings revealed higher rates of patchy/ground-glass opacities (60, 86.96%), pulmonary exudation (25, 36.23%), and pleural effusion (34, 49.27%) in the severe pneumonia group, while pulmonary nodules were more common in the non-severe group (79, 60.31%). Decreased lymphocyte count, a typical feature of COVID-19, was more frequently observed in the severe pneumonia group (74, 56.48 vs. 63, 91.30%, p < 0.0001), Additionally, systolic blood pressure and respiratory rate differed significantly between the two groups (p < 0.05 for both). Thirty-five patients were hospitalized for underlying diseases, such as rheumatologic disorders, of which 32 patients were in the non-severe pneumonia group.
Treatments and outcomes of COVID-19 patients
Commonly used treatment drugs included PAXLOVID, LAGEVRIO, VV116, and Azvudine, and there was no significant difference in medication regimen between patients with severe and non-severe pneumonia (p = 0.1266) (Table 4). PAXLOVID was the most frequently used drug (96, 48%). Mask or nasal cannula oxygen therapy (107, 53.5%) was the most common way of oxygen supply, while mechanical ventilation (invasive: 11, 15.94%, and non-invasive: 6, 8.70%) was more frequently used in the severe pneumonia group. One patient in the non-severe group received extracorporeal membrane oxygenation (ECMO) following cardiac surgery. As for the outcomes of these patients, all deaths occurred in the severe pneumonia group.
Discussion
mNGS is an efficient diagnostic tool because it is high-throughput and has the potential to provide early, actionable diagnoses38. In this study, we retrospectively analysed the variation of SARS-CoV-2 detected by mNGS over approximately six months, as well as the characteristics of co-pathogens and the clinical features of the corresponding patients. Phylogenetic analysis revealed minimal divergence among SARS-CoV-2 sub-lineages, with continuous evolutionary fluctuations mirroring epidemic dynamics. No significant association was observed between SARS-CoV-2 sub-lineages and pneumonia severity. However, severe pneumonia cases predominantly occurred in elderly patients with complex underlying comorbidities. This approach is critical for both tracking Omicron subvariant evolution and identifying co-pathogens in high-risk populations such as immunocompromised cohorts.
Since the COVID-19 outbreak in December 2019, SARS-CoV-2 variants have attracted continuous attention. Several waves of the global pandemic have been driven by SARS-CoV-2 variants of concern (VOCs), such as Omicron, which is currently the most prevalent strain6. The evolution of these variants remains a key area of research39. By late November 2021, Omicron BA.1 had rapidly displaced the prior VOC Delta with Omicron associated with lower severity and mortality40. According to China national center for bioinformation (CNCB) from 1st March to 31st August 2023, a total of 9236 SARS-CoV-2 sequences were uploaded in Shanghai, of which EG.5.1.1 variant was accounted for 22.01%, FU.1 accounted for 20.54%. The Omicron sub-lineages identified in our study demonstrated concordance with national surveillance data. Despite demonstrating attenuated pathogenicity and lacking lineage-specific severity correlations (Supplementary Table 2), Omicron subvariants still caused severe pneumonia in high-risk populations, highlighting the importance of developing broad-spectrum immunity through vaccination strategies targeting multiple subvariants.
SARS-CoV-2 is typically easier to be detected in respiratory tract specimens, such as nasopharyngeal swabs, bronchoalveolar lavage fluid, and sputum. However, we detected SARS-CoV-2 sequences in the blood samples of four patients included in our analysis, as well as in an additional seven blood samples from different patients with respiratory tract specimens already collected (Supplementary Table 1). Seven of these patients were diagnosed with severe pneumonia. Although specific subvariants were identified in these cases, no significant association was observed between viral sub-lineages and viremia occurrence. The development of viremia appeared more closely associated with patients’ underlying comorbidities and immunosuppressed status. In the previous study, SARS-CoV-2 viral loads, particularly plasma viremia, have been associated with an increased risk of mortality41. Additionally, host immunosuppression can result in increased viral shedding and altered SARS-CoV-2 viral decay kinetics42,43. Given the medical history of these patients in our study, the presence of SARS-CoV-2 sequences in the blood of severe pneumonia patients may be due to lung tissue damage, which could allow the release of viral nucleic acid into the bloodstream. In patients with malignant tumours and severe immunosuppression following chemoradiation, the inability to control viral spread may also lead to viremia. Therefore, blood RNAemia could serve as a critical marker for disease severity, especially in immunosuppressed patients44,45.
