
Abstract
α-Glucosidase is an important target in treating type 2 diabetes, and thus, the inhibition of this enzyme could delay sugar digestion and avoid postprandial hyperglycemia. Previous studies revealed that some pyrimidine and piperazine derivatives showed good affinity towards α-glucosidase. In continuing efforts toward the development of α-glucosidase inhibitor, a series of pyrimidinyl-piperazine carboxamide 6–22 containing chiral center have been synthesized. The inhibition activity was evaluated against α-glucosidase from Saccharomyces cerevisiae. All tested compounds exhibit excellent inhibition effects compared to acarbose (IC50 = 817.38 µM). Compounds bearing S-configurations at the chiral center displayed up to 5-fold more active than R-configurations. Among them, compounds 7c, 17c, 21c, and 22c were the top four compounds with IC50 values in a range of 0.4–1.5 µM. A kinetic study revealed that competitive inhibition as their mechanism of action. The results of the computational study indicated that hydrophobic interactions were the key factor in this activity. Moreover, molecular dynamics simulation showed that the 21c/α-glucosidase complex provided more stability by maintaining a consistent binding pose. MM-GBSA analysis revealed that the binding energy of 21c was approximately 11 kcal/mol lower than its counterpart, confirming the superiority of the S-configuration. The cytotoxicity test indicated that the top four compounds were not toxic to normal cells at the given IC50 value. Hence, these compounds are promising candidate as α-glucosidase inhibitors.
Similar content being viewed by others
Introduction
Diabetes mellitus is a group of metabolic disorders characterized by hyperglycemia and carbohydrate, fat, and protein metabolism abnormalities. Diabetes is a life-threatening, chronic disease that could reduce human life expectancy1. According to the International Diabetes Federation (IDF) report, in 2021, the number of adults with diabetes was 537 million worldwide, projected to increase by 45% in 20452. Type 2 diabetes mellitus is the most prevalent type, where approximately 90% of diabetes cases come from this type. In type 2 diabetes, blood contains high levels of sugar due to ineffective or low amounts of insulin in the body, thus causing health complications such as heart attack, kidney problems, and nerve damage3,4.
To date, there is no permanent cure for diabetes, but it can be treated and controlled5. Common medication for type 2 diabetes is by delaying the release of glucose and decreasing postprandial hyperglycemia. α-Glucosidase is responsible for hydrolyzing carbohydrates into glucose; therefore, inhibition of α-glucosidase is an effective way to treat type 2 diabetes6,7. Some α-glucosidase inhibitors such as acarbose, voglibose, and miglitol, are readily available in the market. These drugs are pseudo-carbohydrates that competitively inhibit α-glucosidase; however, they have undesired gastrointestinal side effects such as abdominal pain, flatulence, and diarrhea8. Therefore, finding new α-glucosidase inhibitors is still an interesting topic for many researchers. In this context, yeast α-glucosidase from Saccharomyces cerevisiae has been used extensively in finding potent inhibitor9,10,11. Yeast and human α-glucosidase share similar substrate specificity, pH optimum, and inhibitor sensitivity. Thus, the yeast enzyme serves as a good experimental model to learn more about the structure, substrate specificity, and enzymatic mechanism of human α-glucosidase12.
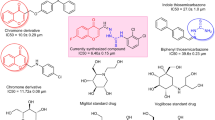
One of the prominent candidates for α-glucosidase inhibitors is heterocycle compounds. There are numerous heterocycles found in bioactive compounds, e.g., pyrimidine, pyrazole, indole, imidazole, piperidine, and piperazine13,14,15,16,17,18. Inspired by heterocyclic containing antidiabetic drug, Gosogliptin, we designed pyrimidinyl-piperazine carboxamide as α-glucosidase inhibitor (Fig. 1). It contained pyrimidine and piperazine heterocycles that have been reported exhibit broad spectrum biological activity e.g., anticancer, antimalarial, antibiotic, antiviruses, antidiabetic agents including as α-glucosidase inhibitor19,20,21,22,23. For example, pyrimidine-containing compound A isolated from leaves of Morus Astropurpurea inhibits yeast α-glucosidase with an IC50 of 10.3 µM and synthetic compound B inhibits both α-glucosidase from yeast and rat intestine with IC50 values of 16.4 and 45 µM, respectively11,24. Moreover, compound C (CHEMBL1411034) containing piperazine carboxamide exhibits promising activity toward human lysosomal α-glucosidase with an IC50 value of 25.1 µM25. Chirality, on the other hand, has been reported to affect the selectivity of a compound in biological systems26,27,28,29. To study this effect on α-glucosidase, we introduced a chiral center to the piperazine moiety of pyrimidinyl-piperazine carboxamide. In this position, a molecule may orient in a proper position in proximity to an active site of an enzyme. Herein, we report the synthesis of chiral pyrimidinyl-piperazine carboxamide and their biological evaluation as the new α-glucosidase inhibitors.

Rational for the design chiral pyrimidinyl-piperazine carboxamide derivatives.
Results and discussion
Chemistry
The synthesis route for pyrimidinyl-piperazine carboxamide 6–22 is outlined in Fig. 2. In general, these reactions gave medium to high yield in the range of 40–95%. The first reaction involved protecting one of the amine groups of 2-methylpiperazine 1 in the presence of di-tert-butyl dicarbonate in mild conditions to produce racemic 1-boc-3-methyl piperazine 2a30. Racemic or commercially available chiral (R/S)-1-boc-3-methyl piperazine 2a-c was further reacted with 2,4-dichloropyrimidine 3 to obtain pyrimidinyl-piperazine intermediates 4a-c via nucleophilic substitution. Intermediate 5 was synthesized by treating the corresponding intermediates 4a-c with various substituted-aniline. Upon reaction in the presence of trifluoroacetic acid, the Boc protection group of piperazine was removed31. Then, without further purification, intermediate 5 reacted with aryl isocyanate, affording the pyrimidinyl-piperazine carboxamide derivatives 6–21 with different configurations at the chiral center32. In addition, compound 22c was obtained by modifying the hydroxyl substituent of compound 21c with benzyl bromide. The structures of newly synthesized compounds were confirmed by their1H NMR13, C NMR, and HRMS spectra, which can be seen in Supplementary Information.

Synthesis route for compounds 6–22. Reagent and conditions: (a) Boc2O, DCM, 0–5 °C, 5 h; (b) Et3N, iPrOH, reflux, 12 h; (c) R-NH2, TFA, iPrOH, reflux, 12 h; (d) R-NCO, Et3N, DCM, r.t. 2 h; (e) Benzyl bromide, K2CO3, Acetone, rt, 3 h.
In vitro α-Glucosidase inhibitory activity
A series of pyrimidinyl-piperazine carboxamide derivatives (6–22) were investigated for their α-glucosidase inhibitory activity. The α-glucosidase from Saccharomyces cerevisiae (EC.3.2.1.20) was used to evaluate the α-glucosidase inhibitory activity. The assay procedure was validated with acarbose as a reference drug, and the result was similar to that reported in the literatures (IC50 = 817.38 ± 6.3 µM)33,34. All synthesized compounds generally showed excellent inhibition values compared to acarbose, ranging from 0.4 µM to 23 µM, as shown in Table 1.
Initially, compound 6a was screened and found to be active as a α-glucosidase inhibitor with an IC50 value of 2.41 µM. Compound 6a possessed racemic chiral carbon; therefore, the importance of chiral configuration was still unknown. To probe the chiral effect of this compound, we prepared four pairs of enantiomers of the compound (6b-c, 7b-c, 8b-c, 9b-c) with different substituents of phenyl urea. Compounds 6c, 7c, 8c, and 9c with S-configuration tended to have better affinity toward α-glucosidase by approximately 3-folds than R-configuration. For instance, compounds 9b and 9c had noteworthy differences with IC50 values of 23.41 and 7.43 µM, respectively. Accordingly, it can be assumed that chirality plays a major role in selectivity.
The substituent of phenyl urea moiety also appeared to affect the inhibition of α-glucosidase. Among compounds 6–9, compounds with halogen groups on phenyl urea exhibit more potent inhibition. Compound 7b bearing -CF3 group at C-4 has an IC50 value of 3.47 µM, slightly better than 6b with -Cl at position C-3 and C-4 that possess an IC50 value of 4.53 µM. Changing the -CF3 to -CH3 group at the same position decreases the activity, although they share some similarities (8b; IC50 = 7.78 µM)35. On top of that, compound 9b with -OCH3 group demolished the activity with an IC50 value of 23.41 µM. These results indicate that a special feature of halogen to form halogen bonding is important for the outstanding inhibition activity toward α-glucosidase36. Additionally, hydrogen bonding acceptor (HBA) properties of the -CF3 group may contributed to forming hydrogen bonding to the enzyme37. The same trend was also observed for those with S-configuration (6c-9c). Altogether, compound 7c was found to be the most potent of all four pairs of enantiomers with an IC50 value of 1.10 µM.
Based on the results above, we prepared 11 additional compounds (10c-20c) to explore the influence of substituents of anilino moiety. S-configuration and -CF3 group on phenyl urea were preserved in this series. The electron-withdrawing group (EWG) effect at different positions can be seen from compounds 7c, 10c, and 11c. We observed that placing -Cl at position C-2 (10c; IC50 = 4.21 µM) or C-3 (11c; IC50 = 3.45 µM) had lower activity compared to position C-4 (7c; IC50 = 1.10 µM). Replacing -Cl with other halogen groups (compounds 12c, 13c, and 14c) is well tolerated as their IC50 values were comparable. The inhibition activity of compound 16c with another -Cl group at the C-3 position had no significant influence, yet introducing the -Cl group at both C-2 and C-4 remarkably reduced the activity (15c; IC50 = 12.0 µM). Compound 17c possessing -NO2 group at position C-3 was on par with 7c with an IC50 value of 1.03 µM. Conversely, the -OH group as an electron-donating group (EDG) in compound 18-20c could not enhance the inhibition activity. Among them, compound 18c, which possesses the -OH group at the C-2 position, appeared as the least active with an IC50 value of 5.13 µM. Shifting -OH group at C-4 enhanced the activity slightly (20c; IC50 = 3.71 µM) and doubled when introduced at the C-3 position (19c; IC50 = 2.05 µM). Compounds 10c, 15c, and 18c demonstrated that any substituent at the C-2 position is disadvantageous. It is possibly caused by conformation alteration between the pyrimidiyl-piperazine and anilino moiety, reducing the ability of 2-anilinopyrimidine to form essential interaction with the enzyme38.
