
Abstract
Combining radiotherapy with immune checkpoint inhibitors is a promising approach to improve the effectiveness of cancer treatment. However, the success rates of these clinical studies are limited. It is essential to determine the optimal irradiation scheme that maximizes the therapeutic effect by taking into account the balance between the positive and negative effects of radiation on immunity. In this context, we developed a mathematical mechanistic model that simulates (1) the balance between effector and exhausted cytotoxic T-lymphocytes (CTLs), (2) the number of neoantigens released by high-dose irradiation, and (3) the impact of radiation on draining lymph nodes (DLNs) for systemic anti-tumor immunity, and tested whether this mathematic model fits in several animal experiments. Our mechanistic model reproduced the anti-tumor effects of several cancer treatment models for combination therapies with radiation, immune checkpoint inhibitors, and/or a metabolic modulator. Furthermore, this mechanistic model simulated that tumor suppression in distant metastatic foci, known as the abscopal effect, was dysregulated by hypofractionated high-dose irradiation or by the direct radiation exposure on DLN. The mechanistic model successfully reproduced tumor control under various treatment conditions with appropriate parameters, indicating that it may be useful for optimizing immunoradiotherapy prescriptions.
Similar content being viewed by others


Introduction
Recent studies have clarified the mechanism through which radiotherapy activates the immune system by promoting the release of tumor antigens from radiation-damaged tumor cells. Research combining radiotherapy and immune checkpoint inhibitors (ICIs) has been reported to improve therapeutic effects1,2,3. In unresectable stage 3 lung cancer, adjuvant therapy with durvalumab following definitive chemoradiotherapy has been shown to extend progression-free survival by 11.2 months and is now adopted as the standard treatment4. Numerous prospective clinical studies have been conducted and are currently underway to expand the application of immunoradiotherapy to other types of cancer2,5,6,7,8,9,10.
However, the success rate of immunoradiotherapy in clinical studies has been low, and the benefit of combination therapy is minimal in certain cases because radiation also suppresses immunity10,11. It is well known that radiation enhances the expression of the immunosuppressive molecule PD-L1 on the surface of cancer cells and eradicates tumor-infiltrated CD8+ cytotoxic T cells3,12. Considering the balance between the positive and negative effects of radiation on immunity, it is essential to determine the number of irradiation fractions and doses that maximize therapeutic effects13.
Cancer-immune cycle is a fundamental concept for ICI cancer immunotherapy where neoantigen-specific cytotoxic T-lymphocytes (CTLs) are generated in draining lymph nodes (DLNs) and traffic to tumor sites14. ICI efficacy is regulated by CTL activity and the amount of tumor neoantigens. CTLs are classified as effector or exhausted T cells with different metabolic states15. ICI becomes less effective when the ratio of effector and exhausted CTLs is low in the tumor microenvironment (TME)16. Therefore, the therapeutic effect is improved with metabolic modulators, such as bezafibrate, that increase the ratio of effector and exhausted T cells17,18. Integrating this immune metabolic logic is essential to develop a combined mechanistic model of radiotherapy and immune checkpoint inhibitors.
In the cancer-immunity cycle, DLN is a vital organ to activate CTLs. Preclinical studies have reported that the ablation or surgical removal of DLNs reduces the effectiveness of immunotherapy17,19. Similarly, in radiotherapy, when DLNs are exposed to a high dose of radiation, the therapeutic effect is reduced19,20,21. Reducing the amount of radiation on DLNs may be important for maintaining the cytotoxic effects of immunity.
Radiation is reported to change the neoantigen amount, which regulates the generation of tumor-reactive CTLs. The expression of Trex1, which degrades broken double-stranded DNA in cancer cells, is enhanced under high-dose irradiation such as 20 Gy/fx. As a result, the number of generated neoantigens decreases by high-dose irradiation22.
Mathematical mechanistic models help determine appropriate prescriptions and improve the success rate of clinical trials. Various mechanistic models of immunoradiotherapy have been developed in recent studies23,24,25,26,27,28,29,30. Ciccolini et al. proposed the mechanistic models that can simulate pathophysiological processes in silico23. By inputting arbitrary irradiation doses, drug dosages, and administration schedules, treatment effects (such as changes in tumor volume during treatment) are well simulated. Frederich et al. developed a model that expresses the immune system using simultaneous differential equations26. By describing the magnitude of the immune effect with virtual factors, their model successfully reproduced combination therapy with anti-CTLA-4 and anti-PD-L1 antibodies with a few parameters. Poleszczuk et al. constructed a mechanistic model focusing on the interaction between immunity and radiation, and the model well simulated the tumor growth in a metastatic cancer treatment27,28.
However, these models have not addressed the above-mentioned classification of T cells and the effects of high-dose irradiation. Although Frederich et al. reported a mechanistic model representing exposure to DLNs26, their model did not consider the amount of radiation. It is required to improve mechanistic models for better prediction in the efficacy of more diverse combination treatments.
In this study, we included the following three biological mechanisms related to the cancer-immunity cycle to propose a mathematic mechanistic model for immunoradiotherapy:
-
1.
The balance between effectors and exhausted CTLs.
-
2.
Variation in the number of neoantigens due to Trex1 gene expression during high-dose irradiation.
-
3.
The role of DLNs in immunoradiotherapy.
We validated our constructed mechanistic model using reported animal experiments17,20,21,22,31,32 and confirmed it greatly simulated the preclinical experimental data. This mechanistic model would be useful to optimize irradiation dose and number of fractions for immunoradiotherapy.
Results
Overview of the mathematical mechanistic model
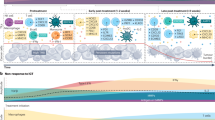
An overview of our mathematical mechanistic model of the cancer-immunity cycle under the effects of radiation is show in Fig. 1. This model acts as a compartment model that represents the activation, loss, and circulation of CTLs in the immune system. The targets and DLNs are the main compartments. When an arbitrary number of targets exist in the body, the number of cancer cells in the ith target (i = 1, 2,…) is expressed as \(\:{n}_{i}^{\left(\text{T}\text{u}\text{m}\text{o}\text{r}\right)}\). The variation in \(\:{n}_{i}^{\left(\text{T}\text{u}\text{m}\text{o}\text{r}\right)}\) per unit time step was expressed by the following formula:
The first and second terms represent the proliferation and decrease of cancer cells in the ith target, respectively. \(\:{C}_{0}\) represents the proliferation rate of the cancer cells, and \(\:{\lambda\:}_{\text{R}\text{T}}\) indicates the survival rate of cancer cells after radiation exposure and is expressed by the LQ model classically used in radiobiology33.