Co-infection and secondary infection remain major challenges in the treatment of COVID-19 pneumonia. Meanwhile, mNGS may have more superiority in diagnosis of mixed infections, especially for intensive care unit (ICU) patients46. Although mNGS demonstrated significantly higher pathogen detection rates compared to conventional microbiological methods, distinguishing microbial colonization from infection cannot be determined solely by mNGS results14,15. We analysed pathogens that were confirmed to have clinical relevance by evaluating characteristics of clinical profiles and assessments of clinicians, despite their possible colonization potential. Previously, bacterial infections commonly occur in patients with prolonged hospital stays, with Pseudomonas aeruginosa, Klebsiella spp., and Staphylococcus aureus being the most frequent pathogens47. Additionally, the mechanisms underlying susceptibility to invasive fungal infections following respiratory viral infections are complex48. COVID-19-associated invasive fungal infections, such as those caused by Aspergillus, Mucorales, and Candida species, are a significant complication in critically ill, hospitalized patients49. Although the other pathogens and SARS-CoV-2 cannot be strictly distinguished as a first infected pathogen because some patients were admission for their underlying diseases, similar bacterial and fungal co-infection patterns were still observed, and broad-spectrum antibacterial and antifungal agents were used in hospitalized patients. Additionally, potential co-infections with Herpesviridae were frequently detected in COVID-19 patients. A meta-analysis of active EBV, CMV, HSV, Varicella Zoster virus (VZV), and HHV-6 infections in the COVID-19 population showed that one pathogen’s activity can trigger the virulence of another50. However, it was also difficult to distinguish by mNGS whether these Herpesviridae were active. After excluding viruses with potential pathogenicity, we still detected other pathogenic viruses alongside SARS-CoV-2. Co-infection with rhinovirus, respiratory syncytial virus, or other respiratory tract viruses occurred easily, which significantly increased the odds of requiring invasive mechanical ventilation and in-hospital mortality51. Similarly, a systematic analysis found that patients with viral co-infections were more likely to experience dyspnoea and had a higher mortality rate (OR 1.66)52. There was no significant difference in co-infection patterns between the severe and non-severe groups in our study, which was likely because these non-severe pneumonia patients also had highly complicated underlying conditions, often involving severe immunosuppression.
It is obvious that the elderly COVID-19 patients require more attention in clinical treatment, especially those with underlying diseases. In this study, severe cases were predominantly observed in older patients, especially those over 65 years of age. Most young and middle-aged patients experienced mild or asymptomatic infections, while greater attention should be given to the elderly, particularly those with underlying conditions. A retrospective observational cohort study in Italy, conducted from March 2020 to June 2022, revealed that 34.7% of patients had severe COVID-19, with the highest mortality rates occurring in older adults across all waves53. Similarly, a retrospective study in Spain found that advanced age and comorbidities were associated with increased hospital mortality54. An international cohort study of COVID-19 patients, spanning 52 countries between January 2020 and January 2022, reported that age was the strongest predictor of mortality risk; each comorbidity nearly doubled the risk of death. Additionally, smoking and obesity were linked to higher mortality risks55. Although the patients enrolled in our study were admitted with complex underlying diseases, these risk factors were noted in our cohort, despite only diabetes mellitus was statistically significant (Table 3).
Conclusion
The bioinformatic analysis of mNGS showed that SARS-CoV-2 infection still occurred commonly in hospitalized patients, perhaps causing severe pneumonia in patients with underlying diseases such as diabetes mellitus. SARS-CoV-2 lineages showed slight distance in the evolutionary branches and no significant evidence in the severity of pneumonia. Potential pathogens detected by mNGS may lead to complex infections, obliging early intervention. The diagnostic capability of mNGS in detecting coinfections makes it particularly valuable for determining causative pathogens in cases of undetermined etiology.
