
Abstract
Acute T cell-mediated rejection (TCMR) is one of the most important causes of kidney graft injury and loss; however, in-depth transcriptomic analyses of relevant tissues and circulating immune cells are limited. In this study, we conducted single-cell RNA sequencing (scRNA-seq) on peripheral blood mononuclear cells and spatial transcriptomics using kidney allograft biopsy samples. The scRNA-seq study included four patients with borderline rejection or biopsy-proven TCMR and two with no evidence of TCMR. For spatial analysis, we compared tissue specimens from one patient diagnosed with borderline rejection and another patient with TCMR to their time-zero protocol biopsies. Six regions of interest per biopsy section were selected, focusing on the evident T-cell infiltration area in the tubulointerstitium and glomeruli. Integrating two methodologies, we pinpointed CD8+ effector memory T cell (TEM) expression profiles and key upregulated genes, including LTB, GZMK, PSME2, UBE2L6, and STAT1. Among them, STAT1 was confirmed as a hub gene through network and pathway analysis. Immunohistochemistry and immunofluorescence on kidney allograft tissue validated the co-expression of STAT1 with CD8, indicating an active inflammatory response. Our findings suggest the presence of a CD8+ STAT1+ TEM subset with features of clonal expansion, providing additional insight into the immunological processes associated with TCMR.
Similar content being viewed by others

Introduction
Acute T cell-mediated rejection (TCMR) is a leading cause of long-term graft loss in kidney transplantation (KT), affecting 5–15% of recipients within the first year and up to 30% when including patients with borderline rejection1,2,3,4. The Banff classification system, used for the histopathological diagnosis of acute rejection, has limitations when facing nonspecific lesions, subjective interpretations, and potential variability in grading between pathologists5,6,7. Molecular analyses have revealed discrepancies between histopathological diagnoses and molecular subtypes8,9,10,11. Recent updates to the Banff classification involve the use of molecular studies, which have improved our comprehension of the immune landscape in rejection and inflammation. However, a more comprehensive approach is still required to better understand the heterogeneity and pathogenesis of TCMR4,12,13.
Single-cell RNA-sequencing (scRNA-seq) has provided unprecedented insights into immune responses during acute rejection. Wu et al. characterized monocytes and endothelial cell groups in a single human kidney allograft biopsy from a patient undergoing mixed rejection14, while Malone et al. distinguished recipient versus donor leukocytes in patients undergoing antibody-mediated rejection15. However, scRNA-seq lacks spatial context, which is crucial for understanding cellular interactions and structural changes within tissue14,15,16. Spatial transcriptomics (ST) has emerged as a complementary tool to capture both gene expression patterns and their spatial distribution throughout tissue, allowing for a more comprehensive understanding of cellular interactions and tissue organization17. Salem et al. were the first to perform ST analysis using specimens from human kidney with chronic T cell-mediated rejection using the GeoMx Digital Space Profiling platform with whole-exome sequencing, providing a broader picture of the various genes involved in TCMR pathogenesis17. While several studies have employed ST technologies to assess the impact of ischemic kidney disease, glomerulopathy, and diabetic chronic kidney disease (CKD), their application in the field of KT has been rather limited18,19,20,21,22. ST analysis of kidney biopsies enables the visualization of gene expression patterns within the context of tissue architecture, providing valuable insights into the spatial organization of immune cells and their interactions during the development of rejection.
In the present study, we aimed to explore the molecular patterns associated with TCMR in kidney tissues and investigate whether any corresponding changes might be detected in circulation by integrating scRNA-seq and ST analyses. We sought to examine gene expression profiles and spatial information from both kidney tissues and peripheral blood mononuclear cells (PBMCs), as an initial step toward evaluating their potential relationship during rejection processes. Further, through our transcriptomic analysis of pre- and post-transplant PBMCs from patients with and without TCMR, we attempted to observe how circulating immune cells might reflect molecular changes in the graft kidney. Taken together, this study was designed to identify potential candidate markers for TCMR by examining expression characteristics across the graft kidney and PBMCs.
Results
Schematic study flow chart
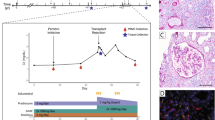
First, we examined paired allograft kidney biopsies from two patients—one with borderline changes (BDR) and one with TCMR grade IIA (i0t0v1)—at time-zero (pre-TCMR(b)) and nine days after KT (TCMR(b)). In addition, we collected blood samples from four patients—three with BDR and one with TCMR grade IIA (i0t1v1)—both before (pre-TCMR(p)) and nine days post-KT (TCMR(p)), as well as from two control patients who did not exhibit rejection, both before (pre-CTRL(p)) and nine days post-KT (post-CTRL(p)) (Fig. 1A and Table S1A). For simplicity, we collectively refer to the borderline and TCMR grade IIA cases as the “TCMR” group throughout the manuscript. A total of 12 blood samples were used for isolating PBMCs. Isolated PBMCs were pooled, and processed using distinct hashtags to enable sample demultiplexing during analysis. After quality control, 48,228 cells, or 4,019 cells per sample, were identified, which underwent manual cell type annotation (Fig. 1B). The distribution of PBMC clusters in individual samples was uniform except for one TCMR(p), and integration between samples was well achieved (Fig. S1A and B). Both patients did not show inflammatory infiltrates comparable to lesions seen in TCMR at the time-zero biopsy, but biopsies at nine days post-KT diagnosed borderline rejection and grade II TCMR, respectively. Only the patient with borderline rejection had elevated serum creatinine levels, and both patients were well-treated with steroid pulse therapy for three days. For spatial transcriptomic analysis, these biopsies underwent processing using NanoString’s Digital Spatial Profiler, targeting up to 18,000 genes on formalin-fixed paraffin-embedded (FFPE) tissue samples. Expert consultation involving pathology, immunology and nephrology guided the selection of six ROIs per biopsy section (Fig. 1C). This included four ROIs with evident T-cell infiltration in the tubulointerstitium and two ROIs dedicated to the glomeruli. A total of 13,189 genes passed quality control and proceeded to downstream analysis.

Study design and workflow. (a) Schematic representation of the study design and workflow. Time-zero kidney biopsy samples (pre-TCMR(b)) and POD-9 protocol biopsy samples (TCMR(b)) were obtained from two KT recipients. PBMCs were prospectively collected from six KT recipients before and after transplant. Kidney biopsies were subjected to spatial transcriptomics analysis using the GeoMx DSP platform, targeting a comprehensive panel of RNA biomarkers. PBMCs were labeled and processed for scRNA-seq using the 10X Genomics platform. (b) The UMAP plot shows the clustering of a total of 12 PBMCs samples. A total of 48,228 cells were annotated into 10 clusters, each representing distinct cell types characterized by specific surface markers. (c) Dot plot represents the gene expression profiles of key markers across the identified PBMC clusters. The size of the dot corresponds to the percentage of cells expressing a given marker within the cluster, whereas the color intensity reflects the average expression level of the marker in that cluster. (d) Comparative histological and immunofluorescent images of time-zero and POD-9 kidney biopsy samples. Histology sections underwent PAS staining, and selected ROIs were analyzed by GeoMx DSP for spatial gene expression. The fluorescence panel illustrates the cellular composition within the graft, using the following morphologic markers: nuclei stained in blue, CD45+ immune cells in green, PanCK+ epithelial cells in yellow, and α-SMA+ myofibroblasts in red. BDR borderline rejection, CTRL control, DSP digital spatial profiling, KT kidney transplantation, PBMC peripheral blood mononuclear cell, POD post-operative day, ROI region of interest, scRNA-seq single-cell RNA sequencing, TCMR acute T-cell mediated rejection, UMAP uniform manifold approximation and projection.
scRNA-seq analysis of PBMCs revealed 10 different cell types
Following data processing, 48,228 cells from the 12 distinct samples of six patients were retained for in-depth analysis. We categorized the cells into 10 distinct clusters based on specific markers: CD4 + T cells (CD3D+ and CD4+), CD8+ T cells (CD3D+ and CD8A+), mucosal-associated invariant T cells (CD3D+, CD8A+, and TRAV1-2+), NKT cells (CD3D+, NKG7+, and GNLY+), NK cells (CD3D−, NKG7+, GNLY+, and FCGR3A+), CD16 + monocytes (FCGR3A+ and HLA-DRA+), CD14 + monocytes (CD14+), dendritic cells (CD1C+), B cells (CD19+ and MS4A1+), and platelets (PPBP+) (Fig. 1B and D). The proportion of the whole clusters and T lymphocyte subsets is illustrated in Figure S2A and B.