\(\:{\lambda\:}_{\text{I}\text{M}\text{M}}\) indicates the survival rate of cancer cells resulting from anti-tumor immunity. The cell-killing effect of immunization generally depends on the number of CTLs infiltrating the tumor. Therefore, the following formula represented \(\:{\lambda\:}_{\text{I}\text{M}\text{M}}\) in this study:
\(\:{n}_{i}^{(\text{C}\text{T}\text{L}-\text{T}\text{M}\text{E})}\) represents the number of CTLs in the the ith target, and \(\:{\gamma\:}_{1}\) is a parameter representing the cytotoxicity per unit number of CTLs.

Mechanistic model of the cancer-immunity cycle for immunotherapy.
The balance between effectors and exhausted CTLs on the mathematical mechanistic model
In Eq. 2, \(\:{n}_{i}^{(\text{C}\text{T}\text{L}-\text{T}\text{M}\text{E})}\) is defined as the sum of the numbers of activated (effector) CTLs \(\:{n}_{i}^{(\text{C}\text{T}\text{L}-\text{T}\text{M}\text{E})\left(1\right)}\) and exhausted CTLs \(\:{n}_{i}^{(\text{C}\text{T}\text{L}-\text{T}\text{M}\text{E})\left(2\right)}\) in the ith target. The variation in \(\:{n}_{i}^{(\text{C}\text{T}\text{L}-\text{T}\text{M}\text{E})\left(1\right)}\) per unit time step is assumed by the following formula:
The first term represents the influx of CTLs from the DLNs into the TME. Cinf indicates the ability of CTLs to infiltrate the TME. \(\:{\lambda\:}_{ind}^{\left(CTL\right)}\) indicates the strength of CTL migration from DLNs to the TME. M indicates the total number of DLNs set in the mechanical model. \(\:{n}_{m}^{\left(\text{C}\text{T}\text{L}-\text{D}\text{L}\text{N}\right)}\) is the number of CTLs included in the mth DLN (m = 1, 2,…). \(\:{\lambda\:}_{RT}^{\left(CTL-DLN\right)}\) indicates the survival rate of CTLs in DLNs that have received radiation and is expressed by the LQ model. In a DLN with ideally spared doses, \(\:{\lambda\:}_{RT}^{\left(CTL-DLN\right)}=1\). The first term in Eq. 3 shows that the increase in the number of CTLs in the target depends on the sum of the number of CTLs \(\:{n}_{m}^{\left(\text{C}\text{T}\text{L}-\text{D}\text{L}\text{N}\right)}\) included in all the DLNs. Thus, CTLs increase even in non-irradiated targets, possibly simulating an abscopal effect in the model.
The second term of Eq. 3 represents the proliferation of CTLs in the TME. It is well known that tertiary lymphoid structures (TLSs) are formed within tumors and CTLs proliferate there34,35. \(\:{C}_{1}\) indicates the proliferation rate of the activated CTLs in the TME.
The third term of Eq. 3 represents the decay of the activated CTLs in the TME. \(\:{\lambda\:}_{RT}^{\left(CTL-TME\right)}\) indicates the survival rate of CTLs in the TME that have received radiation. \(\:{\lambda\:}_{exh}\) is a parameter representing the probability of transitioning from an activated state to an exhausted state. \(\:{\lambda\:}_{CTL}\) indicates the survival rate of CTLs resulting from the immunosuppressive mechanism within the TME.
The variation in the number of exhausted CTLs \(\:{n}_{i}^{(\text{C}\text{T}\text{L}-\text{T}\text{M}\text{E})\left(2\right)}\) is expressed by the following formula:
The first term indicates an increase in exhausted CTLs in the TME and is expressed by the transition from activated CTLs.
The second term indicates a decrease in exhausted CTLs. \(\:{\lambda\:}_{OA}\) indicates the survival rate associated with overactivation when the anti-PD-1/PD-L1 antibody is administered. Although effector T cells with a proliferative capacity rely on both glycolysis and mitochondrial metabolism, such as oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO), exhausted T cells predominantly fuel glycolysis to compensate for impaired mitochondrial function36. Anti-PD-1 antibodies eliminate brake signaling and restore their functions. However, at the same time, PD-1 blockade upregulates glycolysis in T cells and boosts overactivation, resulting in cell death in part of the CTLs17,18.
Variation in the number of neoantigens due to Trex1 gene expression during high-dose irradiation on the mathematical mechanistic model
In Eq. 3, the variation in the number of CTLs in the DLN \(\:{n}_{m}^{\left(CTL-DLN\right)}\) in mth DLN was calculated using the following formula:
The first term indicates the number of CTLs activated by antigen-presenting cells. \(\:{C}_{5}\) represents the number of CTLs activated per unit of naïve T cells and antigen-presenting cells. \(\:{\lambda\:}_{PD-1}\) is a parameter that indicates the suppressive effect of PD-L1 on the surface of normal cells. \(\:{n}_{naive-Tcell}\) indicates the number of naïve T cells entering the DLN and has a fixed value when there is no irradiation. \(\:{n}_{m}^{\left(APC\right)}\) indicates the number of antigen-presenting cells in the DLN. The second term represents the decreasing term. In this mechanistic model, effector T cells that were activated and proliferated in the DLN immediately left the DLN through systemic migration and did not remain in the next time step.
In Eq. 5, \(\:{n}_{m}^{\left(APC\right)}\) is represented by the following formula:
The first term indicates the number of antigen-presenting cells that mature in the TME and migrate to the DLN. \(\:{C}_{7}\) represents the number of immature antigen-presenting cells existing in and around the TME and the probability of maturation of antigen-presenting cells per unit number of antigens. \(\:{I}_{m}\) is the total number of targets to which the mth DLN belongs. \(\:{\lambda\:}_{RT}^{\left(APC-TME\right)}\) represents the survival rate of antigen-presenting cells following irradiation to the ith target. \(\:{\lambda\:}_{CTLA-4}^{\left(TME\right)}\) represents the probability that the maturation of antigen-presenting cells is inhibited by effector T cells in the TME that express CTLA-4. Finally, \(\:{n}_{i}^{\left(Antigen\right)}\) represents the number of neoantigens contained in the ith target.
The second term in Eq. 4 represents the number of antigen-presenting cells lost per unit time in the DLN.\(\:{\:\lambda\:}_{CTLA-4}^{\left(DLN\right)}\) represents the probability that the maturation of antigen-presenting cells is inhibited by effector T cells expressing CTLA-4. \(\:{\lambda\:}_{NL}^{\left(APC\right)}\) is a parameter that indicates the survival rate of antigen-presenting cells due to natural decay. Furthermore, \(\:{\lambda\:}_{RT}^{\left(APC-DLN\right)}\) indicates the survival rate of antigen-presenting cells following irradiation to the DLN.
