
Abstract
Chronic low back pain (cLBP ) is a common condition that affects the quality of life, can be caused by disc degeneration, facet arthropathy, myofascial pain, sacroiliac joint pain, spondyloarthropathies, and nociplastic pain etc. Research suggests that excessive pyroptosis may trigger cytokine release and harmful inflammation, leading to tissue damage and organ dysfunction, which could be a potential molecular mechanism underlying these conditions. Our aim in this research was to look into the effects of genes associated with pyroptosis in cLBP, with the goal of describing possible markers for diagnosis and targets for treatment. A comprehensive analysis of Differentially Expressed Genes (DEGs) was performed utilizing the GSE177034 datasets sourced from the Gene Expression Omnibus (GEO) repository. GSE177034 included 98 patients admitted to the hospital for treatment of low back pain, all of whom had peripheral blood extracted and sequenced. We identified 883 DEGs, including 397 upregulated and 486 downregulated genes, in cLBP. The WGCNA revealed 11 co-expression modules, with the blue module presenting the utmost positive connection to pyroptosis. The GO and KEGG analyses showed that DEGs associated with pyroptosis were substantially enriched in pathways like the NF-kappaB transcription factor activity and the MyD88-dependent signaling pathway of Toll-like receptors. Machine learning approaches identified five pyroptosis relate hub genes (AVIL, PADI4, ARHGAP24, ST3GAL6, and BICDL2) as potential biomarkers. The cLBP diagnostic column chart model was established, demonstrating the accuracy of hub genes as biomarkers. A hub mRNA-related interaction network was constructed to identify potential therapeutic drugs, followed by verification of hub gene expression levels to validate their role in cLBP. Moreover, the investigation into immune cell infiltration revealed substantial disparities in the presence of five distinct immune cell types between the chronic LBP and acute LBP cohorts. Specifically, activated effector memory CD8 T cells (R = − 0.5407, p-value < 0.001) ,and CD56bright natural killer cells (R = − 0.5438, p-value < 0.001) were all found to be significantly different. These findings suggest that immune involvement may play a role in the development of cLBP. This study identified diagnostic biomarkers and therapeutic targets for cLBP, offering promising avenues for improved diagnosis and treatment. The identification of specific biomarkers, including AVIL, PADI4, ARHGAP24, ST3GAL6, and BICDL2, enhances diagnostic precision by enabling the stratification of cLBP subtypes based on underlying pathophysiology.
Similar content being viewed by others


Introduction
Chronic low back pain (cLBP) is a common condition, affecting patients’ living standards. People will always experience low back pain, either mild or severe, throughout their life, with an annual prevalence of chronic low back pain as high as 7% in the United States and a lifetime prevalence of approximately 47% in African countries. Chronic low back pain imposes high social costs on society, both in terms of direct healthcare expenditures and decreased social production due to disability, with $100 billion spent annually in the United States to treat low back pain, and since 2015 low back pain has been the leading cause of disability worldwide1,2. Despite the high prevalence, the pathophysiology of cLBP remains poorly understood, and current treatment modalities offer limited efficacy, highlighting the need for further research3. The pathological causes of low back pain are diverse, ranging from the lumbar spine to the brain from mechanical causes to genetic factors, with approximately 85% being non-specific low back pain4. In-depth research on chronic low back pain has revealed that certain specific genes are not only associated with the initial onset of this pain condition but are also closely related to its persistence5. Intervertebral disc degeneration is one of the causes of chronic low back pain, and studies have shown that low expression of Noggin, lipopolysaccharide-, and inflammatory factors can lead to pyroptosis of the nucleus pulposus, which can lead to intervertebral disc degeneration6,7,8. Recent investigations have drawn attention to the impact of pyroptosis in the development of inflammatory and degenerative disorders, particularly rheumatoid arthritis and osteoarthritis9. The initiation of inflammasomes triggers pyroptosis, leading to the release of inflammatory cytokines such as interleukin (IL)−1β and IL-18, which are crucial in the development of tissue inflammation and damage10. The role of pyroptosis in cLBP remains unclear; however, its documented involvement in other musculoskeletal conditions suggests it may be a treatment target11.