Considering compounds 17c and 19c having polar groups at position C-3 was beneficial for this ring, we introduced the -OH group at C-3 and -Cl at C-4 position to obtain compound 21b-c. The mentioned compound displayed excellent inhibitory activity with IC50 values of 2.08 and 0.44 µM for 21b and 21c, respectively. We further modified the compound 21c by attaching the benzyl group to the -OH, but it turned out that the compound 22c activity decreased with an IC50 value of 1.50 µM. The results suggested that the hydrogen bonding acceptor/donor (HBA/HBD) properties of the hydroxyl group were critical and that bulky substituents were unfavorable.
Kinetic study
To study the mechanism of pyrimidinyl-piperazine carboxamide derivatives in the inhibition of α-glucosidase, an enzymatic kinetic study of compound 21c was carried out. The mode of inhibition was determined by Lineweaver–Burk plots, and then the Ki was calculated with the secondary plot of the Lineweaver–Burk plots as depicted in Fig. 3. The analysis of the obtained Lineweaver–Burk plots showed that with increasing concentrations of Km increased, while the Vmax value remained unchanged. It suggested that compound 21c is a competitive inhibitor of the α-glucosidase. Moreover, out of the secondary plot, the inhibitor constant (Ki) of 21c was determined to be 0.74 µM. Kinetic assay of some other selected compounds also showed the same trend, indicating all S and R configurations had competitive inhibition as a mode of action (see Supplementary Information Fig. S76).

Kinetic study of compound 21c. (A) Lineweaver–Burk plots for the inhibition. (B) The secondary plot between Km and various concentrations.
Computational studies
Molecular docking was performed to predict the plausible binding mode of selected compounds within the active site of α-glucosidase using a Molecular Operating Environment (MOE). Since the crystal structure of α-glucosidase from Saccharomyces cerevisiae used in the experiment has not been reported, we utilized the predicted structure of the enzyme (UniProt: P53341) obtained from the EBI Alpha Fold database (https://alphafold.ebi.ac.uk) for this study39,40. As depicted in Fig. 4, the binding modes of compounds 7c, 9c and 21c share notable similarities. The analysis revealed that these compounds commonly interact with the enzyme through: (a) hydrogen bonding between the pyrimidine heteroatom and the R312 residue; (b) interactions of the piperazine and anilino moieties attached to the pyrimidine ring with F157 via π-alkyl and π-π interactions; and (c) hydrophobic interactions between the phenyl urea group and the A278 and F300 residues.

Binding interactions of compounds (A) 7c, (B) 9c, (C) 21c, and (D) 21b in the active site of α-glucosidase. Key interactions such as hydrophobic, hydrogen, and halogen bonds, as well as π–π stacking and π–cation interactions, are shown in the diagrams. (E) Superimposition of binding modes of compounds 21b and 21c with different configurations at the chiral center.
The comparison of binding modes of compounds 7c and 9c illustrates the importance of the -CF3 group (Fig. 4A and B). It formed multiple halogen bonds with amino acid residues in the active site, E276 and A278. In contrast, the -OCH3 group of 9c had no interaction with any amino acid residues. Halogen bonding is predominantly an electrostatic interaction between the electron-deficient areas of the σ-hole on the halogen and the Lewis base interaction partner. Introducing halogen bonds could improve ligand affinities without disrupting other structurally important interactions36,41. The effect is reflected in their docking S values, which were − 8.32 and − 7.51, respectively, consistent with their respective IC50 values. On the other hand, the binding modes of compound 7c and 21c were similar. Both interated with hydrophobic residues (F157, V277, A278, F300) and charged residues (H239, E276, E304, R312, D349). However, the presence of a hydroxyl group in anilino moiety of 21c resulted in a lower docking S value of -8.41 due to the additional hydrogen bond formation with R312 residue in the binding pocket (Fig. 4C).
To understand better the chirality effect toward α-glucosidase inhibition, compound 21b with R-configuration, i.e., enantiomer pair of 21c, was synthesized. The activity of this compound is indeed lower than 21c with an IC50 value of 2.08 µM and docking S value of -8.28, consistent with other enantiomers. Compared to its pair, 21b binding interactions were notably different. It was predominantly hydrophobic interactions with F157, F158, F177, and F300 (Fig. 4D). It have fewer halogen bonding and essential binding interations with R312 were absent. These could reflect the lower inhibitory effect of R-configuration on α-glucosidase. Superimposition of both compound binding modes revealed that chirality affected their conformation in the active site of α-glucosidase as they showed opposite directions (Fig. 4E). Because assessing the dynamic binding information solely based on static molecular docking is unfeasible, molecular dynamics (MD) simulation was carried out for this enantiomer. Stability, number of contacts, and number of hydrogen bonds (h-bonds) of complex systems upon the simulation time were evaluated. As illustrated in Fig. 5A, the backbone of compound 21b/α-glucosidase complex showed some fluctuation of RMSD values in the first 100 ns and then showed minimal movement in the range of 1.5–2.0 Å before the system was considered to reach equilibrium at 300 ns. Meanwhile, the 21c/α-glucosidase complex reached equilibrium after 78 ns with average movement in the range of 2.0 − 2.2 Å. Ligand fit backbone RMSD was generated to investigate how much the ligand position has varied relative to the protein42. It revealed that 21b had higher fluctuation than 21c with RMSD values of 7.1 Å and 3.6 Å, respectively. Therefore, the 21b binding pose was less preserved during the simulation, indicating the binding interactions were weaker than 21c. Apart from that, the average number of contacts between 21c and amino acid residues within the active site was 307 ± 12 contacts, higher than compound 21b (292 ± 10), despite both compounds consisting of 57 atoms (Fig. 5B). As a result, the probability of ligand-protein interaction formation of 21c was higher during the simulation. In Fig. 5C, only a few h-bonds were detected (1–2 h-bonds), suggesting that the h-bonds type of interaction was not dominantly responsible for the superiority of compound 21c. Highest occupancy of 21b were h-bonds with T307 (37%), and E304 (20%), while from compound 21c h-bonds with D408 (53%), and F157 (12%) were observed.

(A) RMSDs of the backbone and ligand (Lig fit Protein), (B) number of contacts, and (C) number of hydrogen bonds of 21b and 21c in complex with α-glucosidase obtained from 500-ns MD simulation.
A per-residue decomposition analysis was performed to obtain the energetic contribution of the amino acids involved during simulations. Binding free energy decomposition was performed using the gmx_mmpbsa tool43,44. Figure 6A displayed amino acid residues with negative energy per frame from 0 to 500 ns. In the beginning, the 21b diagram displayed energy contribution of -0.5 kcal/mol from L218, H239, H245, and A278, and as time increased, the energy turned to nearly 0 kcal/mol within 220 ns. This explained the high fluctuation of compound 21b in Fig. 5. The losses were compensated by forming stronger interactions with E304, T307 and F300. After 200 ns, energy contribution remained constant. The highest contributed amino acid residues in 21b binding were R312, F177, F300 and T307, with averaged energy − 3.54, -1.66, -1.52, and − 1.46 kcal/mol, respectively. In comparison, 21c contributed amino acid residue energies were relatively constant, meaning the binding interaction was maintained during the whole simulation, except for F300. The R312, F157, T215, and H239 were the top contributed amino acid residues for 21c with energy − 5.1, -2.31, -1.61, and − 1.59 kcal/mol, respectively. Accordingly, these amino acid residues were essential in α-glucosidase inhibition, which agreed well with the previous reports9,39,40,45,46,47. The findings were supported by binding free energy calculation using the MM-GBSA method of 100 snapshots extracted from the last 200 ns, where compound 21c showed better affinity than 21b toward the enzyme with ΔGbind of -27.13 ± 3.2 kcal/mol and − 15.61 ± 3.2 kcal/mol, respectively (Fig. 6B).

(A) Per-residue energy decomposition of compounds 21b and 21c in complex with α-glucosidase during a 500-ns MD simulation, showing interaction energy contributions from key residues. (B) MM-GBSA binding free energy analysis of compounds 21b and 21c.
Cytotoxicity study
Among the pyrimidinyl-piperazine carboxamide derivatives, the most potent compounds 7c, 17c, 21c, and 22c were selected for further evaluation of their cytotoxicity against normal human dermal fibroblast (HDF) cells. Cytotoxicity was assessed using the MTT assay, and the results were expressed as CC50 values (Fig. 7). All four compounds exhibited relatively high CC50 values toward normal cells compared to their IC50 values against the target α-glucosidase, suggesting a favorable therapeutic margin. Specifically, the CC50 values for compounds 7c, 17c, 21c, and 22c were 42.33, 15.15, 23.1, and > 200 µM, respectively, while the IC50 values were much lower, ranging from 0.4 to 1.5 µM. The observed values imply that the active compounds possess potent enzyme inhibitory activity with minimal cytotoxic effects on normal cells. Notably, compound 22c, which contains an additional phenyl ring substitution, demonstrated a significantly higher CC50 value relative to the others, suggesting that this structural feature may contribute to reduced cytotoxicity and improved safety profiles. These findings provide valuable insight for further optimization of the pyrimidinyl-piperazine carboxamide scaffold.

Cytotoxic concentration (CC50) of compounds 7c, 17c, 21c and 22c.
In silico ADMET analysis
The compounds 7c, 17c, 21c, and 22c were selected for further ADMET evaluation based on their top IC₅₀ values against α-glucosidase and low cytotoxicity in normal cell lines. These compounds demonstrated strong inhibitory activity while maintaining favorable safety profiles in preliminary assays. ADMET profiling (see Table S1 in the SI) revealed high human intestinal absorption (85.7–93.1%) and moderate Caco-2 permeability (log Papp between 0.474 and 0.866), supporting their potential for oral administration. All compounds showed low blood-brain barrier permeability (logBB < -1), suggesting minimal CNS-related side effects, and low to moderate volume of distribution (log VDss between − 0.349 and − 0.016). Metabolic analysis indicated that all compounds are substrates and inhibitors of CYP3A4, and inhibitors of CYP2C9, with only 7c identified as a CYP2D6 substrate. None inhibited CYP2D6. Given their interaction with major CYP enzymes, particular attention should be paid to potential drug–drug interactions (DDIs) during further development48. Excretion data showed moderate clearance rates (log mL/min/kg from − 0.312 to -0.021). Toxicity predictions identified 17c as AMES toxic, while all compounds showed hepatotoxic potential but no hERG inhibition, suggesting low cardiotoxicity risk. Overall, the selected α-glucosidase inhibitors exhibit favorable ADMET characteristics, with 21c and 22c showing slightly more favorable safety margins.