The variation in the number of neoantigens in the TME was calculated using the following formula:
The first term of Eq. 7 indicates an increase in neoantigens. \(\:{I}_{i}^{\left(Int\right)}\) and \(\:{I}_{i}^{\left(Rad\right)}\) indicate the number of neoantigens released from living cancer cells and cancer cells killed by radiation, respectively. The second term of Eq. 7 represents the attenuation of neoantigens. \(\:{\lambda\:}_{NL}^{\left(Antigen\right)}\) is a parameter that indicates the survival rate of neoantigens.
\(\:{C}_{Trex1}\) is a factor that indicates variations in the amount of antigen generated in association with Trex1 gene expression. According to a recent study22, Trex1 expression in cells is enhanced by high-dose irradiation, leading to the decomposition of broken double-stranded DNA in the cytoplasm. As a result, the number of generated neoantigens decreases. To implement this mechanism, we used an error function to represent \(\:{C}_{Trex1}\) as shown below:
\(\:{C}_{10}\), \(\:{C}_{11}\), and \(\:{C}_{12}\) are parameters, and \(\:{d}_{i}^{\left(max\right)}\) indicates the maximum dose per fraction irradiated on the target from the start of treatment.
The role of DLNs in immunoradiotherapy on the mathematical mechanistic model
We implanted a mechanism, that radiation exposure of DLNs decreases the number of naïve T cells and induces immunosuppressive effect, into our model.
The number of naïve T cells supplied to the DLN \(\:{n}_{na \ddot i ve-Tcell}\) was assumed by the following equation using the normal tissue complication probability (NTCP) of the DLN.
When the DLN receives ideally spared doses, \(\:{n}_{na \ddot i ve-Tcell}\)=1. In this study, NTCP was calculated using an empirical formula used for OARs, such as the intestine and rectum37. This formula has five parameters: \(\:{TD}_{50}\), \(\:{n}_{p}\), \(\:{m}_{p}\), \(\:\alpha\:\), \(\:\beta\:\).
Comparison with animal experiments
We validated our mechanistic model reflecting the cancer-immunity cycle by comparing the calculated tumor volumes with those reported in animal experiments. The experimental data used for the verification are listed in Table 1. Beam collimators were used in these experiments. Therefore, we assumed that when one ROI was irradiated, exposure to another ROI was completely avoided. In the mechanistic model, the tumor volume was assumed to be proportional to the number of cancer cells \(\:{n}_{i}^{\left(\text{T}\text{u}\text{m}\text{o}\text{r}\right)}\). The proportionality coefficient is shown in \(\:{k}_{i}\).
We compared the results of the animal experiments of Park et al.31 and that of the mechanistic model (Fig. 2A-B). The tumor area was assumed to be proportional to two-thirds power of \(\:{n}_{i}^{\left(\text{T}\text{u}\text{m}\text{o}\text{r}\right)}\). To assess the generalization of the developed model, the model parameters were first optimized using data from the control group, mono-PD-1 antibody therapy (aPD-1), and mono-radiation therapy (RT). Subsequently, the computational results obtained with the optimized parameters were compared against experimental data under conditions involving a combination of anti-PD-1 and RT therapies.
The developed mechanistic model reproduced the efficacy of irradiation and PD-1 antibody therapy in the primary tumor (Fig. 2A). Furthermore, our model reproduced the enhancement of tumor control in distant metastatic lesions, that is, the abscopal effect (Fig. 2B). These results demonstrate that our models is capable of generalization.

Comparisons between the mechanistic model (line) and the experiment values of Park et al. (dots). The horizontal axis represents the number of days since the tumor was inoculated into the mouse, and the vertical axis represents the tumor area. (A) the area of the primary tumor treated with radiation. (B) the abscopal effect indicating the distant tumor area without radiation.
As shown in Fig. 3, the results of the developed mechanistic model were compared with tumor growth in animal experiments reported by Chamoto et al.17. In our mechanistic model, it was assumed that \(\:{\gamma\:}_{1}\) in Eq. 2 was zero after the administration of the anti-CD8 antibody because anti-tumor immunity should be completely lost. We also supposed that anti-CD8 antibody effect was not attenuated throughout the experiment.
Our mechanistic model simulated the result of animal experiment that anti-CD8 antibody administration increases tumor growth compared to that in the untreated control because the anti-tumor immune effect was deactivated (Fig. 3A). Our model also reproduced the results of the tumor growth reduction by anti-PD-L1 antibody administration.
Next, we compared tumor growth under conditions in which a fatty acid oxidation activator, bezafibrate was co-administered with the anti-PD-L1 mAb (Fig. 3B). In the mechanistic model, the effect of bezafibrate was assumed to persist throughout the process. As mentioned previously, bezafibrate activates the mitochondrial metabolism and suppresses CTL exhaustion17,18. The mechanistic model reproduced the animal experiment that bezafibrate synergized with the anti-tumor effect of the anti-PD-L1 antibody but did not show anti-tumor activity alone.

Comparisons between the mechanistic model (line) and experiments of Chamoto et al. (dots). (A) the comparison with Experiment No. 1 shown in Table 1. (B) the comparison with Experiment No. 2. The blue dotted line shows the simulation results for bezafibrate monotherapy. The horizontal axis shows the number of days after tumor inoculation, and the vertical axis shows the tumor volume.
This mechanistic model was also validated in immunoradiotherapy animal models, as reported by V-Box et al. (Fig. 4)22. In these combinatorial experiments of irradiation and anti-CTLA-4 antibody, the radiation effects of 8 Gy × 3 and 20 Gy × 1 fractions were compared for both irradiated and non-irradiated distant metastatic lesions to evaluate the abscopal effect22. Our mechanistic model reproduced the animal experiment result in the immunoradiotherapy effect on both sides of the tumors in the 8 Gy × 3 fractions (Fig. 4A). Notably, our mechanistic model simulated that a single high-dose irradiation (20 Gy × 1 fraction) impaired the abscopal effect (Fig. 4B). There was no significant difference in the primary tumor volume between the two radiation doses (Fig. 4A-1 and B-1).