Differential gene expression in the tubulointerstitium and glomeruli of pre- versus TCMR revealed via ST
In our strategic approach, we selected four ROIs within the tubulointerstitium and two within the glomeruli. We detected marked differences in gene expression between the pre-TCMR(b) and TCMR(b) samples within the tubulointerstitium and glomeruli, separately. Within the tubulointerstitium, there were 270 upregulated and 168 downregulated genes, whereas 11 upregulated and 22 downregulated genes were detected in glomeruli (Fig. 2A and B). Table 1A lists the full names of genes that were significantly upregulated or downregulated in tubulointerstitial regions, and Table S3A lists the exact p-values. Applying the same criteria for DEG identification, we obtained a higher number of DEGs in the tubulointerstitium than in the glomeruli. Pathway enrichment analysis of the 270 upregulated genes from the TCMR(b) interstitial regions highlighted the allograft rejection pathway as the most dominant pathway, followed by the interferon response pathway (Fig. 2C and Table S2). The principal components analysis (PCA) plot illustrated the separation between the pre-TCMR(b) and TCMR(b) conditions, with greater distinction observed in the tubulointerstitium compared to the glomeruli (Fig. 2D). From a pathological standpoint, TCMR primarily occurs in the tubulointerstitium, and immune responses in the glomerular regions are less definitive5,23,24. However, our high-resolution tissue profiling suggested subtle yet significant differences in gene expression profiles between pre-TCMR(b) and TCMR(b) glomeruli. This underscores that gene expression changes occur within glomeruli during acute rejection development, albeit to a lesser extent than in the tubulointerstitium.

DEGs and pathway analysis from GeoMx DSP data. (a) Volcano plot illustrates DEGs within 16 total tubulointerstitial ROIs, comparing TCMR(b) to pre-TCMR(b). 270 upregulated genes and 168 downregulated genes are identified. (b) Volcano plot illustrates DEGs within 8 ROIs of the glomeruli area, comparing TCMR(b) to pre-TCMR(b). 11 upregulated genes and 22 downregulated genes are identified. The x-axis represents the log2 fold change, while the y-axis shows the negative log10 p-value. Red dots, blue dots, and black dots indicate upregulated, downregulated, and non-significant genes, respectively. (c) Bar graph shows the enriched pathway analysis via MSigDB hallmark conducted on the 270 upregulated genes within the interstitial region of TCMR(b). The length of the bar represents the significance of the pathway. Only pathways with p-value < 0.01 are shown, with the allograft rejection pathway being the most significant, followed by interferon gamma response. (d) PCA plot representing the separation of gene expression profiles between pre-TCMR(b) and TCMR(b) within both the glomerulus and tubulointerstitium. The proximity of the points indicates the similarity of gene expression profiles. The colors distinguish the pre- and TCMR states: glomerulus of pre-TCMR(b) in pink, tubulointerstitium of pre-TCMR(b) in blue, glomerulus of TCMR(b) in purple, and tubulointerstitium of TCMR(b) in yellow. DEG differentially expressed gene, DSP digital spatial profiling, PCA principal component analysis, ROI region of interest, TCMR acute T cell-mediated rejection.
CD8+ effector memory T cells predominated in TCMR as revealed by scRNA-seq analysis
The study focused on T cells, as they are well-established drivers of TCMR5,24. We identified 11 distinct T cell clusters, and each cluster was characterized by delineating genes with elevated expression profiles across the clusters (Fig. 3A–C). To further ascertain the identity and role of each cluster, we assessed T-cell receptor (TCR) clonality. Notably, CD8 effector memory T (CD8+ TEM) cells exhibited a pronounced expansion (TCR clonality), emphasizing their critical role in TCMR (Fig. 3D). Over half of the CD8+ TEM cells were largely expanded cells containing more than 10 clones, and hyperexpanded T cells were most prevalent in CD8+ TEM across all T cell subsets (Fig. 3E). Thus, CD8+ TEM cells can be considered the predominant clinically relevant cell type among the PBMCs of patients with TCMR. We then sought to determine genes expressed within this specific cell population.

scRNA-seq analysis of T cell subsets in PBMCs. (a) UMAP plot of 30,197 cells shows the clusters of various T cell subsets. (b) Feature plots overlaid on UMAP coordinates displaying the gene expression of key T cell markers, including CD4, CD8A, TCF7, CCR7, FOXP3, TRAV1-2, PRF1, and GZMB. The intensity of the color indicates the level of gene expression. (c) Heatmap showing the gene expression levels of canonical markers across different T cell clusters. The color gradient from light to dark green represents the scaled expression level from low to high. (d) UMAP plot categorizes clonally expanded T cells based on the size of the TCR clonality. The color coding ranges from light red (single cell, n = 1) to dark red (hyperexpanded clone, 100 < n). (e) Bar plot illustrates the distribution of TCR clonality across various T cell clusters. The clonality is categorized based on the size of the TCR clones: n = 1 (single), 1 < n ≤ 5 (small), 5 < n ≤ 10 (medium), 10 < n ≤ 100 (large), and 100 < n (hyperexpanded). Different colors represent these categories, with the bar heights indicating the number of cells in each clonality category within each T cell cluster. NA non-available, PBMC peripheral blood mononuclear cell, scRNA-seq single-cell RNA sequencing, TCMR acute T cell-mediated rejection, TCR T-cell receptor, UMAP uniform manifold approximation and projection.
Integrative analysis of ST and scRNA-seq revealed six key genes associated with TCMR
From scRNA-seq data, we obtained the DEGs between pre-TCMR(p) and TCMR(p) as well as between post-CTRL(p) and TCMR(p), specifically within CD8+ TEM cells (Table 1B and Table S3B, C). To determine common DEGs in the CD8+ TEM cells among PBMCs and the allograft kidney tissue exhibiting marked T cell infiltration, we analyzed DEGs within the tubulointerstitium of pre-TCMR(b) versus TCMR(b). We identified the concurrent upregulation of six genes in the TCMR state: LTB, STAT1, GBP5, PSME2, GZMK, and UBE2L6 (Fig. 4A–C). Among these, five genes (LTB, STAT1, GBP5, PSME2, and UBE2L6) were uniquely identified as DEGs during the transition from pre-TCMR(p) to TCMR(p), but not from pre-CTRL(p) to post-CTRL(p) (Fig. 4B). Thus, we speculated that these genes were key players in TCMR and focused on them in our subsequent analyses. These findings were validated using the public microarray dataset GSE36059, which comprises samples representing T cell-mediated rejection, antibody-mediated rejection, mixed-type rejection, non-rejection, and nephrectomy25. Consistent with our findings, elevated expression of STAT1, GBP5, PSME2, and UBE2L6 was observed in patients with TCMR compared to those without TCMR (Fig. 4D and Figure S3).

Identification of six commonly upregulated genes in the comparative analysis of kidney allograft tissues and PBMCs. (a) Venn diagram illustrates the overlap of DEGs between pre-TCMR(p) vs. TCMR(p), pre-TCMR(b) vs. TCMR(b), and post-CTRL(p) vs. TCMR(p). The overlapped area of three comparisons identifies six concurrently upregulated genes in both kidney tissue and PBMCs. The table shows average log2 fold change values and associated p-values for six key genes identified across comparisons. (b) Violin plots comparing the expression levels of LTB, STAT1, GBP5, PSME2, GZMK, and UBE2L6 between PBMC samples. The plots depict the distribution and relative expression intensity of each gene. (c) Box plots shows the expression levels of the six key genes comparing pre-TCMR(b) and TCMR(b), highlighting significant upregulation in TCMR(b). (d) Volcano plot shows the DEGs between TCMR (n = 35) vs. non-TCMR (n = 281) samples in the public microarray dataset, GSE36059. The plot identifies significant upregulation of STAT1, GBP5, PSME2, and UBE2L6 in TCMR, with log2FC and -log10(p-value) indicating the magnitude and significance of differential expression. DEG differentially expressed gene, FC fold change, PBMC peripheral blood mononuclear cell, TCMR acute T cell-mediated rejection.