Conclusion
In summary, a new series of pyrimidinyl-piperazine carboxamide (6–22) have been synthesized and evaluated for their inhibition activity toward α-glucosidase. Most of the target compounds showed significant inhibitory potency. Compound 21c with S-configuration at the chiral center was the most potent compound with an IC50 value of 0.44 µM. In addition, this compound has no cytotoxic effect at given IC50 values. A kinetic study revealed that competitive inhibition is the mode of inhibition. Outstanding activity contributed by several essential interactions includes hydrogen bonding, π-stacking, and hydrophobic interactions inside the active site. Molecular dynamic revealed that a compound with S-configuration at the chiral center gives better stability and lower binding free energy toward α-glucosidase. Considering the inhibition potential and safety uses, compounds 21c and 22c should be interesting to develop further as antidiabetic agents.
Methods
Chemistry
All the reagents were purchased from TCI Chemical and used without further purification. Thin-layer chromatography (TLC) was performed using Merck silica gel 60 (GF254) plates and column chromatography (CC) using SiliaFlash® 60–200 μm, 60 Å silica gels1. H and13C NMR spectra were recorded by a JEOL JNM-ECZ500R/S1 spectrometer (500 MHz for1H NMR, 125 MHz for13C NMR) in Chloroform-d, Acetone-d6, and DMSO-d6. All the chemical shifts were reported as parts per million (ppm, δ) values. The MS spectra were recorded using a micrOTOF-Q II. Optical rotation measurements were carried out on Jasco P-1010.
General procedure for synthesizing Boc-protected piperazine
2-methylpiperazine 1 (2 g, 10 mmol) in dichloromethane (20 mL) was added to di-tert-butyl dicarbonate (2.3 mL, 10 mmol) dropwise while stirred in an ice bath. After 5 h, the solvent was removed, and then the residue was washed with cold water to obtain 1-Boc-3-methylpiperazine 2a, which was directly used in the next step without further purification.
General procedure for synthesizing compound 4
A mixture of racemic (2a) or chiral (2b-c) 1-Boc-3-methylpiperazine (2 g, 10 mmol), 2,4-dichloropyrimidine 3 (1.49 g, 10 mmol) and triethylamine (30 mmol) in isopropanol (15 mL) was refluxed for 12 h. The reaction was monitored using n-hexane/ethyl acetate in a proportion of 8:2. The solvent was removed upon completion, and then the residue was purified by column chromatography on silica gel (n-hexane/ethyl acetate in gradient) to obtain compound 4a-c.
tert-butyl 4-(2-chloropyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4a): White solid; yield: 65%;1H NMR (500 MHz, Chloroform-d) δ 8.02 (d, J = 6.2 Hz, 1 H), 6.35 (d, J = 6.2 Hz, 1 H), 4.42 (s, 1 H), 4.21–3.77 (m, 3 H), 3.19 (ddd, J = 13.4, 11.7, 3.9 Hz, 1 H), 3.12 (d, J = 13.4 Hz, 1 H), 3.06–2.80 (m, 1 H), 1.45 (s, 9 H), 1.18 (d, J = 6.7 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 162.5, 160.8, 157.5, 155.0, 101.5, 80.4, 48.1, 47.5, 46.6, 43.6, 42.5, 38.8, 28.4, 14.5.
tert-butyl (R)-4-(2-chloropyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4b): White solid; yield: 77%;1H NMR (500 MHz, Chloroform-d) δ 7.96 (d, J = 6.1 Hz, 1 H), 6.32 (d, J = 6.2 Hz, 1 H), 4.37 (s, 1 H), 4.09–3.77 (m, 3 H), 3.14 (ddd, J = 13.4, 11.7, 3.9 Hz, 1 H), 3.07 (d, J = 13.2 Hz, 1 H), 3.01–2.84 (m, 1 H), 1.40 (s, 9 H), 1.13 (d, J = 6.8 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 162.4, 160.7, 157.5, 155.0, 101.5, 80.3, 48.0, 47.5, 46.6, 43.6, 42.5, 38.7, 28.4, 14.4.
tert-butyl (S)-4-(2-chloropyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4c): White solid; yield: 64%;1H NMR (500 MHz, Chloroform-d) δ 8.02 (d, J = 6.2 Hz, 1 H), 6.34 (d, J = 6.2 Hz, 1 H), 4.41 (s, 1 H), 4.16–3.80 (m, 3 H), 3.19 (ddd, J = 13.4, 11.7, 3.9 Hz, 1 H), 3.12 (d, J = 13.0 Hz, 1 H), 3.03–2.85 (m, 1 H), 1.45 (s, 9 H), 1.18 (d, J = 6.8 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 162.5, 160.8, 157.5, 155.1, 101.5, 80.4, 48.2, 47.5, 46.6, 43.7, 42.5, 38.8, 28.4, 14.5.
General procedure for synthesizing compounds 6–21
Compound 4a-c (313 mg, 1 mmol) and the appropriate substituted aniline (1.1 mmol) were dissolved in isopropanol (3 mL). Trifluoroacetic acid (230 µL, 3 mmol) was added to the mixture and then refluxed for 12 h. The reaction was monitored using n-hexane/ethyl acetate in a proportion of 1:1. Upon completion, the mixture is neutralized by 1 M sodium hydroxide solution. The solvent was removed and then extracted with ethyl acetate as an organic layer. The organic portion was dried using Na2SO4, and then the solvent was removed under in vacuo. The obtained crude product was directly used in the next step without further purification.
To a solution of crude product in dry dichloromethane (3 mL) was added aryl isocyanate (1 mmol), and triethylamine (278 µL, 2 mmol) and then stirred at room temperature under nitrogen atmosfer for 3 h. The reaction was monitored using dicholoromethane/methanol in a proportion of 8:2. Upon completion, the solvent was removed, and then the residue was purified by column chromatography on silica gel (dicholoromethane/methanol in gradient) to obtain product 6a-c, 7b-c, 8b-c, 9b-c, 10-20c, 21b-c.
4-(2-((4-chlorophenyl)amino)pyrimidin-4-yl)-N-(3,4-dichlorophenyl)-3-methylpiperazine-1-carboxamide (6a): Light grey solid; yield: 57%;1H NMR (500 MHz, Acetone-d6) δ 8.81 (s, 1 H), 8.30 (s, 1 H), 7.95 (d, J = 6.1 Hz, 1 H), 7.91 (d, J = 2.5 Hz, 1 H), 7.77 (d, J = 9.0 Hz, 2 H), 7.47 (dd, J = 8.8, 2.5 Hz, 1 H), 7.37 (d, J = 8.8 Hz, 1 H), 7.25 (d, J = 8.9 Hz, 2 H), 6.21 (d, J = 6.2 Hz, 1 H), 4.57 (s, 1 H), 4.17 (d, J = 11.9 Hz, 2 H), 4.06 (td, J = 13.4, 1.4 Hz, 1 H), 3.43–3.31 (m, 2 H), 3.21 (ddd, J = 12.3, 10.9, 3.9 Hz, 1 H), 1.22 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, Acetone-d6) δ 162.1, 159.2, 155.7, 154.9, 140.9, 140.0, 131.5, 130.2, 128.4, 125.4, 124.1, 120.8, 120.4, 119.2, 95.6, 47.9, 47.1, 43.6, 38.6, 14.3; HRMS (m/z): [M + H]+ calcd. for C22H21Cl3N6O, 491.0921; found, 491.0912.
(R)-4-(2-((4-chlorophenyl)amino)pyrimidin-4-yl)-N-(3,4-dichlorophenyl)-3-methylpiperazine-1-carboxamide (6b): Light yellow; yield: 47%; [α]D = -55.5 (c 0.01 in MeOH);1H NMR (500 MHz, Acetone-d6) δ 8.80 (s, 1 H), 8.33 (s, 1 H), 7.94 (d, J = 6.1 Hz, 1 H), 7.89 (d, J = 2.5 Hz, 1 H), 7.76 (d, J = 8.9 Hz, 2 H), 7.47 (dd, J = 8.9, 2.5 Hz, 1 H), 7.36 (d, J = 8.8 Hz, 1 H), 7.24 (d, J = 8.9 Hz, 2 H), 6.20 (d, J = 6.1 Hz, 1 H), 4.56 (s, 1 H), 4.16 (d, J = 14.0 Hz, 2 H), 4.06 (td, J = 13.5, 1.5 Hz, 1 H), 3.45–3.29 (m, 2 H), 3.21 (ddd, J = 12.3, 11.1, 4.3 Hz, 1 H), 1.22 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, Acetone-d6) δ 162.1, 159.8, 156.7, 154.9, 140.9, 140.3, 131.4, 130.2, 128.3, 125.1, 124.0, 120.8, 120.2, 119.2, 95.6, 47.7, 47.2, 43.6, 38.5, 14.2; HRMS (m/z): [M + H]+ calcd. for C22H21Cl3N6O, 491.0921; found, 491.0907.
(S)-4-(2-((4-chlorophenyl)amino)pyrimidin-4-yl)-N-(3,4-dichlorophenyl)-3-methylpiperazine-1-carboxamide (6c): White solid; yield: 54%; [α]D = + 58.7 (c 0.01 in MeOH);1H NMR (500 MHz, Chloroform-d) δ 7.97 (d, J = 6.1 Hz, 1 H), 7.59 (d, J = 2.5 Hz, 1 H), 7.56 (s, 1 H), 7.48 (d, J = 8.9 Hz, 2 H), 7.31 (d, J = 8.8 Hz, 1 H), 7.24 (d, J = 8.9 Hz, 2 H), 7.21 (dd, J = 8.7, 2.5 Hz, 1 H), 6.75 (s, 1 H), 6.00 (d, J = 6.1 Hz, 1 H), 4.42 (s, 1 H), 4.13 (d, J = 13.0 Hz, 1 H), 3.96 (dt, J = 12.3, 3.9 Hz, 1 H), 3.82 (dd, J = 13.8, 3.1 Hz, 1 H), 3.47 (dd, J = 13.4, 4.1 Hz, 1 H), 3.39 (ddd, J = 13.5, 10.5, 3.8 Hz, 1 H), 3.27 ddd, J = 12.0, 10.6, 4.0 Hz, 1 H), 1.26 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 161.9, 159.1, 156.2, 154.9, 138.6, 138.5, 132.6, 130.4, 128.8, 127.0, 126.5, 121.8, 120.7, 119.5, 95.4, 48.2, 47.2, 43.7, 38.5, 15.5; HRMS (m/z): [M + H]+ calcd. for C22H21Cl3N6O, 491.0921; found, 491.0912.