Comparisons between the mechanistic model (line) and the V-Box experiment (dots). The graphs show a combination therapy of the anti-CTLA-4 antibodies with radiation therapy. (A) and (B) show comparisons with Experiment Nos. 3, and 4, respectively in Table 1. A-1 and B-1 show the tumor volumes of the primary tumor, and A-2 and B-2 show the tumor volumes of distant metastatic lesions.
The results of animal experiments conducted by Buchwald et al.20 (Experiment No. 5 in Table 1) were compared to those of the calculation with our mechanistic model. Our model reproduced that DLN irradiation did not alter the effect of irradiation on the primary tumors but decreased the effect on the distant tumor, suppressing the abscopal effect (Fig. 5A and B).

Comparisons between the mechanistic model (line) and experiments of Buchwald et al. (dots). (A) the tumor volume of the primary tumor with radiation. (B) the tumor volume of the secondary tumor without radiation.
We further evaluated this mechanistic model on another animal experiment conducted by Marciscano et al.21. As the data in Marciscano et al. report were plotted for individual mice, it was difficult to read the numerical values. Thus, the calculation results were superimposed on the image data. Our mechanistic model qualitatively reproduced the tumor-controlling effects of radiation, ICI, and a combination of them (Fig. 6B, D, E, G and H). The model also reproduced how DLN irradiation suppressed the effects of cancer immunotherapy (Fig. 6C, F and I). For comparison with Experiment Nos. 8–10 shown in Table 1, please refer to Appendix A3.

Comparison between mechanistic model (red line) and the experiments of Marciscano et al. (black line). The horizontal axis represents the number of days since the tumor was inoculated into the mouse, and the vertical axis represents the tumor volume. (A) control (isotype); (B) radiation to the tumor (Tumor RT); (C) radiation to the tumor and DLN (T + DLN); (D) anti-PD-1 antibody treatment; (E) anti-PD-1 antibody treatment + radiation to the tumor; (F) anti-PD-1 antibody treatment + radiation to the tumor and DLN; (G) anti-CTLA-4 antibody treatment; (H) anti-CTLA-4 antibody treatment + radiation to the tumor; and (I) anti-CTLA-4 antibody treatment + radiation to the tumor and DLN.
Discussion
Considering the balance between the positive and negative effects of radiation on immunity, it is important to determine the number of fractions and irradiation doses that maximize the therapeutic effects of immunotherapy. To this end, we propose a mathematical mechanistic model by considering following three biological mechanisms, which are central in the cancer-immunity cycle under radiotherapy:
-
1.
The balance of effectors and exhausted CTLs.
-
2.
Variation in number of neoantigens due to Trex1 gene expression during high-dose irradiation.
-
3.
The role of DLN in immunoradiotherapy.
Regarding (1), our mechanistic model reproduced the anti-tumor effect of the combination of bezafibrate and anti-PD-L1 antibody. The mechanistic model also showed that bezafibrate alone did not exert anti-tumor effects. For (2), our mechanistic model reproduced not only the combined effects of radiotherapy and anti-PD-1 and anti-CTLA-4 antibodies, but also the attenuated abscopal effect under high-dose irradiation with a single dose of 20 Gy. For (3), our mechanistic model reproduced the impact of DLN radiation on the loss of the abscopal effect.
Our mechanistic model showed that hypofractionated high-dose irradiation exhibited an immunosuppressive effect that may be associated with toxicity on T cells and Trex1 gene upregulation. Consistent with this, hypofractionated irradiation has been reported to form an immunosuppressive TME and promote tumor invasion and spread38. On the other hand, hypofractionated irradiation is reported to induce antitumor immune responses by releasing tumor-associated antigens and inflammatory cytokines, normalizing aberrant tumor vasculature, and enhancing antitumor effects38. Further discussion is required to implement hypofractionated irradiation in mechanistic models.
Although Frederich et al. reported a mechanistic model representing exposure to DLNs26, their model did not consider the amount of radiation. Our mechanistic model regards DLN as an organ at risk (OAR) causing immunosuppressive effect according to the amount of radiation exposure. Radiation exposure of DLN decreases the number of naïve T cells \(\:{n}_{na \ddot i ve-Tcell}\:\)(Eq. 9) and reduces the number of activated CTLs \(\:{n}_{m}^{\left(\text{C}\text{T}\text{L}-\text{D}\text{L}\text{N}\right)}\) (Eq. 5). Here, since the calculation formula for normal tissue complication probability (NTCP) is used to calculate \(\:{n}_{na \ddot i ve-Tcell}\), the decrease in \(\:{n}_{na \ddot i ve-Tcell}\:\)due to radiation is irreversible. However, in the clinic, as reported that radiation induced lymphopenia (RIL) recovers after the end of radiotherapy, the number of lymphocytes, including naïve T cells, is reversible29,39. Therefore, this implementation method cannot necessarily be appropriate. Further discussion is necessary to implement irradiation to DLN in the model.
By setting the appropriate parameters, our mechanistic model could flexibly reproduce the tumor volume under various treatment conditions. The parameters obtained in this study are provided in the appendix. Excluding components related to the LQ model, the parameters remain consistent across each cell line. Therefore, we consider that the obtained parameters exhibit a certain degree of reliability in capturing immunological characteristics. Furthermore, comparisons with the experiments conducted by Park (Fig. 2) and Moore (Figs. A3 and A4) demonstrate that the developed model is generalized, enabling the prediction of combination therapy effects based on parameters determined from monotherapy data. Thus, this mechanistic model may help to optimize prescriptions for immunoradiotherapy and improve the success rate of clinical studies.
Because the TME and immune reactions are heterogeneous in clinical practice, the mechanistic model parameters related to this heterogeneity should be adjusted based on the data based on the patient samples. Recent studies suggest that gene, protein, and metabolite expression for each cell subset within the TME, are involved in the heterogeneity of immune reactions in patients40,41,42,43. Including these clinical data should help develop the fixed model parameters.
It remains to be addressed which prescriptions would be recommended by this mechanistic model, and further in-depth research is needed in the future. As shown in Figs. 5 and 6, our mechanistic model showed that DLN irradiation suppressed tumor control. Avoiding DLN irradiation may increase the immune effects and improve therapeutic efficacy. In conventional treatments, DLNs have been included in the radiation field to prevent recurrence in cases where cancer cells may have metastasized to the DLNs, such as head and neck cancer or stage III lung cancer44,45. Although it is still not realistic to completely spare DLNs from radiation, exploring the appropriate irradiation dose, number of fractions, and timing of irradiation to the DLNs using a mechanistic model may be valuable in improving the therapeutic effects.