The objective of our study was to investigate the role of genes linked to pyroptosis in cLBP and identify possible biomarkers for diagnosis and therapy. In this investigation, we applied a multi-pronged bioinformatics technique, consisting of the Differentially gene expression analysis(DEGs) to identify expressed genes between control (Acute LBP) and experimental groups (Chronic LBP).Weighted correlation network analysis(WGCNA) used hierarchical clustering to identify gene modules and colors to indicate modules. A dynamic tree-cutting approach was used to identify different modules, and during module selection, the adjacency matrix (a measure of topological similarity) was converted to a topological overlay matrix (TOM), and modules were detected by clustering analysis. The GeneMANIA website (http://genemania.org) predicts relationships between functionally similar genes and central genes, including protein–protein, protein-DNA interactions, pathways, physiological and biochemical responses, co-expression and co-localisation. We constructed protein–protein interaction (PPI) networks of pyroptosis-related genes and pathways that could be involved in cLBP through the GeneMANIA website.Additionally, we performed an immune cell infiltration analysis and a receiver operating characteristic curve (ROC) analysis to evaluate the diagnostic value of the identified genes. The ROC is a useful method for assessing the performance of a diagnostic test.The ROC curve is a comprehensive indicator that reflects the sensitivity and specificity of continuous variables, with the most common indicator being the Area Under Curve (AUC), obtained from the operational characteristic map of the subject’s sensitivity and specificity. Gene Ontology (GO) enrichment analysis includes analysis of biological processes (BP), molecular functions (MF), and cellular components (CC). The Kyoto Encyclopedia of Genes and Genomes (KEGG) is a bioinformatics resource used to mine significantly altered metabolic pathways enriched in gene lists. The R package"clusterProfiler (version 4.2.2)"was applied for GO and KEGG enrichment analysis of autophagy related Differentially expressed genes (DEGs) (p-value < 0.05). Hub mRNAs typically serve as critical nodes within gene interaction networks, where their aberrant expression may drive the pathogenesis and progression of deseases.Using the StarBase online database, we identified five hub mRNAs and retrieved their corresponding mRNA/RBP pairs. Our study showed positive correlation between all five hub genes AVIL, PADI4, ARHGAP24, ST3GAL6, BICDL2, suggesting that they show synergistic effects with similar expression patterns and participate in a certain biological process or function together in the cell. The five hub genes can be used as potential biomarkers to identify cLBP. Hub gene PADI4 detected a correlation with drugs and can be used as a potential drug for the treatment of cLBP.
Methods
The dataset employed in this study are available via the GEO database (Gene Expression Omnibus, https://www.ncbi.nlm.nih.gov/geo/)12. By using the“GEOquery”R package (R version 4.1.2), we accessed the entire gene expression data for people presenting with cLBP, recorded in the GEO database. The dataset GSE177034 (GPL11154 Illumina HiSeq 2000) includes approximately 50,000 transcripts from 98 individuals hospitalized for acute low back pain (acute LBP). After 3 months of follow-up treatment, 49 individuals showed relief and were classified as acute LBP patients13. The remaining 49 patients experienced persistent LBP after 3 months of follow-up treatment and were considered as chronic LBP patients. Peripheral blood samples were obtained and sequenced. In this study, individuals with chronic LBP were designated as the experimental cohort, while those with acute LBP were designated as the control cohort.
Pyroptosis-related genes
Genes related to pyroptosis were sourced from earlier investigations10,14,15,16,17,18 (Table 1).
Differentially expression analysis for chronic LBP
The control and experimental groups, namely acute LBP and chronic LBP, respectively, were analyzed to pinpoint Differentially expressed genes (DEGs) utilizing the R package"DESeq2 (version 1.34.0)"19. To identify DEGs, we required an absolute |log2Fold Change|> 0 and an FDR-adjusted p-value below 0.05.