(R)-4-(2-((4-chlorophenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (7b): White solid; yield: 76%; [α]D = -60.3 (c 0.01 in MeOH);1H NMR (500 MHz, Chloroform-d) 7.98 (d, J = 6.1 Hz, 1 H), 7.72 (s, 1 H), 7.52 (d, J = 9.0 Hz, 2 H), 7.49 (d, J = 8.9 Hz, 2 H), 7.48 (d, J = 8.9 Hz, 2 H), 7.23 (d, J = 8.8 Hz, 2 H), 6.88 (s, 1 H), 5.99 (d, J = 6.2 Hz, 1 H), 4.43 (s, 1 H), 4.13 (d, J = 11.6 Hz, 1 H), 3.99 (d, J = 12.1 Hz, 1 H), 3.84 (dd, J = 13.6, 3.4 Hz, 1 H), 3.48 (dd, J = 13.4, 4.2 Hz, 1 H), 3.39 (dd, J = 13.5, 10.5, 3.7 Hz, 1 H), 3.28 (ddd, J = 11.9, 10.5, 3.9 Hz, 1 H), 1.26 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 161.9, 159.3, 156.4, 154.9, 142.1, 138.7, 128.8, 126.9, 126.3 (d, J = 4.1 Hz), 125.0 (d, J = 32.5 Hz), 124.3 (d, J = 270.8 Hz), 120.8, 119.4, 95.4, 48.1, 47.2, 43.7, 38.5, 15.5; HRMS (m/z): [M + H]+ calcd. for C23H22ClF3N6O, 491.1573; found, 491.1568.
(S)-4-(2-((4-chlorophenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (7c): White solid; yield: 71%; [α]D = + 63.6 (c 0.01 in MeOH);1H NMR (500 MHz, Chloroform-d) δ 7.98 (d, J = 6.1 Hz, 1 H), 7.60 (s, 1 H), 7.53 (d, J = 8.9 Hz, 2 H), 7.50 (d, J = 9.0 Hz, 2 H), 7.49 (d, J = 8.8 Hz, 2 H), 7.24 (d, J = 8.8 Hz, 2 H), 6.79 (s, 1 H), 6.01 (d, J = 6.2 Hz, 1 H), 4.44 (s, 1 H), 4.15 (d, J = 12.8 Hz, 1 H), 3.99 (dt, J = 12.2, 3.9 Hz, 1 H), 3.85 (dd, J = 13.3, 3.4 Hz, 1 H), 3.51 (dd, J = 13.4, 4.1 Hz, 1 H), 3.41 (ddd, J = 13.5, 10.4, 3.7 Hz, 1 H), 3.30 (ddd, J = 12.0, 10.5, 3.9 Hz, 1 H), 1.28 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 161.9, 159.2, 156.2, 154.8, 142.1, 138.6, 128.8, 127.0, 126.3 (d, J = 4.1 Hz), 125.1 (d, J = 32.7 Hz), 124.3 (d, J = 270.6 Hz), 120.8, 119.4, 95.4, 48.2, 47.2, 43.7, 38.5, 15.5; HRMS (m/z): [M + H]+ calcd. for C23H22ClF3N6O, 491.1573; found, 491.1564.
(R)-4-(2-((4-chlorophenyl)amino)pyrimidin-4-yl)-3-methyl-N-(p-tolyl)piperazine-1-carboxamide (8b): White solid; yield: 74%; [α]D = -72.9 (c 0.01 in MeOH);1H NMR (500 MHz, Chloroform-d) δ 8.40 (s, 1 H), 7.90 (d, J = 6.2 Hz, 1 H), 7.51 (d, J = 8.8 Hz, 1 H), 7.24 (d, J = 8.4 Hz, 2 H), 7.24 (d, J = 8.9 Hz, 2 H), 7.09 (d, J = 8.1 Hz, 2 H), 6.58 (s, 1 H), 5.98 (d, J = 6.3 Hz, 1 H), 4.41 (s, 1 H), 4.10 (d, J = 13.0 Hz, 1 H), 3.97 (dd, J = 12.3, 4.0 Hz, 1 H), 3.82 (dd, J = 14.3, 2.7 Hz, 1 H), 3.46 (dd, J = 13.4, 4.1 Hz, 1 H), 3.40 (ddd, J = 13.7, 10.5, 3.8 Hz, 1 H), 3.25 (ddd, J = 12.0, 10.4, 4.0 Hz, 1 H), 2.29 (s, 3 H), 1.27 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 161.9, 158.5, 155.7, 154.8, 138.5, 136.1, 133.2, 129.6, 128.8, 127.1, 120.8, 120.6, 95.1, 48.4, 47.2, 43.5, 38.8, 20.9, 15.6; HRMS (m/z): [M + H]+ calcd. for C23H25ClN6O, 437.1857; found, 437.1849.
(S)-4-(2-((4-chlorophenyl)amino)pyrimidin-4-yl)-3-methyl-N-(p-tolyl)piperazine-1-carboxamide (8c): White solid; yield: 73%; [α]D = + 74.1 (c 0.01 in MeOH);1H NMR (500 MHz, Chloroform-d) δ 7.99 (d, J = 6.2 Hz, 1 H), 7.50 (d, J = 8.9 Hz, 2 H), 7.47 (s, 1 H), 7.25 (d, J = 8.6 Hz, 4 H), 7.10 (d, J = 8.3 Hz, 2 H), 6.51 (s, 1 H), 6.00 (d, J = 6.2 Hz, 1 H), 4.42 (s, 1 H), 4.11 (d, J = 13.4 Hz, 1 H), 3.97 (dt, J = 12.8, 4.0 Hz, 1 H), 3.82 (dd, J = 13.4, 3.2 Hz, 1 H), 3.45 (dd, J = 13.3, 4.1 Hz, 1 H), 3.38 (ddd, J = 13.7, 10.6, 3.8 Hz, 1 H), 3.24 (ddd, J = 12.2, 10.5, 3.9 Hz, 1 H), 2.29 (s, 3 H), 1.27 (d, J = 6.7 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 161.9, 159.4, 156.6, 155.6, 138.7, 136.2, 133.2, 129.6, 128.8, 126.8, 120.6, 120.6, 95.4, 48.2, 47.3, 43.6, 38.6, 20.9, 15.5; HRMS (m/z): [M + H]+ calcd. for C23H25ClN6O, 437.1857; found, 437.1851.
(R)-4-(2-((4-chlorophenyl)amino)pyrimidin-4-yl)-N-(4-methoxyphenyl)-3-methylpiperazine-1-carboxamide (9b): White solid; yield: 93%; [α]D = -53.6 (c 0.01 in MeOH);1H NMR (500 MHz, DMSO-d6) δ 9.21 (s, 1 H), 8.37 (s, 1 H), 7.96 (d, J = 6.1 Hz, 1 H), 7.71 (d, J = 9.0 Hz, 2 H), 7.31 (d, J = 9.0 Hz, 2 H), 7.24 (d, J = 8.9 Hz, 2 H), 6.79 (d, J = 9.0 Hz, 2 H), 6.25 (d, J = 6.1 Hz, 1 H), 4.49 (s, 0 H), 4.22–4.00 (m, 2 H), 3.96 (dt, J = 13.2, 2.2 Hz, 1 H), 3.66 (s, 3 H), 3.22–3.10 (m, 2 H), 2.99 (ddd, J = 12.6, 12.2, 3.8 Hz, 1 H), 1.11 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, DMSO-d6) δ 162.1, 159.8, 157.0, 156.1, 155.1, 140.7, 133.8, 128.7, 124.5, 122.3, 120.5, 114.1, 96.2, 55.6, 47.6, 47.5, 43.8, 38.8, 14.9; HRMS (m/z): [M + H]+ calcd. for C23H25ClN6O2, 453.1806; found, 453.1797.
(S)-4-(2-((4-chlorophenyl)amino)pyrimidin-4-yl)-N-(4-methoxyphenyl)-3-methylpiperazine-1-carboxamide (9c): White solid; yield: 88%; [α]D = + 55.0 (c 0.01 in MeOH);1H NMR (500 MHz, DMSO-d6) δ 9.21 (s, 1 H), 8.37 (s, 1 H), 7.96 (d, J = 6.1 Hz, 1 H), 7.72 (d, J = 9.0 Hz, 2 H), 7.32 (d, J = 9.1 Hz, 2 H), 7.24 (d, J = 8.9 Hz, 2 H), 6.79 (d, J = 8.9 Hz, 2 H), 6.25 (d, J = 6.1 Hz, 1 H), 4.49 (s, 1 H), 4.15–4.02 (m, 2 H), 3.96 (dt, J = 13.2, 2.1 Hz, 1 H), 3.66 (s, 2 H), 3.23–3.10 (m, 2 H), 2.99 (ddd, J = 12.4, 11.8, 3.8 Hz, 1 H), 1.11 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, DMSO-d6) δ 162.1, 159.8, 157.0, 156.1, 155.1, 140.7, 133.9, 128.7, 124.5, 122.3, 120.5, 114.1, 96.2, 55.6, 47.6, 47.5, 43.8, 38.8, 14.9; HRMS (m/z): [M + H]+ calcd. for C23H25ClN6O2, 453.1806; found, 453.1800.
(S)-4-(2-((2-chlorophenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (10c): White solid; yield: 72%; [α]D = + 56.6 (c 0.01 in MeOH);1H NMR (500 MHz, Chloroform-d) δ 8.31 (dd, J = 8.3, 1.6 Hz, 1 H), 7.97 (d, J = 6.2 Hz, 1 H), 7.60 (s, 1 H), 7.50 (s, 4 H), 7.35 (dd, J = 8.0, 1.5 Hz, 1 H), 7.21 (td, J = 7.7, 1.4 Hz, 1 H), 7.18 (s, 1 H), 6.92 (td, J = 7.7, 1.5 Hz, 1 H), 6.00 (d, J = 6.1 Hz, 1 H), 4.40 (s, 1 H), 4.10 (d, J = 15.1 Hz, 1 H), 3.98 (dt, J = 12.4, 3.8 Hz, 1 H), 3.84 (dd, J = 13.5, 3.2 Hz, 1 H), 3.43 (dd, J = 13.5, 4.2 Hz, 1 H), 3.36 (ddd, J = 13.6, 10.5, 3.7 Hz, 1 H), 3.24 (ddd, J = 11.4, 10.8, 3.8 Hz, 1 H), 1.23 (d, J = 6.7 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 161.9, 158.9, 156.1, 155.1, 142.3, 136.5, 129.3, 127.2, 126.2 (d, J = 4.3 Hz), 124.9 (d, J = 32.7 Hz), 124.3 (d, J = 271.5 Hz), 123.1, 122.8, 121.1, 119.5, 95.9, 48.1, 47.2, 43.7, 38.5, 15.4; HRMS (m/z): [M + H]+ calcd. for C23H22ClF3N6O, 491.1574; found, 491.1568.