There are limitations in this study: (1) As mentioned earlier, the parameters are consistent across the same cell line. However, the LQ parameters required adjustment according to the irradiation dose even for the same cell line. This is because the LQ model does not adequately support high-dose irradiation. Additionally, discrepancies between experimental and calculated values in Fig. 4B become more pronounced. This discrepancy may also be caused by the current imperfect LQ model under high-dose irradiation without adjustment for the dose rate. Improving the LQ model remains a subject for future investigation. (2) The developed model acts as a compartment model that represents the activation, loss, and circulation of CTLs in the immune system. For compartment models, the precise definition of the network structure is crucial. In this study, we manually defined the network structure, given the simplicity of the mouse model. However, in clinical applications, particularly in cases of metastasis, it is not straightforward to determine which DLN corresponds to each tumor site and where they are located. (3) In this study, we used the results of control and monotherapy treatments as training data and evaluated the model’s generalization performance using combination therapy data. However, for clinical applications, it will be necessary to validate the model using a larger and more diverse dataset because of the heterogeneity.
In summary, mechanistic models to simulate the pathophysiological processes in silico have been proposed to optimize irradiation schemes for immunoradiotherapy. In this study, we proposed a mechanistic model that simulates the balance between effector and exhausted CTLs, variations in the number of neoantigens generated by Trex1 gene expression during high-dose irradiation, and irradiation effect on DLNs. Through comparisons with animal experiments, our model reproduced tumor controls under various experimental conditions by setting the appropriate parameters. Therefore, our mechanistic model may be useful for optimizing immunoradiotherapy regimens and improving the success rates of clinical studies. At the same time, it needs to be further improved for clinical applications using more preclinical data and by establishing a methodology for adjusting the parameters for each patient based on their baseline data.
Materials and methods
In Eq. 3, \(\:{C}_{\text{i}\text{n}\text{f}}\) indicates the ability of CTLs to infiltrate the TME. Under control conditions without treatment, \(\:{C}_{\text{i}\text{n}\text{f}}=1.0\). In irradiated targets, \(\:{C}_{\text{i}\text{n}\text{f}}\) increases to a constant value \(\:{C}_{\text{i}\text{n}\text{f}}^{\left(RT\right)}\). In pancreatic cancer, \(\:{C}_{\text{i}\text{n}\text{f}}\) may need to be adjusted for other immunosuppressive factors, such as hypoxia and stroma. \(\:{\lambda\:}_{ind}^{\left(CTL\right)}\) indicates the strength of CTL migration from DLNs to the TME. Setting \(\:{\lambda\:}_{ind}^{\left(CTL\right)}\) to 1.0 simulates a situation in which chemokines promoting CTL migration are not present in the TME. \(\:{C}_{1}\) indicates the proliferation rate of the activated CTLs in the TME. However, because tertiary lymphoid structure (TLS) formation was not observed in animal experiments, \(\:{C}_{1}=0\) was used in this study. \(\:{\lambda\:}_{exh}\) is a parameter representing the probability of transitioning from an activated state to an exhausted state. When the patient is under the control condition, the following formula applies: \(\:{\lambda\:}_{exh}={\lambda\:}_{exh}^{(\pm\:)}\). When a drug that activates the peroxisome proliferator-activated receptor (PPAR) and suppresses CTL exhaustion, such as bezafibrate, is administered, \(\:{\lambda\:}_{exh}\) increases from \(\:{\lambda\:}_{exh}^{(\pm\:)}\) to \(\:{\lambda\:}_{exh}^{(+)}\).
In Eq. 4, \(\:{\lambda\:}_{OA}\) indicates the survival rate associated with overactivation when the anti-PD-1/PD-L1 antibody is administered. When the patient is under the control condition, \(\:{\lambda\:}_{\text{O}\text{A}}={\lambda\:}_{OA}^{(\pm\:)}\). When the anti-PD-1/PD-L1 antibody was administered, it decreased to \(\:{\lambda\:}_{OA}^{(+)}\).
In Eqs. 3 and 4, \(\:{\lambda\:}_{CTL}\) indicates the survival rate of CTLs resulting from the immunosuppressive mechanism within the TME and is expressed using the following formula:
\(\:{\lambda\:}_{MDSC}\) represent the contribution of myeloid-derived suppressor cells (MDSCs) and M2 macrophages (hereinafter referred to as M2) infiltrating tumors. Similarly, \(\:{\lambda\:}_{Treg}\) represents the contribution from tumor-infiltrating regulatory T cells (Tregs). \(\:{\lambda\:}_{NL}^{\left(CTL\right)}\) represents the rate of decrease in activated CTLs due to natural decay. Molecular biology suggests that CTLs do not have a clear lifespan; however, in this study, we introduce \(\:{\lambda\:}_{NL}^{\left(CTL\right)}\) as a buffer for the unknown immunosuppressive mechanism that is currently difficult to implement in the model.
\(\:{\lambda\:}_{PD-1}\) is a parameter indicating the strength of immunosuppression through the PD-1/PD-L1 mechanism, also appeared in Eqs. 5, 16 and 17. In the developed model, when the patient is under the control condition, \(\:{\lambda\:}_{PD-1}={\lambda\:}_{PD-1}^{(\pm\:)}\). \(\:{\lambda\:}_{PD-1}^{(\pm\:)}\) should be close to zero for patients with high PD-L1 expression in their tumors.
\(\:{\lambda\:}_{PD-1}\) increases at the timing of anti-PD-1/PD-L1 antibody administration. Antibody administration increases drug concentration in patients. This model assumes that the drug concentration decreases at a constant rate per unit time. If the drug concentration exceeds the threshold, the model transitions to a state in which the drug reaches maximal efficacy (State I) and increases \(\:{\lambda\:}_{PD-1}\) to a constant value \(\:{\lambda\:}_{PD-1}^{(++)}\) from that of the control state \(\:{\lambda\:}_{PD-1}^{(\pm\:)}\). After the State I end, the model transitions to a state in which the drug is partially effective (State II) and increases \(\:{\lambda\:}_{PD-1}\) to \(\:{\lambda\:}_{PD-1}^{(+)}\). Conventional anticancer drugs lose their ability to control tumors as their drug concentration decreases. In contrast, the effects of ICIs persist even after their administration is discontinued, and the mechanistic model follows this phenomenon.