Weighted gene co-expression network analysis (WGCNA) and isolating key modules
The R package"WGCNA (version 1.70–3)"was employed to apply the WGCNA algorithm and establish a gene co-expression network20. To evaluate the resemblance of gene expression profiles, Pearson correlation coefficients were used and then raised to a power to achieve a scale-free network. The“PickSoftThreshold”function from the R package was employed to amplify the co-expression similarity to the power of β = 6, thus creating a weighted adjacency matrix. Gene modules, defined as clusters of genes that are closely connected within the co-expression network, were discerned by the WGCNA using hierarchical clustering and then color-coded. The dynamic tree-cutting technique was utilized to pinpoint various modules. During the phase of module selection, the adjacency matrix, indicative of topological similarity, was turned into a TOM, and modules were detected using cluster analysis. To confirm the link between modules and pyroptosis, Pearson correlation analysis was applied to assess the relationship between module eigenvectors (ME, reflecting the collective expression of the module) and pyroptosis. Modules significantly correlated with pyroptosis were obtained. A heatmap was created to illustrate the structure of co-expression modules, using gene network topological overlaps as the basis. Hierarchical clustering trees of feature vectors and corresponding heat maps of feature vectors were generated to summarize the relationships between modules. By comparing DEGs with pyroptosis-linked genes, we identified the pyroptosis-related DEGs.
Gene ontology and Kyoto encyclopedia of genes and genomes pathway enrichment analysis
The gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed on DEGs associated with pyroptosis, focusing on genes with p-values less than 0.0521. This involved the GO analysis for biological process (BP), molecular function (MF), and cellular component (CC), as well as the KEGG pathway analysis22,23. The ‘clusterProfiler’ R package (version 4.2.2) was employed for this task.
GeneMANIA
The GeneMANIA platform (http://genemania.org) was utilized to predict functional relationships and key hub genes among them, considering various interactions including protein–protein, protein-DNA, shared pathways, physiological and biochemical processes, co-expression, and co-localization24. A network depicting the interactions among key proteins was established by leveraging the capabilities of the GeneMANIA website for protein–protein interaction (PPI) analysis.
Utilizing receiver operating characteristic curve analysis for evaluation
The receiver operating characteristic (ROC) curve analysis is a valuable method for evaluating the precision of diagnostic tests. It provides a graphical representation of the balance between sensitivity and specificity, demonstrating how these factors interact with various threshold settings. The area under the curve (AUC) is the key measure for evaluating the diagnostic strength of a test, summarizing its sensitivity and specificity, as shown by the ROC curve. The ‘pROC’ R package was used to create ROC curves and determine AUC values for the feature genes, thus assessing their potential as diagnostic indicators24. The AUC values, which lie within the range of 0.5 to 1, are interpreted such that values approaching 1 signify superior diagnostic accuracy.
Immune infiltration analysis
By providing a quantitative measure of the relative upregulation or downregulation of genes within a defined gene set for individual samples, single sample gene set enrichment analysis extends the concept of gene set enrichment analysis (GSEA). By leveraging the TISIDB database (http://cis.hku.hk/TISIDB/index.php), we calculated the relative enrichment scores for 28 unique immune cell populations based on their gene expression profiles from individual samples. This classification included a comprehensive range of CD8 and CD4 T cell subsets, covering both their activated and memory phases, as well as T follicular helper cells, gamma delta T cells, and various T helper cell types. In addition, the study encompassed a detailed analysis of regulatory T cells, B cells across various stages of maturation, natural killer (NK) cells along with their subtypes, myeloid-derived suppressor cells, natural killer T cells, and dendritic cells exhibiting diverse levels of activation. Furthermore, we included other immune cells, such as macrophages, eosinophils, mast cells, monocytes, and neutrophils25. To illustrate the variations in the levels of immune cell infiltration between chronic LBP and acute LBP groups, we used the ‘ggplot2’ R package, version 3.3.6, for creating the graphical models26.