(S)-4-(2-((3-chlorophenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (11c): White solid; yield: 80%; [α]D = + 53.3 (c 0.01 in MeOH);1H NMR (500 MHz, Chloroform-d) δ 8.01 (d, J = 6.0 Hz, 1 H), 7.93 (s, 1 H), 7.86 (s, 1 H), 7.52 (d, J = 9.2 Hz, 2 H), 7.50 (d, J = 9.2 Hz, 2 H), 7.18 (d, J = 5.5 Hz, 1 H), 6.99–6.92 (m, 1 H), 6.89 (s, 1 H), 6.01 (d, J = 6.1 Hz, 1 H), 4.48 (s, 1 H), 4.00 (dt, J = 12.3, 3.9 Hz, 1 H), 3.85 (dd, J = 13.5, 3.3 Hz, 1 H), 3.50 (dd, J = 13.4, 4.1 Hz, 1 H), 3.42 (ddd, J = 13.6, 10.4, 3.8 Hz, 1 H), 3.31 (ddd, J = 12.0, 10.4, 3.9 Hz, 1 H), 1.30 (d, J = 6.7 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 161.8, 159.3, 156.7, 154.9, 142.1, 141.4, 134.4, 129.8, 126.3 (d, J = 3.6 Hz), 125.0 (d, J = 32.6 Hz), 124.3 (d, J = 271.6 Hz), 121.8, 119.5, 119.3, 117.2, 95.5, 48.2, 47.3, 43.6, 38.7, 15.6; HRMS (m/z): [M + H]+ calcd. for C23H22ClF3N6O, 491.1574; found, 491.1566.
(S)-4-(2-((4-fluorophenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (12c): Grey solid; yield: 62%; [α]D = + 64.1 (c 0.01 in MeOH);1H NMR (500 MHz, Chloroform-d) δ 7.98 (d, J = 6.1 Hz, 1 H), 7.53 (d, J = 8.9 Hz, 2 H), 7.50 (d, J = 8.9 Hz, 2 H), 7.47 (dd, J = 9.0, 4.9 Hz, 2 H) 7.36 (s, 1 H), 6.99 (t, J = 8.7 Hz, 2 H), 6.75 (s, 1 H), 5.99 (d, J = 6.1 Hz, 1 H), 4.44 (s, 1 H), 4.15 (d, J = 11.4 Hz, 1 H), 3.99 (dt, J = 12.2, 4.1 Hz, 1 H), 3.84 (dd, J = 13.4, 3.4 Hz, 1 H), 3.50 (dd, J = 13.4, 4.1 Hz, 1 H), 3.40 (ddd, J = 13.6, 10.6, 3.7 Hz, 1 H), 3.29 (ddd, J = 11.9, 10.6, 3.9 Hz, 1 H), 1.27 (d, J = 6.7 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 161.9, 159.6, 158.4 (d, J = 241.2 Hz), 156.5, 154.8, 142.1, 135.9 (d, J = 2.4 Hz), 126.3 (d, J = 3.9 Hz), 125.1 (d, J = 33.4 Hz), 124.3 (d, J = 271.4 Hz), 121.4 (d, J = 7.9 Hz), 119.3, 115.4 (d, J = 22.7 Hz), 95.2, 48.1, 47.3, 43.7, 38.4, 15.5; HRMS (m/z): [M + H]+ calcd. for C23H22F4N6O, 475.1869; found, 475.1860.
(S)-4-(2-((4-bromophenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (13c): Light brown solid; yield: 76%; [α]D = + 62.2 (c 0.01 M in MeOH);1H NMR (500 MHz, Chloroform-d) δ 8.50 (s, 1 H), 7.88 (d, J = 6.3 Hz, 1 H), 7.50 (s, 4 H), 7.44 (d, J = 8.5 Hz, 2 H), 7.36 (d, J = 8.6 Hz, 2 H), 7.12 (s, 1 H), 5.97 (d, J = 6.3 Hz, 1 H), 4.41 (s, 1 H), 4.11 (d, J = 13.7 Hz, 1 H), 3.99 (d, J = 11.9 Hz, 1 H), 3.85 (dd, J = 13.4, 3.6 Hz, 1 H), 3.48 (dd, J = 13.5, 4.1 Hz, 1 H), 3.39 (d, J = 14.1, 10.8, 3.4 Hz, 1 H), 3.28 (ddd, J = 11.6, 11.5, 3.8 Hz, 1 H), 1.25 (d, J = 6.7 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 161.8, 158.3, 155.0, 154.5, 142.2, 138.9, 131.7, 126.2 (d, J = 3.9 Hz), 125.0 (d, J = 32.8 Hz), 124.3 (d, J = 271.3 Hz), 121.3, 119.5, 114.7, 95.2, 48.4, 47.1, 43.6, 38.7, 15.6; HRMS (m/z): [M + H]+ calcd. for C23H22BrF3N6O, 535.1069; found, 535.1054.
(S)-3-methyl-N-(4-(trifluoromethyl)phenyl)-4-(2-((4-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)piperazine-1-carboxamide (14c): White solid; yield: 91%; [α]D = + 56.9 (c 0.01 in MeOH);1H NMR (500 MHz, Chloroform-d) 8.03 (d, J = 6.1 Hz, 1 H), 7.68 (d, J = 8.5 Hz, 2 H), 7.61 (s, 1 H), 7.54 (d, J = 8.7 Hz, 5 H), 7.50 (d, J = 8.9 Hz, 2 H), 6.71 (s, 1 H), 6.07 (d, J = 6.1 Hz, 1 H), 4.49 (s, 1 H), 4.18 (d, J = 11.9 Hz, 1 H), 4.02 (dt, J = 12.1, 3.1 Hz, 1 H), 3.87 (dd, J = 13.3, 3.4 Hz, 1 H), 3.54 (dd, J = 13.4, 4.1 Hz, 1 H), 3.46 (ddd, J = 13.6, 10.6, 3.8 Hz, 1 H), 3.34 (ddd, J = 12.2, 10.6, 4.0 Hz, 1 H), 1.31 (d, J = 6.7 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 161.9, 159.0, 156.4, 154.8, 143.1, 142.0, 126.3 (d, J = 4.0 Hz), 126.1 (d, J = 4.7 Hz), 125.1 (d, J = 33.0 Hz), 124.5 (d, J = 271.7 Hz), 124.3 (d, J = 272.8 Hz), 123.6 (d, J = 32.6 Hz) 119.3, 118.5, 96.0, 48.3, 47.3, 43.7, 38.6, 15.6; HRMS (m/z): [M + H]+ calcd. for C24H22F6N6O, 525.1837; found, 525.1826.
(S)-4-(2-((2,4-dichlorophenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (15c): White solid; yield: 43%; [α]D = + 55.1 (c 0.01 in MeOH);1H NMR (500 MHz, Chloroform-d) δ 8.39 (d, J = 8.9 Hz, 1 H), 8.03 (d, J = 6.1 Hz, 1 H), 7.53 (d, J = 9.0 Hz, 2 H), 7.50 (d, J = 8.8 Hz, 2 H), 7.36 (d, J = 2.4 Hz, 1 H), 7.31 (s, 1 H), 7.20 (dd, J = 8.9, 2.4 Hz, 1 H), 6.79 (s, 1 H), 6.06 (d, J = 6.1 Hz, 1 H), 4.44 (s, 1 H), 4.16 (d, J = 13.4 Hz, 1 H), 4.00 (dt, J = 12.1, 4.1 Hz, 1 H), 3.85 (dd, J = 13.2, 3.4 Hz, 1 H), 3.51 (dd, J = 13.3, 4.1 Hz, 1 H), 3.41 (ddd, J = 13.5, 10.6, 3.7 Hz, 1 H), 3.31 (ddd, J = 12.1, 10.6, 3.8 Hz, 1 H), 1.28 (d, J = 6.7 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 161.9, 159.0, 156.7, 154.8, 142.1, 135.4, 128.8, 127.3, 126.5, 126.3 (d, J = 3.9 Hz), 125.1 (d, J = 32.6 Hz), 124.3 (d, J = 271.4 Hz), 123.0, 121.2, 119.4, 96.3, 48.1, 47.2, 43.7, 38.5, 15.5; HRMS (m/z): [M + H]+ calcd. for C23H21Cl2F3N6O, 525.1184; found, 525.1172.
(S)-4-(2-((3,4-dichlorophenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (16c): White solid; yield: 75%; [α]D = + 53.6 (c 0.01 in MeOH);1H NMR (500 MHz, Acetone-d6) δ 8.58 (s, 1 H), 8.40 (s, 1 H), 8.29 (d, J = 2.5 Hz, 1 H), 8.00 (d, J = 6.1 Hz, 1 H), 7.74 (d, J = 8.5 Hz, 2 H), 7.61 (dd, J = 8.8, 2.6 Hz, 1 H), 7.54 (d, J = 8.6 Hz, 2 H), 7.39 (d, J = 8.8 Hz, 1 H), 6.26 (d, J = 6.1 Hz, 1 H), 4.60 (s, 1 H), 4.23–4.13 (m, 2 H), 4.09 (dt, J = 13.4, 2.0 Hz, 1 H), 3.41–3.32 (m, 2 H), 3.24 (ddd, J = 12.0, 11.5, 3.5 Hz, 1 H), 1.25 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, Acetone-d6) δ 162.1, 159.5, 156.7, 155.0, 144.4, 141.5, 131.5, 130.2, 125.7 (d, J = 3.7 Hz), 124.9 (d, J = 270.4 Hz), 123.0 (d, J = 31.6 Hz), 122.6, 119.8, 119.0, 118.5, 96.2, 47.7, 47.2, 43.6, 38.6, 14.3; HRMS (m/z): [M + H]+ calcd. for C23H21Cl2F3N6O, 525.1184; found, 525.1170.