Radiation exposure increases PD-L1 expression in cancer cells31. Thus, in the developed mechanistic model, when a target was irradiated, a PD-L1 overexpression flag was generated for a certain period. When this flag is raised, the model transitions to the PD-L1 overexpression state and decreases \(\:{\lambda\:}_{PD-1}\) to a constant value \(\:{\lambda\:}_{PD-1}^{(-)}\). According to31, this process also applies to distant metastatic tumors that have not been irradiated. Here, when \(\:{\lambda\:}_{PD-1}\) at the control state is \(\:{\lambda\:}_{PD-1}^{(\pm\:)}\), then \(\:{\lambda\:}_{PD-1}^{(++)}>{\lambda\:}_{PD-1}^{(+)}>{\lambda\:}_{PD-1}^{(\pm\:)}>{\lambda\:}_{PD-1}^{(-)}\). All these parameters were fixed but might be adjusted for each animal experiment. Please refer to the appendix for the behavior of \(\:{\lambda\:}_{PD-1}\) when radiation and drugs are combined.
\(\:{\lambda\:}_{MDSC}\) in Eq. 10 denotes the immunosuppressive effects of MDSCs and M2s that infiltrate the target. \(\:{\lambda\:}_{MDSC}\) is expressed using the number of MDSCs \(\:{n}_{i}^{\left(\text{M}\text{D}\text{S}\text{C}\right)}\) and M2s \(\:{n}_{i}^{\left(\text{M}2\right)}\) in the target.
.
Here, \(\:{\gamma\:}_{2}\) is a parameter representing the immunosuppressive strength per unit number of MDSCs and M2s.
\(\:{\lambda\:}_{Treg}\) in Eq. 10 indicates the contribution of Tregs infiltrating the target and is assumed in the following equation using the number of Tregs in the target \(\:{n}_{i}^{\left(\text{T}\text{r}\text{e}\text{g}\text{-TME}\right)}\).
.
Here, \(\:{\gamma\:}_{3}\) is a parameter representing the immunosuppressive strength per unit number of Tregs.
For the numbers of MDSCs \(\:{n}_{i}^{\left(\text{M}\text{D}\text{S}\text{C}\right)}\) and M2s \(\:{n}_{i}^{\left(\text{M}2\right)}\), the variation per unit time step was assumed using the following formula:
.
The first term of each formula indicates the number of MDSCs and M2s induced by the cancer cells. The second term is a decreasing term, where \(\:{\lambda\:}_{RT}^{\left(MDSC\right)}\:\)and \(\:{\lambda\:}_{RT}^{\left(M2\right)}\) are the cell survival rate as a result of radiation, and \(\:{\lambda\:}_{NL}^{\left(\text{M}\text{D}\text{S}\text{C}\right)}\) and \(\:{\lambda\:}_{NL}^{\left(\text{M}2\right)}\) are the survival rates as a result of natural decay.
For the number of Tregs, \(\:{n}_{i}^{\left(\text{T}\text{r}\text{e}\text{g}-\text{T}\text{M}\text{E}\right)}\), the variation per unit time step was assumed using the following formula:
.
The first term in this formula indicates the number of Tregs induced by cancer cells. The second term represents the influx of Tregs from DLN into the tumor environment. \(\:{\lambda\:}_{ind}^{\left(Treg\right)}\) indicates the strength of Treg migration from the DLN to the tumor environment. \(\:{\lambda\:}_{RT}^{\left(Treg-DLN\right)}\) indicates the survival rate of Tregs in DLNs that have received radiation. Similar to CLTs, the increase in Tregs in the TME depends on the sum of the number of Tregs \(\:{n}_{m}^{\left(\text{T}\text{r}\text{e}\text{g}-\text{D}\text{L}\text{N}\right)}\) included in all DLNs (see Eq. 3). Thus, the number of Tregs increased even in the unirradiated targets.
The third term represents a reduction in Tregs in the tumor environment. \(\:{\lambda\:}_{RT}^{\left(Treg-TME\right)}\) indicates the survival rate of Tregs in the TME that have received radiation. \(\:{\lambda\:}^{\left(Treg\right)}\) indicates the survival rate of Treg. and is expressed by the following formula.
.
\(\:{\lambda\:}_{NL}^{\left(\text{T}\text{r}\text{e}\text{g}\right)}\) are the survival rates as a result of natural decay. \(\:{\lambda\:}_{PD-1}\) is a parameter indicating the strength of immunosuppression through the PD-1/PD-L1 mechanism.
In this model, the variation in the number of Tregs in DLN \(\:{n}_{m}^{\left(\text{T}\text{r}\text{e}\text{g}-\text{D}\text{L}\text{N}\right)}\) is assumed by the following formula:
.
The first term indicates the number of Tregs activated by antigen-presenting cells. \(\:{C}_{6}\) indicates the number of effector T cells obtained per unit of naïve T cells and antigen presenting cells. Similar to CTLs. When the anti-CTLA-4 antibody was administered and the effect was maintained, \(\:{C}_{6}\) increased to a constant value \(\:{C}_{6}^{(++)}\) (see Eq. 5). \(\:{\lambda\:}_{PD-1}\) is a parameter indicating the immunosuppressive effects of PD-L1 expressed normal cells. The second term represents the decreasing term. In this mechanistic model, effector T cells that were activated and proliferated in the DLN immediately left the DLN through systemic migration and did not remain in the next time step.
In Eqs. 13, 14 and 15, \(\:{I}_{i}^{\left(\text{M}\text{D}\text{S}\text{C}\right)}\), \(\:{I}_{i}^{\left(\text{M}2\right)}\) and \(\:{I}_{i}^{\left(Treg\right)}\) are assumed as follows.
.
\(\:{C}_{2}\), \(\:{C}_{3}\) and \(\:{C}_{4}\) represent the number of MDSCs, M2s and Tregs induced in the TME per unit number of cancer cells, respectively, and \(\:{I}_{max}^{\left(\text{M}\text{D}\text{S}\text{C}\right)}\), \(\:{I}_{max}^{\left(\text{M}2\right)}\) and \(\:{I}_{max}^{\left(\text{T}\text{r}\text{e}\text{g}\right)}\) are the upper limits. In this study, p was fixed at 0.3.
In Eq. 5, \(\:{C}_{5}\) represents the number of CTLs activated per unit of naïve T cells and antigen-presenting cells. When the patient is under the control condition, \(\:{C}_{5}={C}_{5}^{(\pm\:)}\). In this model, \(\:{C}_{5}\) increased to a constant value \(\:{C}_{5}^{(++)}\) when the anti-CTLA-4 antibody was administered, decreased to \(\:{C}_{5}^{(+)}\) after a certain amount of time, and the effect of the drug attenuated. Here, \(\:{C}_{5}^{(++)}>{C}_{5}^{(+)}>{C}_{5}^{(\pm\:)}\), and all are set as parameters. The mechanism of anti-CTLA-4 antibodies has not been fully understood; in this study, we hypothesized that one of the effects of the anti-CTLA-4 antibody would be CTL reactivation in the DLN.