RBP-mRNA network construction
We used the StarBase Platform (https://starbase.sysu.edu.cn/tutorialAPI.php#RBPTarget) to explore the interactions among non-coding RNAs and to scrutinize the linkages between mRNA molecules and RNA-binding proteins (RBPs). This analysis was conducted using data from high-throughput sequencing technologies such as CLIP-seq, degradome-seq, and RNA-RNA interaction datasets. To pinpoint critical mRNA-RBP interactions under disease conditions, the following thresholds were established: a p-value of < 0.05, a minimum of five clusters (clusterNum ≥ 5), and at least five CLIP-seq experiments (clipExpNum ≥ 5). The criteria established served to evaluate both the statistical significance and robustness of mRNA-RBP interactions. After pinpointing the crucial mRNA-RBP pairs, we employed Cytoscape software to construct a thorough RBP-mRNA interaction network.
Statistical analysis
Statistical analysis was performed using R software, version 4.1.2. The Spearman correlation test was selected to ascertain the correlation between two variables. Comparisons between the two groups were made with the Wilcoxon test. Comparisons between three or more groups were made with the Kruskal–Wallis test. A two-tailed p-value of < 0.05 was indicative of statistical significance.
Results
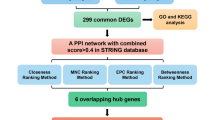
The study’s workflow diagram is depicted in Fig. 1.

Workflow diagram.
Differentially gene expression
Comparing chronic LBP against acute LBP groups revealed a total of 883 DEGs (|log2Fold Change|> 0, FDR corrected p-value < 0.05). Within the cLBP samples, 397 and 486 genes were up- and downregulated, respectively (Table 2). The DEGs are presented using a volcano plot in Fig. 2A. Furthermore, a heatmap was created to showcase the expression patterns of the top five upregulated genes, which included ADAMTS2, TBRG1, ACBD7-DCLRE1CP1, OLAH, and RNFT1, and the top five downregulated genes, namely EPOP, SDF2L1, HDDC3, CALR, and NAXE, with their categorization based on p-value ranking (Fig. 2B). The GO enrichment analysis outcomes highlighted significant enrichment in several pathways, including the aerobic respiration (GO:0,009,060), oxidative phosphorylation (GO:0,006,119) and cellular respiration (GO:0,045,333) within BP. Additionally, there was enrichment in the mitochondrial protein − containing complex (GO:0,098,798), mitochondrial inner membrane (GO:0,005,743), inner mitochondrial membrane protein complex (GO:0,098,800) in CC, as well as in MF such as structural constituent of ribosome (GO:0,003,735), oxidoreduction − driven active transmembrane transporter activity (GO:0,015,453) and electron transfer activity (GO:0,009,055) (Fig. 2C, Table 3). The KEGG pathway analysis demonstrated considerable enrichment in a selection of pathways,including Parkinson disease (hsa05012), Oxidative phosphorylation (hsa00190), Prion disease (hsa005020), and other related pathways (Fig. 2D, Table 4).

Visualization of Differentially expressed genes (DEGs). (A) Volcano map of DEGs in the chronic LBP and acute LBP groups. (B) Heatmap of the top five up-regulated and down-regulated genes. (C) Gene Ontology analysis of DEGs. (D) KEGG analysis of DEGs.
Weighted gene coexpression network analysis and module identification
The WGCNA revealed that a soft threshold (β) of 6 (Fig. 3A) led to an average connectivity close to 0 and scale independence over 0.85. Eleven coexpression modules were detected, with unrelated genes categorized into a grey module, which was subsequently excluded from analysis (Fig. 3B). The correlation of module eigengenes (ME) was computed to discern the intermodular relationships and to assess their significance. A visual representation of the gene network characteristics was created using a heatmap (Fig. 3C). Topological overlap was illustrated in a heatmap of the gene network (Fig. 3D). Eleven MEs were correlated with pyroptosis, showing relationships with the highest significance. The blue module gene clustering (n = 699, Table 5) demonstrated the most pronounced positive correlation (Fig. 3E) with pyroptosis (r = 0.5053, P < 0.05), prompting detailed analyses.Therefore, we subsequently consider mainly the blue module, as this one may indicate pyroptosis more accurately.By cross-referencing the DEGs with the pyroptosis-related modules, 48 key genes were discovered, highlighting their significance for subsequent investigations (Table 6, Fig. 3F).