STAT1 emerges as a molecular marker for TCMR
To explore potential correlations among the DEGs between pre-TCMR(p) versus TCMR(p) as well as post-CTRL(p) versus TCMR(p), and to determine the significance of specific genes, we conducted both network and pathway analysis. Among the six target genes examined, given the extent of interaction, our analysis highlighted STAT1 as a primary and influential gene. In the comparative analysis of DEGs between pre-TCMR(p) and TCMR(p) (Table S3B), we identified several pivotal pathways, including “type II interferon response”, “antigen processing and presentation”, “NF-kB signaling pathway”, “cytokine receptor binding”, and “immune response” from all identified DEGs (Fig. 5A and Table 2A). Further, DEGs between post-CTRL(p) and TCMR(p) listed in Table S3C were enriched in a cluster of pathways associated with specific cell types, including “splenocyte in red pulp,” “non-germinal center cells in lymph node,” and “non-germinal center cells in tonsil” from all identified DEGs (Fig. 5B and Table 2B).

Network analysis and pathway analysis. Network analysis representing the interactions among the DEGs between (a) pre-TCMR(p) and TCMR(p) and (b) between post-CTRL(p) and TCMR(p). Nodes indicate genes, with STAT1 highlighted as a red circle out of other five genes (LTB, GBP5, PSME2, GZMK, and UBE2L6), emphasizing its central role within the network. The thickness of the lines indicates extent of interaction. The size of nodes indicates p-value. The color of nodes indicates \(\:{log}_{2}FC\). Genes within the same pathway are shown in the same color along the circular rim. CTRL control, TCMR acute T cell-mediated rejection.
Co-expression of STAT1 and CD8 in allograft kidney tissue confirmed via IHC and IF
To confirm the expression of STAT1 as well as phosphorylated STAT1 (pSTAT1) in allograft kidney tissues, we employed IHC. Notably, STAT1 and pSTAT1 co-localized with regions where a significant accumulation of inflammatory cells was detected via periodic acid–Schiff (PAS) staining. Furthermore, these areas exhibited an abundance of CD3-positive cells, whereas neither STAT1 nor CD3 were noted in pre-TCMR(b) specimens (Fig. 6A and Figure S4. The observed pattern implies that T cells infiltrating these regions trigger inflammation and contribute to acute rejection. These rejection-associated T cells were found to express STAT1 and the expressed STAT1 appeared to be phosphorylated, functioning as a transcription factor. Upon subsequent examination of CD8 and STAT1 using IF, it was evident that CD8 and STAT1 co-localized and were specifically expressed at sites of extensive T-cell infiltration (Fig. 6B). This underscores the fact that CD8+ T cells present in rejection-affected areas exhibit high STAT1 expression. As these CD8+ T cells circulate systemically in the bloodstream, STAT1-expressing CD8+ T cells and the immune responses during TCMR.

Localization and co-expression of STAT1 and CD8 in kidney allograft tissue TCMR in IHC and IF images. (a) Longitudinal tissue sections stained for PAS and IHC images stained for CD3, STAT1 and pSTAT1 are shown in the left. The images on the right are zoomed-in views of stained area, with PAS staining detecting general tissue structure. STAT1 and pSTAT1 expression coincided with regions where an accumulation of inflammatory cells and CD3 + cells was detected, as shown in IHC images. (b) Immunofluorescence images showing co-localization of CD8 (green) and STAT1 (red) in kidney tissue TCMR. DAPI (blue) is used to stain nuclei, providing a reference for cell localization. White arrows indicate areas where CD8 and STAT1 co-expression (yellow) is particularly pronounced, suggesting active involvement of CD8+ STAT1+ T cells in the rejection process. Scale bars indicate 100 μm. (c) Representative immunofluorescence images of PBMCs from two TCMR patients (one borderline and one grade II TCMR) and one post-CTRL patients. PBMCs were cyto-spun onto slides and stained using DAPI (blue), and CD8 (green) and pSTAT1 (red) antibodies. Merged images demonstrate the colocalization of pSTAT1 and CD8 (yellow). pSTAT1 expression was higher in both BDR(p) and TCMR(p) than in post-CTRL(p), with a greater number of cells showing colocalization of pSTAT1 and CD8. pSTAT1 was expressed at similar levels in both BDR(p) and grade II TCMR (TCMR(p)) samples. Scale bars indicate 50 μm. BDR borderline rejection, CTRL control, IHC immunohistochemistry, PAS periodic acid–Schiff, PBMC peripheral blood mononuclear cell, pSTAT1 phosphorylated STAT1, TCMR acute T cell-mediated rejection.
To evaluate the expression of pSTAT1 in CD8+ T cells from PBMC samples, we conducted immunofluorescence staining on PBMCs from 3 samples using Cytospin (Fig. 6C). Quantification of IF staining revealed distinct differences in the proportion of CD8+ and CD8+ pSTAT1+ cells among patients. In the post-CTRL sample, a total of 99 cells were identified, of which 59 were CD8+, and 9 were CD8+ pSTAT1+ cells. Thus, CD8+ cells accounted for 59.6% of all present cells, and CD8+ pSTAT1+ cells represented 15.25% of the CD8+ population. In the borderline TCMR (BDR(p)) sample, 203 total cells were detected, including 173 CD8+ cells and 94 CD8+ pSTAT1+ cells, corresponding to 85.2% CD8+ cells among all present cells and 54.3% CD8+ pSTAT1+ cells among CD8 + cells. In the TCMR(p) sample, 345 total cells were counted, with 280 CD8+ cells and 121 CD8+ pSTAT1+ cells, yielding 81.2% CD8+ cells of total cells and 43.2% CD8+ pSTAT1+ cells among CD8+ cells. In conclusion, pSTAT1 expression was markedly higher in the TCMR group (including one borderline rejection (post-BDR(p)) and one grade II TCMR) than in the post-CTRL(p), with a significant overlap observed between pSTAT1 and CD8 signals. Additionally, pSTAT1 was expressed at similar levels in both post-BDR(p) and TCMR(p).
Discussion
In this study, we integrated ST analysis of kidney allografts with scRNA-seq of human PBMCs to investigate potential gene expression patterns between the kidney tissue and systemic circulation of patients experiencing TCMR. This study represents one of the early comparative analyses utilizing both ST and scRNA-seq in KT recipients with TCMR and borderline rejection.
Our ST analysis suggested differences in DEG profiles between the tubulointerstitium and glomeruli at the onset of TCMR. The gene expression changes observed within the tubulointerstitium during TCMR, along with the upregulation of allograft rejection and interferon response pathways, align with the physiological prominence of this region in TCMR pathogenesis5,12,23. While the tubulointerstitium might be the primary site of TCMR-related immune responses, our ST data suggest that the glomeruli may also exhibit molecular alterations during rejection. These findings are in line with previous reports showing that various forms of glomerular involvement can occur in both T cell- and antibody-mediated rejection26,27,28. Although all TCMR(b) samples in our study had a Banff g-score of zero (Table S1B), we observed differential gene expression in the glomerular ROIs, suggesting their potential role in the immune mechanisms of TCMR25,28. However, caution is needed when interpreting these results. Because GeoMx ROIs are rectangular but glomeruli are spherical, the selected glomerular ROIs may include some surrounding tubulointerstitial tissue. As such, some of the observed transcriptional changes may reflect overlapping signals from adjacent compartments. Nonetheless, the presence of glomerular gene expression differences, even if modest, suggests that the glomeruli may not be entirely spared from immune-related processes during TCMR, highlighting the complexity and possible discordance between histology and molecular findings.