Effector T cells in DLN, such as CTLs and Tregs, traffic to the tumor environment by chemokines released from the tumor tissue (tumor cells and tumor-infiltrated CTLs). In Eqs. 3 and 15, the strength of migration from the DLN to the tumor environment \(\:{\lambda\:}_{ind}^{\left(CTL\right)}\) and \(\:{\lambda\:}_{ind}^{\left(Treg\right)}\) are assumed by the following formulae:
.
\(\:{\gamma\:}_{4}\) is a parameter indicating the strength of CTL migration per unit number of cancer cells and CTLs, and \(\:{\gamma\:}_{5}\) indicates the strength of Treg migration per unit number of cancer cells.
In Eq. 6, the factors \(\:{\lambda\:}_{CTLA-4}^{\left(TME\right)}\) and \(\:{\lambda\:}_{CTLA-4}^{\left(DLN\right)}\) represent a loss of antigen-presenting cell activity in TME and DLNs due to binding CTLA-4 on the surface of effector T cells to CD80/CD86 on antigen-presenting cells. They are represented by the following formula:
.
Here, \(\:{\gamma\:}_{6}\) and \(\:{\gamma\:}_{7}\) are parameters representing the inactivation status of antigen-presenting cells per unit number of effector T cells expressing CTLA4. In the developed model, when the patients are under the control condition, \(\:{\gamma\:}_{6}={\gamma\:}_{6}^{(\pm\:)}\) and \(\:{\gamma\:}_{7}={\gamma\:}_{7}^{(\pm\:)}\). When the anti-CTLA-4 antibody is administered, the parameters increase and then remain at a constant value \(\:{\gamma\:}_{6}^{(++)}\) and \(\:{\gamma\:}_{7}^{(++)}\). Following a certain amount of time and reduction of the effect of the drug, they decrease to \(\:{\gamma\:}_{6}^{(+)}\) and \(\:{\gamma\:}_{7}^{(+)}\). Here, \(\:{\gamma\:}_{6}^{(\pm\:)}>{\gamma\:}_{6}^{(+)}>{\gamma\:}_{6}^{(++)}\) and \(\:{\gamma\:}_{7}^{(\pm\:)}>{\gamma\:}_{7}^{(+)}>{\gamma\:}_{7}^{(++)}\) and all are set as parameters.
In Eq. 7, \(\:{I}_{i}^{\left(Int\right)}\) and \(\:{I}_{i}^{\left(Rad\right)}\) indicate the number of neoantigens released from living cancer cells and cancer cells killed by radiation, respectively, and are expressed as follows:
.
\(\:{C}_{8}\) and \(\:{C}_{9}\) indicate the number of antigens released per unit number of cancer cells, and \(\:{I}_{max}^{\left(Int\right)}\) and \(\:{I}_{max}^{\left(Rad\right)}\) indicate the upper limit of antigens released per unit time step. In this study, p was fixed at 0.3.
As shown in Eq. 27, the LQ model was used for calculating \(\:{\lambda\:}_{\text{R}\text{T}},\:\:{\lambda\:}_{RT}^{\left(CTL-TME\right)},\:\:{\lambda\:}_{RT}^{\left(CTL-DLN\right)}\), \(\:{\lambda\:}_{RT}^{\left(Treg-TME\right)},\:\:{\lambda\:}_{RT}^{\left(Treg-DLN\right)}\), \(\:{\lambda\:}_{RT}^{\left(APC-TME\right)},\:{\:\lambda\:}_{RT}^{\left(APC-DLN\right)},\:\:{\lambda\:}_{RT}^{\left(M2\right)}and\:\)\(\:{\lambda\:}_{RT}^{\left(MDSC\right)}.\)
.
\(\:{d}_{i}\) is irradiation dose to the compartment in each time step. Specific α and β were used for each cell type (tumor, CTL, Treg, APC, M2 and MDSC).
The mechanical model parameters were fitted to reproduce the experimental values. In this study, we used a grid search to optimize the parameters for simplicity. The developed model has a large number of parameters, and it is not realistic to search all the parameters at once. Therefore, we fixed some parameters based on the experimental conditions (for example, if beam radiation is not performed, the parameters related to radiobiological effect are fixed and other parameters were searched) and determined the parameters sequentially. The fixed and changing parameters are presented in the appendix.
Data availability
The data that supports the findings of this study are available from the corresponding author, Kenji Chamoto, upon reasonable request.
References
Formenti, S. C. & Demaria, S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J. Natl. Cancer Inst. 105(4), 256–265 (2013).
Liu, Y. et al. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J. Hematol. Oncol. 11(1), 104 (2018).
Zhao, X. & Shao, C. Radiotherapy-mediated immunomodulation and anti-tumor abscopal effect combining immune checkpoint Blockade. Cancers 12(10), 2762 (2020).
Antonia, S. J. et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl. J. Med. 377(20), 1919–1929 (2017).
McLaughlin, M. et al. Inflammatory microenvironment remodeling by tumour cells after radiotherapy. Nat. Rev. Cancer. 20(4), 203–217 (2020).
Rückert, M. et al. Radiotherapy and the immune system: more than just immune suppression. Stem Cells. 39(9), 1155–1165 (2021).
Janopaul-Naylor, J. R. et al. The abscopal effect: A review of pre-clinical and clinical advances. Int. J. Mol. Sci. 22(20), 11061 (2021).
Zhai, D. et al. Radiotherapy: brightness and darkness in the era of immunotherapy. Transl Oncol. 19, 101366 (2022).
Zhang, Z. et al. Radiotherapy combined with immunotherapy: the dawn of cancer treatment. Signal. Transduct. Target. Ther. 7(1), 258 (2022).
Galluzzi, L., Aryankalayil, M. J., Coleman, C. N. & Formenti, S. C. Emerging evidence for adapting radiotherapy to immunotherapy. Nat. Rev. Clin. Oncol. 20(8), 543–557 (2023).
Mell, L. K. et al. Radiotherapy with durvalumab vs. cetuximab in patients with locoregionally advanced head and neck cancer and a contraindication to cisplatin: phase II results of NRG-HN004. Int. J. Radiat. Oncol. Biol. Phys. 114(5), 1058 (2022).
Wen, L. et al. The research progress of PD-1/PD-L1 inhibitors enhancing radiotherapy efficacy. Front. Oncol. 11, 799957 (2021).
Demaria, S. et al. Radiation dose and fraction in immunotherapy: one-size regimen does not fit all settings, so how does one choose? J. Immunother Cancer. 9(4), e002038 (2021).