(S)-3-methyl-4-(2-((3-nitrophenyl)amino)pyrimidin-4-yl)-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (17c): Yellow solid; yield: 86%; [α]D = + 75.8 (c 0.01 in MeOH);1H NMR (500 MHz, Acetone-d6) δ 9.13 (t, J = 2.3 Hz, 1 H), 8.92 (s, 1 H), 8.53 (s, 1 H), 8.01 (d, J = 6.1 Hz, 1 H), 7.86 (dd, J = 8.2, 2.1 Hz, 1 H), 7.74 (d, J = 8.7 Hz, 2 H), 7.73 (dd, J = 8.3, 2.5 Hz, 1 H), 7.53 (d, J = 8.6 Hz, 2 H), 7.47 (t, J = 8.1 Hz, 1 H), 6.27 (d, J = 6.1 Hz, 1 H), 4.66 (s, 1 H), 4.33–4.17 (m, 2 H), 4.12 (dt, J = 13.6, 2.2 Hz, 1 H), 3.50–3.35 (m, 2 H), 3.25 (ddd, J = 13.1, 11.0, 3.9 Hz, 1 H), 1.25 (d, J = 6.7 Hz, 3 H);13C NMR (125 MHz, Acetone-d6) δ 162.1, 159.5, 156.8, 155.2, 148.7, 144.4, 142.6, 129.4, 125.7 (q, J = 4.1 Hz), 124.9 (q, J = 270.4 Hz), 124.3, 123.0 (q, J = 32.2 Hz), 119.2, 115.2, 112.6, 96.2, 47.9, 47.3, 43.6, 38.8, 14.2; HRMS (m/z): [M + H]+ calcd. for C23H22F3N7O3, 502.1814; found, 502.1801.
(S)-4-(2-((2-hydroxyphenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (18c): Orange solid; yield: 39%; [α]D = + 74.4 (c 0.01 in MeOH);1H NMR (500 MHz, Acetone-d6) δ 8.39 (s, 1 H), 8.06 (s, 1 H), 7.93 (d, J = 6.3 Hz, 1 H), 7.75 (d, J = 8.5 Hz, 2 H), 7.55 (d, J = 8.7 Hz, 2 H), 7.50 (d, J = 7.9 Hz, 1 H), 6.93–6.85 (m, 2 H), 6.76 (ddd, J = 8.6, 6.8, 1.8 Hz, 1 H), 6.23 (d, J = 6.3 Hz, 1 H), 4.59 (s, 1 H), 4.28–4.14 (m, 2 H), 4.08 (dt, J = 13.7, 2.3 Hz, 1 H), 3.40–3.32 (m, 2 H), 3.22 (ddd, J = 12.6, 11.0, 3.7 Hz, 1 H), 1.23 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, Acetone-d6) δ 162.3, 159.9, 155.0, 154.7, 148.5, 144.4, 129.1, 125.7 (d, J = 3.6 Hz), 124.9 (d, J = 270.4 Hz), 123.9, 123.0 (d, J = 32.6 Hz), 121.6, 119.6, 119.0, 118.9, 118.1, 95.3, 47.8, 47.2, 43.6, 38.4, 14.4; HRMS (m/z): [M + H]+ calcd. for C23H23F3N6O2, 473.1913; found, 473.1898.
(S)-4-(2-((3-hydroxyphenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (19c): Light brown solid; yield: 40%; [α]D = + 61.1 (c 0.01 in MeOH);1H NMR (500 MHz, Acetone-d6) δ 8.38 (s, 1 H), 8.22 (s, 1 H), 7.95 (d, J = 6.1 Hz, 1 H), 7.75 (d, J = 8.5 Hz, 2 H), 7.54 (d, J = 8.5 Hz, 2 H), 7.45 (t, J = 2.2 Hz, 1 H), 7.15 (dt, J = 8.6, 1.1 Hz, 1 H), 7.04 (t, J = 8.1 Hz, 1 H), 6.41 (ddd, J = 8.0, 2.5, 0.9 Hz, 1 H), 6.18 (d, J = 6.1 Hz, 1 H), 4.58 (s, 1 H), 4.25–4.14 (m, 2 H), 4.08 (dt, J = 13.3, 2.2 Hz, 1 H), 3.39–3.29 (m, 2 H), 3.21 (ddd, J = 12.6, 11.1, 4.0 Hz, 1 H), 1.22 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, Acetone-d6) δ 162.1, 159.8, 157.8, 156.4, 154.9, 144.4, 142.4, 129.2, 125.7 (d, J = 4.8 Hz), 124.9 (d, J = 270.3 Hz), 122.9 (d, J = 32.5 Hz), 119.0, 110.3, 108.3, 106.0, 95.2, 47.7, 47.3, 43.7, 38.5, 14.1; HRMS (m/z): [M + H]+ calcd. for C23H23F3N6O2, 473.1913; found, 473.1903.
(S)-4-(2-((4-hydroxyphenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (20c): Brown solid; yield: 42%; [α]D = + 67.7 (c 0.01 in MeOH);1H NMR (500 MHz, Acetone-d6) δ 8.44 (s, 1 H), 8.15 (s, 1 H), 7.90 (d, J = 6.1 Hz, 1 H), 7.74 (d, J = 8.5 Hz, 2 H), 7.54 (d, J = 8.5 Hz, 2 H), 7.50 (d, J = 8.8 Hz, 2 H), 6.75 (d, J = 8.8 Hz, 2 H), 6.10 (d, J = 6.1 Hz, 1 H), 4.54 (s, 1 H), 4.20–4.12 (m, 2 H), 4.06 (dt, J = 13.5, 2.2 Hz, 1 H), 3.36–3.25 (m, 2 H), 3.18 (ddd, J = 15.6, 11.5, 3.9 Hz, 1 H), 1.19 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, Acetone-d6) δ 162.2, 160.2, 156.6, 155.1, 152.4, 144.4, 133.2, 125.7 (q, J = 4.2 Hz), 124.9 (d, J = 270.4 Hz), 122.9 (q, J = 32.5 Hz), 121.2, 119.1, 115.0, 94.5, 47.6, 47.3, 43.7, 38.4, 14.1; HRMS (m/z): [M + H]+ calcd. for C23H23F3N6O2, 473.1913; found, 473.1900.
(R)-4-(2-((4-chloro-3-hydroxyphenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (21b): White solid; yield: 73%; [α]D = -55.7 (c 0.01 in MeOH);1H NMR (500 MHz, DMSO-d6) δ 9.90 (s, 1 H), 9.08 (s, 1 H), 8.94 (s, 1 H), 7.95 (d, J = 6.0 Hz, 1 H), 7.69 (d, J = 8.6 Hz, 2 H), 7.56 (d, J = 8.5 Hz, 2 H), 7.48 (t, J = 1.4 Hz, 1 H), 7.10 (d, J = 1.3 Hz, 2 H), 6.23 (d, J = 6.2 Hz, 1 H), 4.51 (s, 1 H), 4.15–4.06 (m, 2 H), 4.01 (dt, J = 13.4, 2.2 Hz, 1 H), 3.26–3.11 (m, 2 H), 3.06 (ddd, J = 12.6, 12.2, 3.8 Hz, 1 H), 1.10 (d, J = 6.6 Hz, 3 H)13. C NMR (125 MHz, DMSO-d6) δ 162.1, 159.9, 157.1, 155.4, 153.2, 144.9, 141.5, 129.6, 126.2, 123.0 (d, J = 285.7 Hz), 122.2 (d, J = 31.7 Hz), 119.5, 111.7, 111.3, 107.3, 96.0, 47.8, 47.4, 44.0, 38.7, 14.8. HRMS (m/z): [M + H]+ calcd. for C23H22ClF3N6O2, 507.1523; found, 507.1512.
(S)-4-(2-((4-chloro-3-hydroxyphenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (21c): White solid; yield: 64%; [α]D = + 53.4 (c 0.01 in MeOH);1H NMR (500 MHz, DMSO-d6) δ 9.89 (s, 1 H), 9.07 (s, 1 H), 8.93 (s, 1 H), 7.95 (d, J = 6.0 Hz, 1 H), 7.68 (d, J = 8.6 Hz, 2 H), 7.56 (d, J = 8.9 Hz, 2 H), 7.47 (s, 1 H), 7.09 (s, 2 H), 6.24 (d, J = 6.3 Hz, 1 H), 4.51 (s, 1 H), 4.17–4.07 (m, 2 H), 4.00 (d, J = 14.3 Hz, 1 H), 3.22 (dd, J = 13.5, 4.0 Hz, 1 H), 3.19–3.13 (m, 1 H), 3.06 (td, J = 12.6, 3.7 Hz, 1 H), 1.10 (d, J = 6.6 Hz, 3 H)13. C NMR (125 MHz, DMSO-d6) δ 161.6, 159.4, 156.6, 155.0, 152.7, 144.4, 141.0, 129.2, 125.7, 122.5, (d, J = 285.7 Hz), 121.7 (d, J = 31.7 Hz), 119.1, 111.3, 110.8, 106.9, 95.5, 47.3, 46.9, 43.5, 39.5, 38.2, 14.4. HRMS (m/z): [M + H]+ calcd. for C23H22ClF3N6O2, 507.1523; found, 507.1516.
General procedure for synthesizing compound 22
Compound 21c (101 mg, 0.2 mmol), benzyl bromide (24 µL, 0.2 mmol), and potassium carbonate (55 mg, 0.4 mmol) in acetone (5 mL) was heated at room temperature for 3 h. Upon the completion of the reaction, the mixture was diluted with water and then extracted using an ethyl acetate. Organic portion was dried using Na2SO4 and then solvent was removed under in vacuo. The residue was purified by column chromatography on silica gel (n-hexane/ethyl acetate, 1:1) to obtain compound 22c.