In our scRNA-seq analysis of PBMCs, we observed the hyperexpanded populations of CD8+ TEM cells and cytotoxic CD4+ T cells. Our attention was drawn particularly to CD8+ TEM cells because there was upregulation of GZMB, GNLY, PRF1, and GZMK expression, which are known as functional T cell markers29,30,31,32. When examining the dynamics of T cell clonal expansion, we observed an increase in clonality in the CD8+ TEM cells as well as in the proliferating CD8+ T and cytotoxic CD4+ T cell clusters33,34,35. Their MHC class I allorecognition functionality appeared to be substantially augmented during TCMR progression, potentially implying their central role in its pathogenesis36,37. These observations appear consistent with previous studies that have highlighted the role of CD8+ TEM cells in allograft rejection35,38,39,40. The concept that CD8+ T cells recognize allogeneic MHC class I may support their possible involvement in the TCMR process37,41,42,43.
It is important to acknowledge that our TCMR group included borderline rejection cases and isolated v-lesions, which represents a limitation in interpreting our findings. Mengel et al. demonstrated that the molecular phenotype of early protocol biopsies often reflects injury-repair responses rather than true rejection, with borderline cases frequently representing a subclinical TCMR or injury-repair response44. Similarly, Buxeda et al. reported that early isolated v + show primarily an early injury signature when compared to late isolated v + .45 Although our study includes such cases, our methodological approach differs substantially from these previous studies. Unlike the bulk RNA analysis used by Buxeda et al. that profiled a 770-gene set by nCounter Banff-Human Organ Transplantation (B-HOT) Panel, we performed whole transcriptome spatial profilingg (~ 18,000 genes) specifically on inflamed regions, preserving spatial information. Most importantly, the six genes we identified differ from the injury-related genes reported in these previous studies, supporting potentially distinct biological processes being captured in our analysis.
One of the important finding from our study is that we observed some common gene expression changes between kidney tissue and circulating PBMCs, with a set of six genes emerging as altered in our TCMR samples: LTB, STAT1, GBP5, PSME2, GZMK, and UBE2L6. The rationale behind focusing on these six genes is based on the following observations. Genes highly expressed in TCMR(b) compared to pre-TCMR(b) may be associated with the TCMR phenomenon. If these genes are also highly expressed in the CD8+ TEM cells of TCMR(p) compared to pre-TCMR(p) and in TCMR(p) compared to post-CTRL(p), they might represent potential signals of TCMR. Moreover, if the genes that appear altered in TCMR(b) are also upregulated in the CD8+ TEM cells of TCMR(p), it suggests that the T cells involved in the pathogenesis of TCMR at the tubulointerstitial region (TCMR(b)) have likely entered the circulating blood and been detected as PBMCs (TCMR(p)). These six genes were selected for further downstream analyses as they were upregulated in our TCMR samples across different comparisons and sample types, demonstrating their potential involvement in the TCMR process. These concurrent expression changes might be part of a coordinated immune response against acute rejection, which might eventually lead to surrogate markers of rejection. However, we must acknowledge that these molecular signatures might represent aspects of both rejection and injury-repair responses, particularly given the inclusion of borderline cases and isolated v-lesions in our TCMR group. Notably, these six genes differ from both the injury-related gene sets identified by Mengel et al. (AMAT1, ENDAT, IRITD3, and IRITD5) and the early injury genes (LTF, SERPINA3 and ADAMTS1) reported by Buxeda et al., suggesting potentially distinct biological processes being captured in our study44,45.
In our network and pathway analyses, STAT1, or a signal transducer and activator of transcription 1, occupied the central or hub position in both network and pathway analyses (Fig. 5). STAT1 activation has been associated with kidney injury in mouse models and glomerulopathies in humans46,47,48,49,50. Its role in promoting transcription associated with heightened immunity and driving T cell differentiation has been previously documented51,52,53, with accumulating evidence showing its involvement in the clonal expansion of CD8+ T cells54,55. Furthermore, our ST data were cross-validated against public microarray datasets, suggesting increased expression of STAT1 in patients with TCMR (Fig. 4D). Our IF findings indicated the presence of STAT1-expressing CD8+ T cells in both TCMR kidney tissues and PBMCs, revealing their differentiation and clonal expansion (Fig. 6B and C). In PBMCs, CD8+ cells constituted more than half of the total cells across all patient groups. However, their proportion was markedly higher in TCMR group including BDR patient, exceeding 80%. Notably, the frequency of CD8+ pSTAT1+ cells among the total CD8+ T cells was low in the post-CTRL(p) at only 15%. Whereas in TCMR(p), approximately half of the CD8+ T cells expressed pSTAT1. These findings suggest that a significant fraction of the CD8+ T cell population in TCMR patient consists of pSTAT1+ effector cells. These CD8+ T cells might transition from the graft into the systemic circulation, possibly initiating a more widespread immune response. The cross-compartment validation of STAT1 and our six-gene signature between tissue and peripheral blood supports a coordinated immune response rather than simply localized injury56.
By providing a spatial context to transcriptomic data, we attempted to capture localized changes in the graft kidney while also obtaining a more general view of changes in peripheral immune cells. Analyzing both blood and tissue samples collected at the same time point allows us to understand the relationship between localized immune responses occurring within the graft and circulating immune cells, offering insight into the molecular pathogenesis of TCMR. Our exploratory approach demonstrates that multi-omics approaches can be valuable in investigating molecular signatures and potential biomarkers of TCMR. The identification of a distinct module of genes, including LTB, STAT1, GBP5, PSME2, GZMK, and UBE2L6, working together to drive TCMR highlights the importance of considering the coordinated expression and functional relationships among genes rather than focusing on individual genes or pathways. The specific roles and interactions of this group of genes in the context of TCMR remain largely unexplored; however, our study provides initial insights that warrant further investigation. This perspective suggests the potential for future research into non-invasive monitoring strategies for acute rejection.
Our study has several limitations that must be acknowledged. First, the limited sample size with borderline rejection cases in the TCMR group means our findings should be considered preliminary and hypothesis-generating rather than conclusive. Although we focused on T cell-infiltrated areas in the ST analysis to minimize differences between borderline rejection and true TCMR, their heterogeneity might affect the common DEGs identified. Future studies with substantially larger cohorts and a comprehensive comparison of borderline rejection and TCMR samples are essential to validate our findings and provide insights into the molecular differences and similarities across these histological entities. The small sample size prevents definitive distinction between rejection-specific mechanisms and the stereotyped injury-repair response previously characterized by Mengel et al. Second, further validation of our findings in larger human and animal cohorts is warranted before clinical conclusions can be drawn. The potential causal relationship between STAT1 upregulation and acute rejection requires investigation using robust in vitro or in vivo assays. Third, the use of frozen PBMCs for the scRNA-seq study might have biased the results by affecting some immune cell subsets more than others and not capturing neutrophils. Fourth, our study investigated an early time point after KT, which may not be generalizable to TCMR episodes occurring later in the post-KT period.
In conclusion, our exploratory comparative study using ST and scRNA-seq provides a glimpse into the immunological landscape of TCMR, from localized immune responses within the graft to systemic changes in peripheral immune cells. While acknowledging the limitations of including borderline cases, our multi-omics approach has identified common DEGs among tissue and circulating immune cells which may reflect an interplay between localized and systemic immune responses during the development of TCMR. These findings suggest directions for future research into TCMR pathogenesis at the molecular level and highlight the potential value of multi-level approaches in identifying possible transcriptomic markers for the early detection of acute rejection. Future studies with larger cohorts and clearer stratification between borderline and definitive TCMR are needed to validate our findings and clarify whether the identified molecular signatures reflect specific rejection mechanisms or injury-repair responses.