Chen, D. S. & Mellman, I. Oncology meets immunology: the cancer-immunity cycle. Immunity 39(1), 1–10 (2013).
Jiang, W. et al. Exhausted CD8 + T cells in the tumor immune microenvironment: new pathways to therapy. Front. Immunol. 11, 622509 (2021).
Yost, K. E. et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 25(8), 1251–1259 (2019).
Chamoto, K. et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. U S A. 114(5), E761–E770 (2017).
Chowdhury, P. S., Chamoto, K., Kumar, A. & Honjo, T. PPAR-Induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8+ T cells and facilitates Anti-PD-1 therapy. Cancer Immunol. Res. 6(11), 1375–1387 (2018).
Takeshima, T. et al. Local radiation therapy inhibits tumor growth through the generation of tumor-specific CTL: its potentiation by combination with Th1 cell therapy. Cancer Res. (2010).
Buchwald, Z. S. et al. Tumor-draining lymph node is important for a robust abscopal effect stimulated by radiotherapy. J. Immunother Cancer. 8(2), e000867 (2020).
Marciscano, A. E. et al. Elective nodal irradiation attenuates the combinatorial efficacy of stereotactic radiation therapy and immunotherapy. Clin. Cancer Res. 24(20), 5058–5071 (2018).
Vanpouille-Box, C. et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 8, 15618 (2017).
Ciccolini, J. et al. Mechanistic learning for combinatorial strategies with Immuno-oncology drugs: can model-informed designs help investigators?? JCO Precis Oncol. 4, 486–491 (2020).
Sotolongo-Grau, O. et al. Immune system-tumour efficiency ratio as a new oncological index for radiotherapy treatment optimization. Math. Med. Biol. 26(4), 297–307 (2009).
Kosinsky, Y. et al. Radiation and PD-(L)1 treatment combinations: immune response and dose optimization via a predictive systems model. J. Immunother Cancer. 6(1), 17 (2018).
Friedrich, T., Scholz, M. & Durante, M. A predictive biophysical model of the combined action of radiation therapy and immunotherapy of cancer. Int. J. Radiat. Oncol. Biol. Phys. 113(4), 872–884 (2022).
Poleszczuk, J. T. et al. Abscopal benefits of localized radiotherapy depend on activated T-cell trafficking and distribution between metastatic lesions. Cancer Res. 76(5), 1009–1018 (2016).
Poleszczuk, J. & Enderling, H. The optimal radiation dose to induce robust systemic Anti-Tumor immunity. Int. J. Mol. Sci. 19(11), 3377 (2018).
Sung, W. et al. A tumor-immune interaction model for hepatocellular carcinoma based on measured lymphocyte counts in patients undergoing radiotherapy. Radiother Oncol. 151, 73–81 (2020).
Sung, W. et al. Mathematical modeling to simulate the effect of adding radiation therapy to immunotherapy and application to hepatocellular carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 112(4), 1055–1062 (2022).
Park, S. S. et al. PD-1 restrains radiotherapy-Induced abscopal effect. Cancer Immunol. Res. 3(6), 610–619 (2015).
Moore, C. et al. Personalized ultrafractionated stereotactic adaptive radiotherapy (PULSAR) in preclinical models enhances single-agent immune checkpoint blockade. Int. J. Radiat. Oncol. Biol. Phys. 110(5), 1306–1316 (2021).
van Leeuwen, C. M. et al. The alfa and beta of tumours: a review of parameters of the linear-quadratic model, derived from clinical radiotherapy studies. Radiat. Oncol. 13(1), 96 (2018).
Schrama, D. et al. Targeting of lymphotoxin-alpha to the tumor elicits an efficient immune response associated with induction of peripheral lymphoid-like tissue. Immunity 14(2), 111–121 (2001).
Kirk, C. J. et al. The dynamics of the T-cell antitumor response: chemokine-secreting dendritic cells can prime tumor-reactive T cells extranodally. Cancer Res. 61(24), 8794–8802 (2001).
Scharping, N. E. et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol 22(2), 205–215 .
Lyman, J. T. Complication probability as assessed from dose-volume histograms. Radiat. Res. 8, S13–S19 (1985).
Wang, Y. Advances in hypofractionated irradiation-induced immunosuppression of tumor microenvironment. Front. Immunol. 11, 612072 (2021).
Wang, X. et al. Low lymphocyte count is associated with radiotherapy parameters and affects the outcomes of esophageal squamous cell carcinoma patients. Front. Oncol. 10, 997 (2020).
Aldea, M. et al. Overcoming resistance to tumor-targeted and immune-targeted therapies. Cancer Discov. 11(4), 874–899 (2021).
Meng, L. et al. The combination of radiotherapy with immunotherapy and potential predictive biomarkers for treatment of non-small cell lung cancer patients. Front. Immunol. 12, 723609 (2021).
Darvin, P. et al. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp. Mol. Med. 50(12), 1–11 (2018).
Li, N. et al. Biomarkers related to immune checkpoint inhibitors therapy. Biomed. Pharmacother. 147, 112470 (2022).
Alfouzan, A. F. Radiation therapy in head and neck cancer. Saudi Med. J. 42(3), 247–254 (2021).
Mercieca, S., Belderbos, J. S. A. & van Herk, M. Challenges in the target volume definition of lung cancer radiotherapy. Transl Lung Cancer Res. 10(4), 1983–1998 (2021).
Acknowledgements
This work was supported by JSPS KAKENHI (Grant Number 19H03591).
Funding
This work was supported by JSPS KAKENHI under Grant Number 19H03591 (H.S.) and Yanai Fund (K.C.).
Author information
Authors and Affiliations
Contributions
T.T. and K.C. wrote the main manuscript. H.S contribution conceptualization of this study. T.T., K.M., T.Y., T.K.,Y.O. and Kana Yamasaki contributed to building a methodology. T.T., H.S. and Kana Yamasaki contributed to investigation and data curation and T.T., K.M., H.T. and Kana Yamasaki contributed to validation. K.M., T.Y. and T.K. contributed to building software. F.E., K.K., H.S., and K.C. contributed to funding acquisition. K.K., H.A., N.K., T.H., K.C. and Koichi Yasuda supervised and administrated this project. All authors participated in the study and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Takayanagi, T., Miyazaki, K., Yamasaki, K. et al. Mathematical mechanistic model representing the cancer immunity cycle under radiation effects. Sci Rep 15, 19940 (2025). https://doi.org/10.1038/s41598-025-04715-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-04715-9