(S)-4-(2-((3-(benzyloxy)-4-chlorophenyl)amino)pyrimidin-4-yl)-3-methyl-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (22c): White solid; yield: 95%; [α]D = + 52.1 (c 0.01 in MeOH);1H NMR (500 MHz, Chloroform-d) δ 7.96 (d, J = 6.1 Hz, 1 H), 7.86 (s, 1 H), 7.50 (d, J = 9.0 Hz, 2 H), 7.46 (d, J = 8.4 Hz, 3 H), 7.45 (d, J = 8.8 Hz, 1 H), 7.40 (d, J = 2.4 Hz, 1 H), 7.36 (t, J = 7.4 Hz, 2 H), 7.34–7.28 (m, 1 H), 7.25 (d, J = 8.6 Hz, 1 H), 7.07 (dd, J = 8.7, 2.4 Hz, 1 H), 6.98 (s, 1 H), 5.97 (d, J = 6.1 Hz, 1 H), 5.09 (s, 2 H), 4.39 (s, 1 H), 3.91 (dt, J = 12.4, 4.0 Hz, 1 H), 3.79 (dd, J = 13.5, 3.5 Hz, 1 H), 3.40 (dd, J = 13.4, 4.1 Hz, 1 H), 3.33 (ddd, J = 13.7, 10.6, 3.8 Hz, 1 H), 3.19 (ddd, J = 14.6, 11.3, 4.0 Hz, 1 H)), 1.22 (d, J = 6.6 Hz, 3 H);13C NMR (125 MHz, Chloroform-d) δ 161.9, 159.3, 156.4, 155.0, 154.3, 142.2, 140.0, 136.5, 130.0, 128.8, 128.2, 127.3, 126.2 (d, J = 3.8 Hz), 125.0 (d, J = 32.6 Hz), 124.3 (d, J = 271.3 Hz), 119.5, 115.9, 112.7, 106.0, 95.4, 71.0, 48.1, 47.2, 43.6, 38.4, 15.4; HRMS (m/z): [M + H]+ calcd. for C30H29F3N6O2, 597.1992; found, 597.1983.
α -Glucosidase Inhibition assay
α-Glucosidase from Saccharomyces cerevisiae (EC.3.2.1.20) and p-nitrophenyl-α-D-glucopyranoside (p-NPG) from Sigma-Aldrich were used in this assay. The assay was carried out using the protocol as reported by Supasuteekul et al. with minor modification49. Briefly, α-glucosidase (0.1 U/mL) was prepared in phosphate buffer (0.1 M, pH 6.9) and the samples dissolved in DMSO (2% final concentration). A mixture of 10 µL of samples in various concentrations and 40 µL of enzyme in a 96-well plate was pre-incubated at 37 °C for 10 min. Then, 50 µL of p-NPG (3 mM) was added and further incubated at 37 °C for 20 min. The reaction was stopped by adding 100 µL of 1 M Na2CO3. The amount of p-nitrophenol released was measured using a microplate reader (Allsheng AMR-100) to determine the absorbance at 405 nm.
% Inhibition = (1 - As /Ac) × 100.
As is the absorbance value of the sample, Ac is the absorbance value of the negative control. Each experiment was performed in triplicate. The IC50 value was measured using nonlinear regression analysis of the dose-response curves.
Kinetic study
Compounds 21c were subjected to kinetic experiments to determine the mode of inhibition on α-glucosidase. The experiments were conducted in the presence and absence of the inhibitor at different concentrations of p-NPG and the enzyme concentration was maintained at 0.02 U/mL. A Lineweaver–Burk plot was generated to identify the type of inhibition. The inhibitory constant Ki was determined from the slope versus inhibitor concentrations.
Molecular docking
Molecular docking studies were carried out using Molecular Operating Environment (MOE) software. Yeast α-glucosidase structure prediction, entry P55341, from EBI AlphaFold protein structure database (https://alphafold.ebi.ac.uk/entry/P53341) was used for this study39,40. The protein structure was prepared using the QuickPrep option. Ligands were prepared using a MOE molecule builder, and geometry was optimized. Ligands were docked in the active site pocket using the AMBER10:EHT forcefield. Docking poses were scored by initial rescoring methodology (London dG) and the final rescoring methodology (GBVI/WSA) by placement using Triangle Matcher protocol and Rigid Receptor as post-placement refinement. The binding interactions were visualized by the Protein Ligand Interaction Profiler (PLIP) web tool and PoseView50,51.
Molecular dynamics (MD) simulation
MD simulations of selected compound complexes were performed using GROMACS version 202452. Topology parameters files of each ligand were generated by AnteChamber Python Parser interfacE (ACPYPE) server53,54. The AMBER99-ILDN force field is employed in MD simulations for each tested compound. The system was solvated using a TIP3P water model and neutralized with Na+ and Cl− ions. The Particle Mesh Ewald (PME) method was used to evaluate the long-range electrostatic interaction. The system undergoes energy minimization to stabilize the overall structure and avoid steric clashes, followed by two steps of equilibration of 500 ps of NVT equilibration and 500 ps of NPT equilibration. Finally, the MD production was performed at 300 K and 1 bar pressure for 500 ns with 2 fs timestep. Structural and conformational analysis of all system was conducted using various analysis modules implemented in GROMACS package, while binding free energy and decomposition analysis were determined using gmx_mmpbsa tool43,44.
Cytotoxicity study
MTT assay was used to determine the effects of potential compounds on the viability of human dermal fibroblast cells. Cells were cultured in complete DMEM supplementing 10% FBS, 100 units/mL of penicillin, 100 µg/mL of streptomycin, and 0.25 µg/mL of amphotericin B (Gibco, USA). Cells (2 × 103 cells/well) were plated onto 96-well plates and allowed cell adherent by incubating at 37 °C, 5% CO2 overnight. Cells were treated with varying doses of each compound for 24 h, and 0.5% DMSO was used as an untreated control. Afterward, each well was replaced by 1 mg/mL MTT solution (Invitrogen, USA) and incubated for 4 h at 37 °C. After removing the MTT solution, formazan crystal was solubilized with DMSO, and absorbance was further measured at wavelength 570 nm using a BioTek Synergy H1 microplate reader (BioTek, USA). Cell viability was quantified as the percentage of the control.
ADMET prediction
In silico ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties of the selected compounds were predicted using freely available computational web server pkCSM55. The SMILES representations of each compound were used as input for structure-based prediction. Key pharmacokinetic parameters analyzed included human intestinal absorption (HIA), Caco-2 permeability, blood-brain barrier (BBB) penetration, steady state volume of distribution (VDss), substrate identification, and cytochrome P450 (CYP) enzyme inhibition profiles (including CYP2C9, CYP2D6, and CYP3A4). Total clearance was predicted to estimate compound elimination potential. Toxicological endpoints, including AMES toxicity, hERG I inhibition (cardiotoxicity) and hepatotoxicity were also assessed to evaluate potential safety concerns. All computational predictions were performed using default parameters, and the results were summarized in Supplementary Information Table S1.
Statistical analysis
Statistical analysis was performed using GraphPad Prism Version 8 (San Diego, CA, USA). The one-way ANOVA with Tuckey’s multiple comparison post hoc test was used to show differences between synthesized compounds. Data are presented as mean ± SE. A value of p < 0.05 was considered statistically significant.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Tachkov, K. et al. Life expectancy and survival analysis of patients with diabetes compared to the Non diabetic population in Bulgaria. PLoS One. 15, e0232815. https://doi.org/10.1371/journal.pone.0232815 (2020).
Sun, H. et al. IDF diabetes atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 183, 109119. https://doi.org/10.1016/j.diabres.2021.109119 (2022).
Gopisetty, D. et al. How does diabetes affect daily life?? A Beyond-A1C perspective on unmet needs. Clin. Diabetes. 36, 133–137. https://doi.org/10.2337/cd17-0093 (2018).
Tomic, D., Shaw, J. E. & Magliano, D. J. The burden and risks of emerging complications of diabetes mellitus. Nat. Rev. Endocrinol. 18, 525–539. https://doi.org/10.1038/s41574-022-00690-7 (2022).
Aloke, C. et al. Current advances in the management of diabetes mellitus. Biomedicines 10 https://doi.org/10.3390/biomedicines10102436 (2022).
Dimitriadis, G. D., Tessari, P., Go, V. L. & Gerich, J. E. alpha-Glucosidase Inhibition improves postprandial hyperglycemia and decreases insulin requirements in insulin-dependent diabetes mellitus. Metabolism 34, 261–265. https://doi.org/10.1016/0026-0495(85)90010-1 (1985).
Mwakalukwa, R., Amen, Y., Nagata, M. & Shimizu, K. Postprandial hyperglycemia Lowering effect of the isolated compounds from Olive mill Wastes – An inhibitory activity and kinetics studies on α-Glucosidase and α-Amylase enzymes. ACS Omega. 5, 20070–20079. https://doi.org/10.1021/acsomega.0c01622 (2020).
Reuser, A. J. & Wisselaar, H. A. An evaluation of the potential side-effects of alpha-glucosidase inhibitors used for the management of diabetes mellitus. Eur. J. Clin. Invest. 24 (Suppl 3), 19–24. https://doi.org/10.1111/j.1365-2362.1994.tb02251.x (1994).
Nguyen, D. V. et al. Structure—yeast α-glucosidase inhibitory activity relationship of 9-O-berberrubine carboxylates. Sci. Rep. 13, 18865. https://doi.org/10.1038/s41598-023-45116-0 (2023).
Xu, J. et al. Identification of yeast α-glucosidase inhibitors from Pueraria lobata by ligand fishing based on magnetic mesoporous silicon combined with knock-out/knock-in technology. Food Funct. 14, 1952–1961. https://doi.org/10.1039/D2FO03475A (2023).
Peytam, F. et al. Design, synthesis, molecular docking, and in vitro α-glucosidase inhibitory activities of novel 3-amino-2,4-diarylbenzo[4,5]imidazo[1,2-a]pyrimidines against yeast and rat α-glucosidase. Sci. Rep. 11, 11911. https://doi.org/10.1038/s41598-021-91473-z (2021).
Barker, M. K. & Rose, D. R. Specificity of processing α-glucosidase I is guided by the substrate conformation: crystallographic and in Silico studies. J. Biol. Chem. 288, 13563–13574. https://doi.org/10.1074/jbc.M113.460436 (2013).
Alghamdi, S. S., Suliman, R. S., Almutairi, K., Kahtani, K. & Aljatli, D. Imidazole as a promising medicinal scaffold: current status and future direction. Drug Des. Devel Ther. 15, 3289–3312. https://doi.org/10.2147/dddt.S307113 (2021).
Ebenezer, O., Shapi, M. & Tuszynski, J. A. A review of the recent development in the synthesis and biological evaluations of pyrazole derivatives. Biomedicines 10 https://doi.org/10.3390/biomedicines10051124 (2022).
Frolov, N. A. & Vereshchagin, A. N. Piperidine derivatives: recent advances in synthesis and Pharmacological applications. Int. J. Mol. Sci. 24 https://doi.org/10.3390/ijms24032937 (2023).