Strengths and limitations
The application of metagenomic next-generation sequencing (mNGS) in clinical laboratories facilitated the analysis of co-infective pathogens, a task that was challenging for conventional laboratory methods. It provided more complete infection patterns of COVID-19 patients, especially the individuals with complex underlying conditions. The findings of this study have to be seen in light of two limitations. First, mNGS was not a routine laboratory examination and was recommended only for patients with complex conditions; as a result, data on mild cases were limited in our study. Secondly, the outcomes of these patients were more significantly influenced by underlying diseases, particularly malignancies and immunosuppression. In the future, we would conduct more bioinformatics analysis and to explore the application of mNGS in more infection styles.
Data availability
Sequence data that support the findings of this study have been deposited in the SRA database of National Center for Biotechnology Information with the primary accession code PRJNA1241481. Our SRA records will be accessible with the following link after the indicated release date: https://www.ncbi.nlm.nih.gov/sra/PRJNA1241481.
Abbreviations
- mNGS:
-
Metagenomic next-generation sequence
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- SP:
-
Severe pneumonia
- SCAP:
-
Severe community-acquired pneumonia
- COVID-19:
-
Coronavirus disease 2019
- WBC:
-
White blood cell
- UMSI:
-
Unique molecular sample index
- BALF:
-
Bronchoalveolar lavage fluid
- NCBI:
-
National center of biotechnology information
- BWA:
-
Burrows-Wheeler aligner
- MAF:
-
Minor allele frequency
- VEP:
-
Variant effect predictor
- HQM:
-
High-quality mapped
- IQR:
-
Interquartile range
- CI:
-
Confidence interval
- ECMO:
-
Extracorporeal membrane oxygenation
- TTV:
-
Torque teno virus
- EBV:
-
Epstein-Barr virus
- HHV-7:
-
Human herpesvirus-7
- HSV-1:
-
Herpes simplex virus-1
- CMV:
-
Cytomegalovirus
- HHV-6:
-
Human herpesvirus-6
- RV-A/C:
-
Rhinovirus A/C
- HCoV-OC43:
-
Human coronavirus-OC43
- HMPV:
-
Human metapneumovirus
- HRSV-B:
-
Human respiratory syncytial virus B
- VZV:
-
Varicella zoster virus
- ICU:
-
Intensive care unit
- VOC:
-
Variant of concern
- CNCB:
-
China national center for bioinformation
References
Liu, D. et al. Multicenter assessment of shotgun metagenomics for pathogen detection. EBioMedicine 74, 103649 (2021).
Diao, Z. et al. Validation of a metagenomic next-generation sequencing assay for lower respiratory pathogen detection. Microbiol. Spectr. 11 (1), e0381222 (2023).
Deng, X. et al. Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nat. Microbiol. 5 (3), 443–454 (2020).
Chen, L. et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg. Microbes Infect. 9 (1), 313–319 (2020).
Carbo, E. C. et al. A comparison of five illumina, ion torrent, and nanopore sequencing technology-based approaches for whole genome sequencing of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis. 42 (6), 701–713 (2023).
Wei, D. et al. Pathogen evolution, prevention/control strategy and clinical features of COVID-19: experiences from China. Front. Med. 17 (6), 1030–1046 (2023).
Wei, D. et al. Sequential reinfection with Omicron variants elicits broader neutralizing antibody profiles in booster vaccinees and reduces the duration of viral shedding. J. Med. Virol. 95 (10), e29151 (2023).
Cao, Y. et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by omicron infection. Nature 608 (7923), 593–602 (2022).
Krause, P. R. et al. SARS-CoV-2 variants and vaccines. N. Engl. J. Med. 385 (2), 179–186 (2021).
Suleiman, A. S. et al. A meta-meta-analysis of co-infection, secondary infections, and antimicrobial resistance in COVID-19 patients. J. Infect. Public Health 16 (10), 1562–1590 (2023).
Miao, Q. et al. Microbiological diagnostic performance of metagenomic next-generation sequencing when applied to clinical practice. Clin. Infect. Dis. 67 (2), S231–S240 (2018).
Lin, T. et al. Microbiological diagnostic performance of metagenomic next-generation sequencing compared with conventional culture for patients with community-acquired pneumonia. Front. Cell Infect. Microbiol. 13, 1136588 (2023).