Methods
Study population and clinical data collection
This study was approved by the Institutional Review Board (IRB) of the Seoul National University Hospital (IRB # 2205-175-1329). The study adhered to the ethical guidelines of the Declaration of Helsinki. We confirm that informed consent was obtained from all participants and their legal guardians. Patients included in this study underwent kidney transplantation at the Seoul National University Hospital between April and September 2022. We excluded patients who received multiple organ transplants, focusing solely on living donor KT recipients. Then we included only adult patients aged between 40 and 70 years who have no donor-specific antibodies and no evidence of infection, including cytomegalovirus or BK virus infection. Patients were selected based on the availability of both pre- and post-KT blood samples and kidney biopsy specimens. The study population consisted of patients with either borderline rejection or TCMR grade IIA on protocol biopsies performed nine days after KT. Although these cases do not represent classic TCMR, they exhibit histological features consistent with T cell-mediated immune activation and are referred to as the “TCMR group” throughout the manuscript for simplicity. The control group included patients who did not reject protocol biopsies. Detailed information on the study population is presented in Table S1. PBMCs samples were collected from six different patients. The PBMC analysis included four patients with biopsy-proven TCMR or borderline rejection and two control patients. For spatial transcriptomics analysis, four biopsy samples were collected at time-zero (T0) biopsy and 2w protocol biopsy from two TCMR patients. In the protocol biopsy, one patient exhibited borderline rejection and the other showed TCMR.
Sample collection and histopathological evaluation
PBMCs were collected from the patients at two time points: first, during the pre-KT evaluation when the KT recipients were hospitalized, which occurred prior to the T0 biopsies; and second, on the day of the post-KT protocol kidney biopsies. All patients underwent the standard clinical practice of our center, which includes obtaining T0 kidney biopsies from the allograft immediately after organ procurement but before perfusion. These T0 biopsies were collected as part of the standard clinical protocol to assess the baseline histological characteristics of the graft prior to any potential recipient-mediated changes. It should be noted that we did not perform separate donor biopsies in this study. Following transplantation, planned protocol biopsies were performed two weeks post-KT, usually at post-operative day 9 or 10, to monitor the graft function and detect any signs of subclinical rejection. Once the kidney biopsy was done, three biopsy cores were obtained using a 14-gauge needle under sonographic guidance. The first two cores were subjected to formalin fixation and paraffin embedding for conventional histopathological evaluation. The third core was designated for spatial transcriptomics. All biopsy samples were diagnosed based on the internationally standardized Banff lesion scores5. All patients received basiliximab (Simulect®) at the time of KT, followed by immunosuppression via tacrolimus, mycophenolic acid, and prednisone. Among the TCMR group for scRNA-seq, three patients were diagnosed with borderline rejection, and one was diagnosed with grade II TCMR.
PBMC isolation
Blood samples were collected using BD Vacutainer CPT Mononuclear Cell Preparation Tubes (BD Biosciences, CPT tube)57. Within 2 h post-collection, these were centrifuged at 1800 g for 20 min at 20 ℃. After centrifugation, PBMCs, identified as a whitish layer beneath the plasma, were promptly collected and transferred into a 15-ml conical tube containing 1 ml of RPMI supplemented with 10% FBS. The tube was filled with RPMI to a total volume of 15 ml and centrifuged at 500 g for 5 min at 20 ℃ for washing. The isolated PBMCs were then suspended in freezing medium (AMSBIO, Cambridge, MA, USA; CELLBANKER2) and stored in liquid nitrogen.
In-situ hybridization slide Preparation for ST
GeoMx Digital Spatial Profiler (DSP; NanoString, Seattle, WA, USA) is an ST profiling tool. Slide preparation was performed following the manufacturer’s guidelines (NanoString GeoMx DSP Manual Slide Preparation, MAN-10150-01). For formalin-fixed paraffin-embedded (FFPE) blocks, 5-\(\:{\upmu\:}\text{m}\)-thick kidney biopsy sections were mounted on charged slides (Leica BOND Plus slides, Germany). The slides were baked at 60 ℃ for 30 min, deparaffinized with CitriSolv, and rehydrated through 100% and 95% ethanol, followed by 1X phosphate-buffered saline (PBS) wash for 5 min each. Target epitopes were retrieved and exposed by incubating in 1X Tris-EDTA, pH 9, and preheated 1\(\:{\upmu\:}\text{l}\)/ml proteinase K. To preserve kidney tissue morphology, the slides were immersed in 10% neutral buffered formalin (NBF) and then washed with 1X PBS. To validate the results, immunohistochemistry (IHC) and immunofluorescence (IF) were performed as described above. Consequently, the slides for spatial transcriptomics were incubated with RNA hybridization probes that consist of a target complementary sequence and a DNA oligo barcode for NGS readout using WTA (Whole Transcriptome Atlas), overnight at 37 ℃. On the next day, the slides were stained with the following antibodies: SYTO 13 (NanoString, 121300303, 1:10), CD45 (Cell Signaling Technology, 14579, 1:100), PanCK (Novus, NBP2-33200AF594, 1:100), and alpha Smooth Muscle Actin (Abcam, ab202296, 1:5,000) for 1 h at room temperature. After staining, the slides were loaded into the GeoMx DSP instrument to select and collect regions of interest (ROI). For the pre-TCMR(b) sample, four tubulointerstitial regions with normal histology and two glomeruli were selected. In the TCMR(b) sample, four tubulointerstitial regions with confirmed T cell infiltration, excluding v-lesions and two histologically normal glomeruli were profiled. Higher-magnification images of representative ROIs of glomeruli are provided in Supplementary Figure S5. In the control sample, all selected areas—four tubulointerstitial regions and two glomeruli—showed no histological abnormalities. Oligonucleotides linked to their target gene were cleaved and released from the selected ROI, with an average area of 69,024.5\(\:{{\upmu\:}\text{m}}^{2}\)/ROI collected into a DSP collection plate. Primer pairs, i5/i7 dual-indexing sequences, were used to index oligonucleotides during the polymerase chain reaction (PCR).
Single-cell Preparation and library construction
A total of twelve samples were utilized, encompassing both pre- and post-transplantation samples. These samples were derived from two patients without TCMR and four patients diagnosed with TCMR. After the PBMCs were carefully thawed, 12 different hashtag-oligos were used for de-multiplexing. Single-cell capturing and library construction for scRNA-seq analysis were performed following the manufacturer’s guidelines (10X Genomics, Pleasanton, CA; Chromium Next GEM Single Cell 5’ HT Reagent Kits v2, CG000424). Gel bead-in-emulsions (GEMs) were generated by combining PBMCs samples and gel beads onto Chromium Next GEM Chip N. Following GEM generation, both complementary DNA (cDNA) poly-adenylated mRNA and DNA from hashtag-oligos were simultaneously formed during polymerase chain reaction (PCR). Synthesized full-length cDNA was then subjected to a cleanup process using a Cleanup Mix solution containing Dynabeads MyOne SILANE (PN 2000048, 10X Genomics) in each sample. After the cleanup step, cDNA was amplified to construct libraries for gene expression, T cells, and hashtag-oligo libraries. All the final libraries contained the P5 and P7 priming sites compatible with Illumina sequencers. To assess the yield and purity of the cDNA and three types of libraries, High Sensitivity D5000 and D1000 ScreenTape were employed, respectively, on a 4150 TapeStation instrument (Agilent Technologies, CA, USA).
ST data processing and analysis
GeoMx DSP Analysis Suite (GEOMX-0094, v2.4.2.2) was utilized to analyze ST data. Target genes with 5% of the segment above the threshold were retained in the datasets, and gene expression lower than the limit of quantitation was initially excluded. The limit of quantitation was determined as the negative probe geomean multiplied by the geometric standard deviation of the negative probes, yielding a total of 13,183 genes for further analysis. The read counts of these filtered genes were normalized via the Q3 normalization method. To identify DEGs, the ROIs were discerned between glomeruli and tubulointerstitium, and subsequent comparisons between ROIs from zero-time allograft kidney biopsies of TCMR patients (pre-TCMR(b)) and their biopsies on post-operative day 9 (TCMR(b)) were performed using paired t-tests with Benjamini-Hochberg multiple test correction. The STRING database was then used to explore the interaction networks of the identified DEGs.