Nammalwar, B. & Bunce, R. A. Recent advances in Pyrimidine-Based drugs. Pharmaceuticals (Basel). 17. https://doi.org/10.3390/ph17010104 (2024).
Rizwan, M. et al. A comprehensive review on the synthesis of substituted piperazine and its novel bio-medicinal applications. Chem. Inorg. Mater. 2, 100041. https://doi.org/10.1016/j.cinorg.2024.100041 (2024).
Saleem, F. & Khan, K. M. Indole derivatives: unveiling new frontiers in medicinal and synthetic organic chemistry. Molecules 28 https://doi.org/10.3390/molecules28145477 (2023).
Gong, Z., Xie, Z., Qiu, J., Wang, G. & Synthesis Biological evaluation and molecular Docking study of 2-Substituted-4,6-Diarylpyrimidines as α-Glucosidase inhibitors. Molecules 22, 1865 (2017).
Mallidi, K., Gundla, R., Makam, P., Katari, N. K. & Jonnalagadda, S. B. Dual active pyrimidine-based carbocyclic nucleoside derivatives: synthesis, and in Silico and in vitro anti-diabetic and anti-microbial studies. RSC Adv. 14, 9559–9569. https://doi.org/10.1039/D4RA00304G (2024).
Bassyouni, F. et al. Promising antidiabetic and antimicrobial agents based on fused pyrimidine derivatives: molecular modeling and biological evaluation with histopathological effect. Molecules 26 https://doi.org/10.3390/molecules26082370 (2021).
Koumpoura, C. L., Robert, A., Athanassopoulos, C. M. & Baltas, M. Antimalarial inhibitors targeting epigenetics or mitochondria in plasmodium falciparum: recent survey upon synthesis and biological evaluation of potential drugs against malaria. Molecules 26, 5711 (2021).
Elumalai, K., Shanmugam, A., Devaraji, M. & Srinivasan, S. Synthesis and molecular Docking of pyrimidine derivatives as antibacterial agents. Carbon Resour. Convers. 7, 100222. https://doi.org/10.1016/j.crcon.2024.100222 (2024).
Tang, B. et al. Chemical constituents in leaves of Morus Atropurpurea and their α-glucosidase activity. Chin. Traditional Herb. Drugs. 44, 3109–3113 (2013).
Kim, S. et al. PubChem 2023 update. Nucleic Acids Res. 51, D1373–D1380. https://doi.org/10.1093/nar/gkac956 (2022).
Brooks, W. H., Guida, W. C. & Daniel, K. G. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 11, 760–770. https://doi.org/10.2174/156802611795165098 (2011).
Wang, Z. et al. Enhancing monoamine oxidase B inhibitory activity via chiral fluorination: Structure-activity relationship, biological evaluation, and molecular Docking study. Eur. J. Med. Chem. 228, 114025. https://doi.org/10.1016/j.ejmech.2021.114025 (2022).
Meier, K., Arús-Pous, J. & Reymond, J. L. A potent and selective Janus kinase inhibitor with a chiral 3D-Shaped triquinazine ring system from chemical space. Angew. Chem. Int. Ed. 60, 2074–2077. https://doi.org/10.1002/anie.202012049 (2021).
Han, S. J. et al. Identification of highly selective type II kinase inhibitors with chiral peptidomimetic Tails. J. Enzyme Inhib. Med. Chem. 37, 1257–1277. https://doi.org/10.1080/14756366.2022.2068148 (2022).
Wang, K. M. et al. Design, synthesis and antibacterial evaluation of novel fluoroquinolone and its derivatives. Asian J. Chem. 26, 209–215. https://doi.org/10.14233/ajchem.2014.15572 (2014).
Li, Y. et al. Discovery of 4-piperazinyl-2-aminopyrimidine derivatives as dual inhibitors of JAK2 and FLT3. Eur. J. Med. Chem. 181, 111590. https://doi.org/10.1016/j.ejmech.2019.111590 (2019).
Anglin, J. et al. Discovery and optimization of aspartate aminotransferase 1 inhibitors to target redox balance in pancreatic ductal adenocarcinoma. Bioorg. Med. Chem. Lett. 28, 2675–2678. https://doi.org/10.1016/j.bmcl.2018.04.061 (2018).
Peytam, F. et al. Imidazo[1,2-c]quinazolines as a novel and potent scaffold of alpha-glucosidase inhibitors: design, synthesis, biological evaluations, and in Silico studies. Sci. Rep. 13, 15672. https://doi.org/10.1038/s41598-023-42549-5 (2023).
Taha, M. et al. Synthesis, alpha-glucosidase inhibitory, cytotoxicity and Docking studies of 2-aryl-7-methylbenzimidazoles. Bioorg. Chem. 65, 100–109. https://doi.org/10.1016/j.bioorg.2016.02.004 (2016).
Abula, A. et al. Substitution effect of the trifluoromethyl group on the bioactivity in medicinal chemistry: statistical analysis and energy calculations. J. Chem. Inf. Model. 60, 6242–6250. https://doi.org/10.1021/acs.jcim.0c00898 (2020).
Cavallo, G. et al. The halogen bond. Chem. Rev. 116, 2478–2601. https://doi.org/10.1021/acs.chemrev.5b00484 (2016).
Chaudhari, S. R., Mogurampelly, S. & Suryaprakash, N. Engagement of CF3 group in N–H···F–C hydrogen bond in the solution state: NMR spectroscopy and MD simulation studies. J. Phys. Chem. B. 117, 1123–1129. https://doi.org/10.1021/jp310798d (2013).
Hanson, J. R., Hitchcock, P. B. & Jones, A. B. The conformation of some ortho substituted Stilbenes. J. Chem. Res. 2005, 138–140. https://doi.org/10.3184/0308234054497092 (2005).
Phan, T. H. T. et al. Designing potent α-Glucosidase inhibitors: A synthesis and QSAR modeling approach for biscoumarin derivatives. ACS Omega. 8, 26340–26350. https://doi.org/10.1021/acsomega.3c02868 (2023).
Ardiansah, B. et al. Synthesis, α-Glucosidase inhibitory activity and molecular Docking study of chalcone derivatives bearing a 1H-1,2,3-Triazole unit. Chem. Pharm. Bull. 71, 342–348. https://doi.org/10.1248/cpb.c22-00844 (2023).
Wilcken, R., Zimmermann, M. O., Lange, A., Joerger, A. C. & Boeckler, F. M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 56, 1363–1388. https://doi.org/10.1021/jm3012068 (2013).
Owoloye, A. J.et al. Molecular docking, simulation and binding free energy analysis of small molecules as PfHT1 inhibitors. bioRxiv, 2022.2004.2027.489756 (2022). https://doi.org/10.1101/2022.04.27.489756.
Valdés-Tresanco, M. S., Valdés-Tresanco, M. E., Valiente, P. A., & Moreno, E. A new tool to perform End-State free energy calculations with GROMACS. J. Chem. Theory Comput. 17, 6281–6291. https://doi.org/10.1021/acs.jctc.1c00645 (2021).
MillerB. R. et al. (ed 3rd) MMPBSA.py: An efficient program for End-State free energy calculations. J. Chem. Theory Comput. 8 3314–3321 https://doi.org/10.1021/ct300418h (2012).
Ali, S. et al. Novel 5-(Arylideneamino)-1H-Benzo[d]imidazole-2-thiols as potent Anti-Diabetic agents: synthesis, in vitro α-Glucosidase inhibition, and molecular Docking studies. ACS Omega. 7, 43468–43479. https://doi.org/10.1021/acsomega.2c03854 (2022).
Abdjan, M. I. et al. Structure-based approach: molecular insight of Pyranocumarins against α-glucosidase through computational studies. RSC Adv. 13, 3438–3447. https://doi.org/10.1039/D2RA07537G (2023).
Shayegan, N. et al. Synthesis, in vitro α-glucosidase inhibitory activities, and molecular dynamic simulations of novel 4-hydroxyquinolinone-hydrazones as potential antidiabetic agents. Sci. Rep. 13, 6304. https://doi.org/10.1038/s41598-023-32889-7 (2023).
Wu, Z. et al. ADMET evaluation in drug discovery. 19. Reliable prediction of human cytochrome P450 Inhibition using artificial intelligence approaches. J. Chem. Inf. Model. 59, 4587–4601. https://doi.org/10.1021/acs.jcim.9b00801 (2019).
Supasuteekul, C. et al. DNA damage protective, neuroprotective, and α-glucosidase inhibitory activities of a flavonoid glycoside from leaves of Garcinia gracilis. Revista Brasileira De Farmacognosia. 26, 312–320. https://doi.org/10.1016/j.bjp.2016.01.007 (2016). Antioxidant.
Adasme, M. F. et al. PLIP 2021: expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 49, W530–W534. https://doi.org/10.1093/nar/gkab294 (2021).
Stierand, K. & Rarey, M. Drawing the PDB: Protein – Ligand complexes in two dimensions. ACS Med. Chem. Lett. 1, 540–545. https://doi.org/10.1021/ml100164p (2010).
Abraham, M. J. et al. High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2. GROMACS, 19–25. https://doi.org/10.1016/j.softx.2015.06.001 (2015).
Sousa da Silva, A. W. & Vranken, W. F. ACPYPE - AnteChamber PYthon parser interface. BMC Res. Notes. 5, 367. https://doi.org/10.1186/1756-0500-5-367 (2012).
Kagami, L., Wilter, A., Diaz, A. & Vranken, W. The ACPYPE web server for small-molecule MD topology generation. Bioinformatics 39 https://doi.org/10.1093/bioinformatics/btad350 (2023).
Pires, D. E. V., Blundell, T. L. & Ascher, D. B. PkCSM: predicting Small-Molecule Pharmacokinetic and toxicity properties using Graph-Based signatures. J. Med. Chem. 58, 4066–4072. https://doi.org/10.1021/acs.jmedchem.5b00104 (2015).
Acknowledgements
This research project is supported by the Second Century Fund (C2F), Chulalongkorn University.
Author information
Authors and Affiliations
Contributions
NH: conducted the experiments and wrote the original draft, PM: evaluated the cytotoxic activity of some selected compounds, NF and TR: analyzed computational studies, WC: supervised and designed the study. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Herfindo, N., Mikled, P., Frimayanti, N. et al. Chiral pyrimidinyl-piperazine carboxamide derivatives as potent yeast α-glucosidase inhibitors. Sci Rep 15, 23241 (2025). https://doi.org/10.1038/s41598-025-06104-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-06104-8