Qu, J. et al. Aetiology of severe community acquired pneumonia in adults identified by combined detection methods: a multi-centre prospective study in China. Emerg. Microbes Infect. 11 (1), 556–566 (2022).
Lv, M. et al. Clinical values of metagenomic next-generation sequencing in patients with severe pneumonia: a systematic review and meta-analysis. Front. Cell Infect. Microbiol. 13, 1106859 (2023).
Liu, M. et al. The etiological diagnostic value of metagenomic next-generation sequencing in suspected community-acquired pneumonia. BMC Infect. Dis. 24 (1), 626 (2024).
Amar, Y. et al. Pre-digest of unprotected DNA by benzonase improves the representation of living skin bacteria and efficiently depletes host DNA. Microbiome 9 (1), 123 (2021).
Wang, C. et al. Toward efficient and high-fidelity metagenomic data from sub-nanogram DNA: evaluation of library preparation and decontamination methods. BMC Biol. 20 (1), 225 (2022).
Chen, H. et al. Clinical utility of in-house metagenomic next-generation sequencing for the diagnosis of lower respiratory tract infections and analysis of the host immune response. Clin. Infect. Dis. 71 (4), S416–S426 (2020).
Metsky, H. C. et al. Capturing sequence diversity in metagenomes with comprehensive and scalable probe design. Nat. Biotechnol. 37 (2), 160–168 (2019).
Liu, Y. et al. Diagnostic value of metagenomic next-generation sequencing of lower respiratory tract specimen for the diagnosis of suspected Pneumocystis jirovecii pneumonia. Ann. Med. 55 (1), 2232358 (2023).
Xu, Y. et al. Dynamics of severe acute respiratory syndrome coronavirus 2 genome variants in the feces during convalescence. J. Genet. Genom. 47 (10), 610–617 (2020).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760 (2009).
Fiorini, N., Lipman, D. J. & Lu, Z. Towards PubMed 2.0. Elife 6, e28801 (2017).
Miller, S. et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 29 (5), 831–842 (2019).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i890 (2018).
Jung, Y. & Han, D. BWA-MEME: BWA-MEM emulated with a machine learning approach. Bioinformatics 38 (9), 2404–2413 (2022).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079 (2009).
Ma, W. et al. Genomic perspectives on the emerging SARS-CoV-2 omicron variant. Genom. Proteom. Bioinformat. 20 (1), 60–69 (2022).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17 (1), 122 (2016).
Colson, P. et al. Role of SARS-CoV-2 mutations in the evolution of the COVID-19 pandemic. J. Infect. 88 (5), 106150 (2024).
O’Toole, Á., Pybus, O. G., Abram, M. E., Kelly, E. J. & Rambaut, A. Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. BMC Genom. 23 (1), 121 (2022).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49 (W1), W293–W296 (2021).
Lim, W. S. et al. Defining community acquired pneumonia severity on presentation to hospital: an international derivation and validation study. Thorax 58 (5), 377–382 (2003).
Fine, M. J. et al. A prediction rule to identify low-risk patients with community-acquired pneumonia. N. Engl. J. Med. 336 (4), 243–250 (1997).
España, P. P. et al. Development and validation of a clinical prediction rule for severe community-acquired pneumonia. Am. J. Respir. Crit. Care Med. 174 (11), 1249–1256 (2006).
Charles, P. G. P. et al. SMART-COP: a tool for predicting the need for intensive respiratory or vasopressor support in community-acquired pneumonia. Clin. Infect. Dis. 47 (3), 375–384 (2008).
Hogan, C. A. et al. Clinical impact of metagenomic next-generation sequencing of plasma cell-free DNA for the diagnosis of infectious diseases: A multicenter retrospective cohort study. Clin. Infect. Dis. 72 (2), 239–245 (2021).
Guan, M., Sun, N. & Yau, S. S. T. Geometric analysis of SARS-CoV-2 variants. Gene 909, 148291 (2024).
Ali, K. M. et al. Clinical outcomes and phylogenetic analysis in reflection with three predominant clades of SARS-CoV-2 variants. Eur. J. Clin. Invest. 53 (9), e14004 (2023).