scRNA-seq data processing and analysis
Cell Ranger (version 7.0.1, 10X Genomics) was utilized to to align into reference genome and demultiplex cellular barcodes. We performed 4-plex sequencing, treating each 4-plex as a single sample for quality control (QC). In the raw data, the number of cells for each studied sample was 26,189 cells, 28,590 cells, and 28,125 cells, totaling 82,904 cells. The processed data were then extracted from the “filtered_gene_bc_matrices” directory and imported into R using the ‘read10X’ function from the Seurat package (version 4.3.0). This step enabled the generation of a count matrix based on unique molecular identifiers (UMIs), which provided detailed information on gene counts for each individual cell. To enhance data accuracy, cells presumed to be doublets formed during cell capture were removed using the computational doublet detection tool, DoubletFinder. Quality control measures were applied, and cells with nFeature_RNA > 2500, nFeature_RNA < 200, and percent.mt > 5 were filtered out. As a result, we used 10,964 cells, 15,607 cells, and 21,657 cells per studied sample, with a total of 48,288 cells for further analysis. The final cell counts for each patient were as follows: pre-CTRL(p)-1 = 3,884 cells, post-CTRL(p)-1 = 4,486, pre-TCMR(p)-1 = 2,578, TCMR(p)-1 = 16, pre-CTRL(p)-2 = 4,009, post-CTRL(p)-2 = 4,302, pre-TCMR(p)-2 = 3,433, TCMR(p)-2 = 3,863, pre-TCMR(p)-3 = 5,506, TCMR(p)-3 = 5,173, pre-TCMR(p)-4 = 6,494, and TCMR(p)-4 = 4,484 (Figure S1A and S1B). After integrating all the cells and performing dimensional reduction using UMAP, we confirmed that the cells from each patient were evenly distributed. The sequencing saturation for the three 4-plexed samples was set to 60–70%.
Normalization of the feature expression for each cell by total expression was performed using the LogNormalize method. The data were scaled using the ScaleData function, and principal component analysis (PCA) was performed with the RunPCA function to achieve linear dimensionality reduction. To address batch effects, the Harmony integration method was implemented. Subsequently, the integrated data matrix underwent Uniform Manifold Approximation and Projection (UMAP) visualization of clustering using the RunUMAP, FindNeighbors, and FindClusters functions. We annotated each cluster with the differentially expressed genes (DEGs) found through the FindAllMarkers function (with a threshold of min.pct = 0.25). The distribution of these genes was visualized using the FeaturePlot function, and the DEGs among the groups within a cluster were further delineated using the FindMarkers function.
The protein-protein interaction (PPI) network for DEGs was constructed using the Cytoscape software (version 3.10.0). A cut-off value of 0.2 was applied, and nodes within the network were color-coded according to the log2 fold changes of DEGs derived from the scRNA-seq data. The node sizes were scaled in accordance with their respective p-values, illustrated by the representation of 29 out of 32 genes when comparing pre- to post-KT blood samples of the patients with TCMR (pre-TCMR(p) and TCMR(p) in short, respectively), and 189 out of 213 genes when comparing the TCMR(p) to the post-KT blood samples of patients who did not experience TCMR (post-CTRL(p)) (Fig. 5; Table 2). DEGs were then functionally annotated using the STRING database, focusing on Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses. The resultant plots were color-graded to reflect the significance levels, as determined by the p-values from GO terms.
Immunofluorescence and immunohistochemistry
For immunofluorescence (IF) staining, allograft kidney biopsies were probed with the following primary antibodies diluted in 2% bovine serum albumin (BSA)/Tris-buffered saline (TBST) overnight at 4 ℃: anti-CD8B (sc-1177; Santa Cruz Biotechnology, dilution 1:50, [32-M4]) and anti-STAT1 (#14994; Cell Signaling Technology, dilution 1:200, [D1K9Y]). Tissue samples were then incubated with secondary antibodies for 1 h at room temperature (RT), including an Alexa Fluor 488-conjugated goat anti-mouse for CD8 (A21202; Thermo Fisher Scientific, dilution 1:600) and an Alexa Fluor 555-conjugated goat anti-rabbit for STAT1 (A21428, Thermo Fisher Scientific, dilution 1:600). Furthermore, 4′, 6-diamidino-2-phenylindole (DAPI, Invitrogen) was used to counterstain the nuclei. IF images were acquired using a confocal microscope (Leica TCS SP8; Leica Microsystem GmbH, Wetzlar, Germany). For cytospin immunofluorescence, cells were resuspended in cold 1X PBS and then fixed by adding an equal volume of 4% paraformaldehyde (PFA), followed by incubation for 15 min at room temperature. The 100\(\:{\upmu\:}\text{l}\) of fixed cells were loaded to cytospin funnels. The cells were spun onto the slides at 1,200 rpm for 3 min. Following cytospinning, the slides were air-dried 10 min at room temperature. The cells were permeabilized by incubating the slides in 0.1% Triton X-100 in 1X PBS for 10 min at room temperature and then washed three times with 1X PBS. To block non-specific binding sites, the slides were incubated in 5% BSA in PBS for 1 h at room temperature. Primary antibodies, anti-CD8 (CF802079; OriGene, dilution 1:100, [OTI3H6]) and anti-pSTAT1 (9167 S; Cell Signaling TECHNOLOGY, dilution 1:50, [58D6]) were diluted in 2% BSA in PBS. Subsequently the slides were incubated in a humid chamber overnight at 4 ℃. Fluorescently labeled secondary antibodies, Alexa Fluor 488-conjugated anti-mouse and Alexa Fluor 555-conjugated anti-rabbit, were used to stain CD8 and pSTAT1, respectively.
For immunohistochemistry (IHC) staining, anti-STAT1, the previously mentioned anti-STAT1 antibody, anti-CD3E (790–4341; Roche), and anti-pSTAT1 antibody (9167 S; Cell Signaling TECHNOLOGY, dilution 1:100) was used. The signal was developed using DAB. Counter staining was performed using hematoxylin (MA0101081MIRA01, BBC biochemical). IHC images were captured using a scanner (ZEISS Axioscan 7; ZEISS, German). The stained tissue was observed under an Olympus microscope (BX51F, Olympus, Tokyo, Japan).
Statistical analysis and image generation
Differences in gene expression between pre-TCMR and TCMR samples were statistically evaluated using paired t-tests. Statistical analyses were performed using GraphPad Prism software (v.9.5.0; GraphPad Prism Software, La Jolla, CA, USA). p-<0.05 was considered significant, and expression was measured in arbitrary units. All figures were created with BioRender (BioRender.com) or R studio.
Data availability
The scRNA-seq data and spatial transcriptomic data in this study are available Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE248789) under accession number “GSE248789”. The dataset used for validation is accessible within the GSE36059.
Abbreviations
- BDR:
-
Borderline rejection
- CKD:
-
Chronic kidney disease
- DEG:
-
Differentially expressed gene
- DSP:
-
Digital spatial profiling
- ESKD:
-
End-stage kidney disease
- FFPE:
-
Formalin-fixed paraffin-embedded
- IF:
-
Immunofluorescence
- IHC:
-
Immunohistochemistry
- KT:
-
Kidney transplantation
- PBMC:
-
Peripheral blood mononuclear cell
- PCA:
-
Principal component analysis
- post-CTRL(p):
-
Post-operative PBMCs samples from patients without acute rejection
- post-CTRL(b):
-
Post-operative kidney allograft biopsy samples from patients without acute rejection
- pre-CTRL(p):
-
Pre-operative PBMCs samples from patients without acute rejection
- pre-CTRL(b):
-
Time-zero kidney allograft biopsy samples from patients without acute rejection
- pre-TCMR(p):
-
Pre-operative PBMCs samples from TCMR patients
- pre-TCMR(b):
-
Time-zero kidney allograft biopsy samples from TCMR patients
- ROIs:
-
Regions of interest
- scRNA-seq:
-
Single-cell RNA sequencing
- ST:
-
Spatial transcriptomics
- T0:
-
Time-zero
- TCMR:
-
Acute T cell-mediated rejection
- TCMR(p):
-
Post-operative PBMCs samples from TCMR patients
- TCMR(b):
-
Post-operative kidney allograft biopsy samples from TCMR patients
- TCR:
-
T-cell receptor
- TEM :
-
Effector memory T cell
- UMI:
-
Unique molecular identifier
References
Bouatou, Y. et al. Response to treatment and long-term outcomes in kidney transplant recipients with acute T cell-mediated rejection. Am J. Transplant Jul. 19 (7), 1972–1988. https://doi.org/10.1111/ajt.15299 (2019).
Hoffman, W. et al. The impact of early clinical and subclinical T Cell-mediated rejection after kidney transplantation. Transplantation Jul. 103 (7), 1457–1467. https://doi.org/10.1097/TP.0000000000002560 (2019).
Lefaucheur, C. et al. T cell-mediated rejection is a major determinant of inflammation in scarred areas in kidney allografts. Am J. Transplant Feb. 18 (2), 377–390. https://doi.org/10.1111/ajt.14565 (2018).
Rampersad, C. et al. The negative impact of T cell-mediated rejection on renal allograft survival in the modern era. Am J. Transplant Mar. 22 (3), 761–771. https://doi.org/10.1111/ajt.16883 (2022).
Loupy, A. et al. The Banff 2019 kidney meeting report (I): updates on and clarification of criteria for T cell- and antibody-mediated rejection. Am J. Transplant Sep. 20 (9), 2318–2331. https://doi.org/10.1111/ajt.15898 (2020).
Mengel, M. et al. Banff 2019 meeting report: molecular diagnostics in solid organ transplantation-Consensus for the Banff human organ transplant (B-HOT) gene panel and open source multicenter validation. Am J. Transplant Sep. 20 (9), 2305–2317. https://doi.org/10.1111/ajt.16059 (2020).
Madill-Thomsen, K. et al. Discrepancy analysis comparing molecular and histology diagnoses in kidney transplant biopsies. Am. J. Transpl. 20 (5), 1341–1350. https://doi.org/10.1111/ajt.15752 (May 2020).
Christakoudi, S. et al. Development of a multivariable gene-expression signature targeting T-cell-mediated rejection in peripheral blood of kidney transplant recipients validated in cross-sectional and longitudinal samples. EBioMedicine Mar. 41, 571–583. https://doi.org/10.1016/j.ebiom.2019.01.060 (2019).
Smith, R. N. In-silico performance, validation, and modeling of the Nanostring Banff human organ transplant gene panel using archival data from human kidney transplants. BMC Med. Genomics Mar. 19 (1), 86. https://doi.org/10.1186/s12920-021-00891-5 (2021).
Zhou, H. et al. Diagnostic biomarkers and immune infiltration in patients with T Cell-Mediated rejection after kidney transplantation. Front. Immunol. 12, 774321. https://doi.org/10.3389/fimmu.2021.774321 (2021).
Rosales, I. A. et al. Banff human organ transplant transcripts correlate with renal allograft pathology and outcome: importance of capillaritis and subpathologic rejection. J Am. Soc. Nephrol Dec. 33 (12), 2306–2319. https://doi.org/10.1681/ASN.2022040444 (2022).
Loupy, A., Mengel, M. & Haas, M. Thirty years of the international Banff classification for allograft pathology: the past, present, and future of kidney transplant diagnostics. Kidney Int Apr. 101 (4), 678–691. https://doi.org/10.1016/j.kint.2021.11.028 (2022).
Ho, J. et al. Effectiveness of T cell-mediated rejection therapy: A systematic review and meta-analysis. Am J. Transplant Mar. 22 (3), 772–785. https://doi.org/10.1111/ajt.16907 (2022).
Wu, H. et al. Single-Cell transcriptomics of a human kidney allograft biopsy specimen defines a diverse inflammatory response. J. Am. Soc. Nephrol. 29 (8), 2069–2080. https://doi.org/10.1681/ASN.2018020125 (Aug 2018).
Malone, A. F. et al. Harnessing expressed single nucleotide variation and single cell RNA sequencing to define immune cell chimerism in the rejecting kidney transplant. J. Am. Soc. Nephrol. 31 (9), 1977–1986. https://doi.org/10.1681/asn.2020030326 (2020).
Dangi, A. et al. Single cell transcriptomics of mouse kidney transplants reveals a myeloid cell pathway for transplant rejection. JCI Insight. 5 (20). https://doi.org/10.1172/jci.insight.141321 (2020).
Salem, F. et al. The spatially resolved transcriptional profile of acute T cell-mediated rejection in a kidney allograft. Kidney Int Jan. 101 (1), 131–136. https://doi.org/10.1016/j.kint.2021.09.004 (2022).
Melo Ferreira, R. et al. Integration of Spatial and single-cell transcriptomics localizes epithelial cell-immune cross-talk in kidney injury. JCI Insight Jun. 22 (12). https://doi.org/10.1172/jci.insight.147703 (2021).
Dixon, E. E., Wu, H., Muto, Y., Wilson, P. C. & Humphreys, B. D. Spatially resolved transcriptomic analysis of acute kidney injury in a female murine model. J. Am. Soc. Nephrol. 33 (2), 279–289. https://doi.org/10.1681/ASN.2021081150 (Feb 2022).
Smith, K. D. et al. Digital Spatial profiling of collapsing glomerulopathy. Kidney Int May. 101 (5), 1017–1026. https://doi.org/10.1016/j.kint.2022.01.033 (2022).
Lamarthee, B. et al. Transcriptional and Spatial profiling of the kidney allograft unravels a central role for FcyRIII + innate immune cells in rejection. Nat Commun Jul. 19 (1), 4359. https://doi.org/10.1038/s41467-023-39859-7 (2023).
Chen, D. et al. Single-cell RNA-seq with Spatial transcriptomics to create an atlas of human diabetic kidney disease. FASEB J Jun. 37 (6), e22938. https://doi.org/10.1096/fj.202202013RR (2023).
Callemeyn, J. et al. Allorecognition and the spectrum of kidney transplant rejection. Kidney Int Apr. 101 (4), 692–710. https://doi.org/10.1016/j.kint.2021.11.029 (2022).
Filippone, E. J. & Farber, J. L. The Histological Spectrum and Clinical Significance of T Cell-mediated Rejection of Kidney Allografts. Transplantation. May 1. ;107(5):1042–1055. (2023). https://doi.org/10.1097/TP.0000000000004438
Reeve, J. et al. Molecular diagnosis of T cell-mediated rejection in human kidney transplant biopsies. Am J. Transplant Mar. 13 (3), 645–655. https://doi.org/10.1111/ajt.12079 (2013).
Sentis, A. et al. The prognostic significance of glomerular infiltrating leukocytes during acute renal allograft rejection. Transpl Immunol Nov. 33 (3), 168–175. https://doi.org/10.1016/j.trim.2015.10.004 (2015).
Randhawa, P. T-cell-mediated rejection of the kidney in the era of donor-specific antibodies: diagnostic challenges and clinical significance. Curr Opin. Organ. Transplant Jun. 20 (3), 325–332. https://doi.org/10.1097/MOT.0000000000000189 (2015).
Halloran, P. F., Reeve, J. P., Pereira, A. B., Hidalgo, L. G. & Famulski, K. S. Antibody-mediated rejection, T cell-mediated rejection, and the injury-repair response: new insights from the genome Canada studies of kidney transplant biopsies. Kidney Int Feb. 85 (2), 258–264. https://doi.org/10.1038/ki.2013.300 (2014).
Youngblood, B. et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature Dec. 21 (7685), 404–409. https://doi.org/10.1038/nature25144 (2017).
Abdelsamed, H. A. et al. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp. Med Jun. 5 (6), 1593–1606. https://doi.org/10.1084/jem.20161760 (2017).
Jonsson, A. H. et al. Granzyme K < sup>+ CD8 T cells form a core population in inflamed human tissue. Sci. Transl. Med. 14 (649), eabo0686. https://doi.org/10.1126/scitranslmed.abo0686 (2022).
Lagumdzic, E. et al. Gene expression of peripheral blood mononuclear cells and CD8(+) T cells from gilts after PRRSV infection. Front. Immunol. 14, 1159970. https://doi.org/10.3389/fimmu.2023.1159970 (2023).
Dziubianau, M. et al. TCR repertoire analysis by next generation sequencing allows complex differential diagnosis of T cell-related pathology. Am J. Transplant Nov. 13 (11), 2842–2854. https://doi.org/10.1111/ajt.12431 (2013).