Fajnzylber, J. et al. SARS-CoV-2 viral load is associated with increased disease severity and mortality. Nat. Commun. 11 (1), 5493 (2020).
Li, Y. et al. Immune status and SARS-CoV-2 viral dynamics. J. Infect. Dis. 228 (2), S111–S116 (2023).
Roy-Vallejo, E. et al. SARS-CoV-2 viremia precedes an IL6 response in severe COVID-19 patients: Results of a longitudinal prospective cohort. Front. Med. 9, 855639 (2022).
Lawrence Panchali, M. J. et al. SARS-CoV-2 RNAemia and disease severity in COVID-19 patients. Viruses 15 (7), 1560 (2023).
Giacomelli, A. et al. SARS-CoV-2 viremia and COVID-19 mortality: A prospective observational study. PLoS ONE 18 (4), e0281052 (2023).
Zhou, J. J. et al. Diagnostic value of metagenomic next-generation sequencing for pulmonary infection in intensive care unit and non-intensive care unit patients. Front. Cell. Infect. Microbiol. 12, 929856 (2022).
Westblade, L. F., Simon, M. S. & Satlin, M. J. Bacterial coinfections in coronavirus disease 2019. Trends Microbiol. 29 (10), 930–941 (2021).
Salazar, F., Bignell, E., Brown, G. D., Cook, P. C. & Warris, A. Pathogenesis of respiratory viral and fungal coinfections. Clin. Microbiol. Rev. 35 (1), e0009421 (2022).
Hoenigl, M. et al. COVID-19-associated fungal infections. Nat. Microbiol. 7 (8), 1127–1140 (2022).
Banko, A., Miljanovic, D. & Cirkovic, A. Systematic review with meta-analysis of active herpesvirus infections in patients with COVID-19: Old players on the new field. Int. J. Infect. Dis. 130, 108–125 (2023).
Swets, M. C. et al. SARS-CoV-2 co-infection with influenza viruses, respiratory syncytial virus, or adenoviruses. Lancet 399 (10334), 1463–1464 (2022).
Krumbein, H. et al. Respiratory viral co-infections in patients with COVID-19 and associated outcomes: A systematic review and meta-analysis. Rev. Med. Virol. 33 (1), e2365 (2023).
Trecarichi, E. M. et al. Evolution of in-hospital patient characteristics and predictors of death in the COVID-19 pandemic across four waves: are they moving targets with implications for patient care?. Front. Public Health 11, 1280835 (2023).
Peláez, A. et al. Clinical characteristics and outcomes among hospitalised COVID-19 patients across epidemic waves in Spain: An unCoVer analysis. Med. Clin. 162, 523–531 (2024).
Kartsonaki, C. et al. Characteristics and outcomes of an international cohort of 600 000 hospitalized patients with COVID-19. Int. J. Epidemiol. 52 (2), 355–376 (2023).
Acknowledgements
This study was funded by Shanghai Municipal Science and Technology Major Project (ZD2021CY001), National Key Research and Development Program of China (2024YFC3044400) and Shanghai Targeted Biomedical Emergency Project (23DX1900300). We thank all the participants involved in this study.
Funding
This study was funded by Shanghai Municipal Science and Technology Major Project (ZD2021CY001), National Key Research and Development Program of China (2024YFC3044400) and Shanghai Targeted Biomedical Emergency Project (23DX1900300). We thank all the participants involved in this study.
Author information
Authors and Affiliations
Contributions
X.Z., M.Z., J.M, J.L. and L.H. had the idea for and designed the study. X.L., X.Y. and C.T. were responsible for collecting and summarizing the clinical data. X.L. and X.Y. performed the experimental studies. X.L. and X.Y. carried out the analysis. X.L. and C.T. drafted the manuscript. X.Z. revised the manuscript. All authors reviewed and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Ruijin Hospital in accordance with the Helsinki Declaration. The Review Board exempted requirement for informed consent since this retrospective study used only the laboratory samples and did not have any negative impact on the patients.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, X., Tang, C., Zhou, M. et al. Characteristics of SARS-CoV-2 variants and potential co-infected pathogens in hospitalized patients based on metagenomic next-generation sequencing. Sci Rep 15, 18923 (2025). https://doi.org/10.1038/s41598-025-04111-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-04111-3