Tian, G., Li, M. & Lv, G. Analysis of T-Cell receptor repertoire in transplantation: fingerprint of T Cell-mediated alloresponse. Front. Immunol. 12, 778559. https://doi.org/10.3389/fimmu.2021.778559 (2021).
Dunlap, G. S. et al. Clonal dynamics of alloreactive T cells in kidney allograft rejection after anti-PD-1 therapy. Nat Commun Mar. 21 (1), 1549. https://doi.org/10.1038/s41467-023-37230-4 (2023).
Alachkar, H. et al. Quantitative characterization of T-cell repertoire and biomarkers in kidney transplant rejection. BMC Nephrol Nov. 21 (1), 181. https://doi.org/10.1186/s12882-016-0395-3 (2016).
Burrack, A. L. et al. Simultaneous recognition of allogeneic MHC and cognate autoantigen by autoreactive T cells in transplant rejection. J Immunol Feb. 15 (4), 1504–1512. https://doi.org/10.4049/jimmunol.1700856 (2018).
Schenk, A. D., Nozaki, T., Rabant, M., Valujskikh, A. & Fairchild, R. L. Donor-reactive CD8 memory T cells infiltrate cardiac allografts within 24-h posttransplant in Naive recipients. Am J. Transplant Aug. 8 (8), 1652–1661. https://doi.org/10.1111/j.1600-6143.2008.02302.x (2008).
Du, G. et al. CD8(+) effector memory T cells induce acute rejection of allogeneic heart retransplants in mice possibly through activating expression of inflammatory cytokines. Exp Cell. Res Jun. 1 (1), 1–8. https://doi.org/10.1016/j.yexcr.2017.02.036 (2017).
Ko, E. J. et al. Phenotype and molecular signature of CD8 + T cell subsets in T cell- mediated rejections after kidney transplantation. PLoS One. 15 (6), e0234323. https://doi.org/10.1371/journal.pone.0234323 (2020).
Harper, S. J. et al. CD8 T-cell recognition of acquired alloantigen promotes acute allograft rejection. Proc Natl. Acad. Sci. U S A Oct. 13 (41), 12788–12793. https://doi.org/10.1073/pnas.1513533112 (2015).
Siu, J. H. Y., Surendrakumar, V., Richards, J. A. & Pettigrew, G. J. T cell allorecognition pathways in solid organ transplantation. Front. Immunol. 9, 2548. https://doi.org/10.3389/fimmu.2018.02548 (2018).
Carnel, N., Lancia, H. H., Guinier, C. & Benichou, G. Pathways of antigen recognition by T cells in allograft rejection. Transplantation Apr. 1 (4), 827–837. https://doi.org/10.1097/TP.0000000000004420 (2023).
Mengel, M. et al. The molecular phenotype of 6-week protocol biopsies from human renal allografts: reflections of prior injury but not future course. Am J. Transplant Apr. 11 (4), 708–718. https://doi.org/10.1111/j.1600-6143.2010.03339.x (2011).
Buxeda, A. et al. Clinical and molecular spectrum of v-lesion. Am J. Transplant Nov. 24 (11), 2007–2021. https://doi.org/10.1016/j.ajt.2024.07.025 (2024).
Yang, N. et al. Blockage of JAK/STAT signalling attenuates renal ischaemia-reperfusion injury in rats. Nephrol. Dialysis Transplantation. 23 (1), 91–100. https://doi.org/10.1093/ndt/gfm509 (2008).
Kemmner, S. et al. STAT1 regulates macrophage number and phenotype and prevents renal fibrosis after ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. Feb 1 (2), F277–F291. https://doi.org/10.1152/ajprenal.00004.2018 (2019).
Tao, J. et al. JAK-STAT signaling is activated in the kidney and peripheral blood cells of patients with focal segmental glomerulosclerosis. Kidney Int Oct. 94 (4), 795–808. https://doi.org/10.1016/j.kint.2018.05.022 (2018).
Ying, W. Z. et al. Immunoglobulin light chains generate Proinflammatory and profibrotic kidney injury. J Clin. Invest Jun. 17 (7), 2792–2806. https://doi.org/10.1172/JCI125517 (2019).
Tao, J. et al. JAK-STAT activity in peripheral blood cells and kidney tissue in IgA nephropathy. Clin. J. Am. Soc. Nephrol. Jul 1 (7), 973–982. https://doi.org/10.2215/CJN.11010919 (2020).
Toubiana, J. et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood Jun 23. 127 (25), 3154–3164. https://doi.org/10.1182/blood-2015-11-679902 (2016).
Zhang, Y. et al. PD-L1 up-regulation restrains Th17 cell differentiation in STAT3 loss- and STAT1 gain-of-function patients. J. Exp. Med. 214 (9), 2523–2533. https://doi.org/10.1084/jem.20161427 (2017).
Meyer Zu Horste, G. et al. Fas promotes T helper 17 cell differentiation and inhibits T helper 1 cell development by binding and sequestering transcription factor STAT1. Immunity. Mar 20 ;48(3):556–569e7. (2018). https://doi.org/10.1016/j.immuni.2018.03.008
Quigley, M., Huang, X. & Yang, Y. STAT1 signaling in CD8 T cells is required for their clonal expansion and memory formation following viral infection in vivo. J Immunol Feb. 15 (4), 2158–2164. https://doi.org/10.4049/jimmunol.180.4.2158 (2008).
Ramana, C. V. et al. Inflammatory impact of IFN-gamma in CD8 + T cell-mediated lung injury is mediated by both Stat1-dependent and -independent pathways. Am J. Physiol. Lung Cell. Mol. Physiol Apr. 1 (7), L650–L657. https://doi.org/10.1152/ajplung.00360.2014 (2015).
Baan, C. C., Kannegieter, N. M., Felipe, C. R. & Tedesco Silva, H. Jr. Targeting JAK/STAT signaling to prevent rejection after kidney transplantation: A reappraisal. Transplantation Sep. 100 (9), 1833–1839. https://doi.org/10.1097/TP.0000000000001226 (2016).
Corkum, C. P. et al. Immune cell subsets and their gene expression profiles from human PBMC isolated by vacutainer cell Preparation tube (CPT™) and standard density gradient. BMC Immunol. 16 (1). https://doi.org/10.1186/s12865-015-0113-0 (2015).
Acknowledgements
We would like to acknowledge the SNUH Transplantation Center and Transplantation Research Institute of Seoul National University Medical Research Center for their hard work. Part of this work was presented at the plenary session at the annual meeting of the Asian Transplantation Week 2023.
Funding
This work was supported by the Bio & Medical Technology Development Program of the National Research Foundation funded by the Ministry of Science & ICT (No. 2022M3A9D3016848), SNUH Research Fund (No. 0320230390). This work was partly supported by Institute of Information & communications Technology Planning & Evaluation (IITP) grant funded by the Korea government (MSIT) (NO.RS-2021-II211343, Artificial Intelligence Graduate School Program (Seoul National University)). We thank all participants, their families, surgeons, nursing staff, and other supporters who made these studies possible.
Author information
Authors and Affiliations
Contributions
J.S., M.K., Y.J., C.G.P., S.H.Y., H.J.K., and Y.C.K. conceived and designed the study. M.K., J.S., Y.J., S.C., Y.H.K., J.Y.B., C.S.J., H.Y.S., S.M.L., H.K., H.S.C., and S.H.Y. performed the experiments and collected data. M.K., J.Y.B., C.S.J., H.Y.S., H.S.C., K.H.K., and J.H.M. analyzed and interpreted the data. A.H., S.I.M., J.H., S.P., S.S.H., D.K.K., K.H.O., K.W.J., Y.S.K., K.C.M., H.L., and Y.C.K. provided resources required for the study. S.H.Y., H.J.K., and Y.C.K. supervised the study and manuscript editing. J.S. and M.K. drafted the manuscript. All authors have reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, J., Kang, M., Jang, Y. et al. Discovering molecular signatures in kidney transplant biopsies with borderline changes and isolated V-lesions: single-cell RNA-sequencing analysis of human blood and tissue Spatial transcriptomics. Sci Rep 15, 23770 (2025). https://doi.org/10.1038/s41598-025-05191-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-05191-x