WGCNA analysis (A) soft threshold β = 6, scale-free (R2). (B) The dendrogram illustrates the clustering of co-expressed genes in chronic LBP. (C) Relationships between modules. The correlation heatmap displays the characteristic gene networks. Characteristic genes of each module are depicted by color-coded rows and columns within the heatmap. High and low adjacency levels are represented by red and blue, respectively, with the meta-module shown as a red square on the diagonal. (D) Genes in the heatmap are depicted by each row and column, with lighter shades for low topological overlap and darker reds for high topological overlap, and the module is represented by a darker square on the diagonal. The gene tree and module assignments are depicted on the left and at the top, respectively. (E) Relationship between consistent module characteristic genes and pyroptosis. The table organizes data with rows representing consistency modules and columns for features. Each module’s gene-trait correlation is shown by numerical values. (F) The Venn diagram illustrates the genes that are common to both the set of DEGs and genes most significantly associated with a particular module.
Gene ontology pathway enrichment analysis
The GO enrichment analysis (Table 7) outcomes highlighted significant enrichment in several pathways, including the positive regulation of NF-kappaB transcription factor activity (GO:0,051,092), the MyD88-dependent toll-like receptor signaling pathway (GO:0,002,755), and the regulation of cytokine production involved in immune response (GO:0,002,718) within BP. Additionally, there was enrichment in the phagocytic vesicle membrane (GO:0,030,670) in CC, as well as in MF such as NAD(P) + nucleosidase activity (GO:0,050,135), NAD + nucleotidase, cyclic ADP-ribose generating (GO:0,061,809), and NAD + nucleosidase activity (GO:0,003,953) (Fig. 4A, 4B).

Enrichment analysis based on pyroptosis-associated Differentially expressed genes. (A) GO enrichment result lollipop plot demonstration. (B) Chord diagram of BP enrichment results.
Machine learning screening for hub genes
The LASSO regression identified six essential DEGs related to pyroptosis. (Fig. 5A, 5B). By employing the random forest algorithm, the most influential genes were prioritized, with the top 30 genes being identified. The selection criterion included feature weights like MDA and MDG, classifying these genes as key DEGs associated with pyroptosis (Fig. 5C, 5D). The SVM-RFE method identified 39 key pyroptosis-related DEGs (Fig. 5E). Finally, the intersection of DEGs detected by each method resulted in five key pyroptosis-related DEGs as hub genes for subsequent investigation. The hub genes included AVIL, PADI4, ARHGAP24, ST3GAL6, and BICDL2.

The application of machine learning algorithms aids in the discovery of biomarkers linked to chronic LBP for diagnostic purposes. (A) LASSO regression was used to regress the trajectory of the independent variables, with the horizontal coordinate indicating the logarithm of the lambda of the independent variables and the vertical coordinate indicating the independently accessible coefficients. (B) The LASSO regression model identifies confidence intervals for every lambda value. (C) The error rate of the random forest is contrasted against the number of classification trees it comprises. (D) The top 30 pyroptosis-associated DEGs for both importance rankings in the random forest algorithm. (E) SVM-RFE technique helps select the optimal feature genes. (F) Venn diagram provides a visual representation of the intersection for three machine learning methods.
Hub gene interaction analysis
A PPI network for the hub genes was established using GeneMANIA, showing that all five hub genes were part of the network’s interactions (Fig. 6A). The GO and KEGG analyses were performed on 25 genes in total, encompassing five pivotal genes and 20 associated genes. The GO enrichment findings indicated substantial enrichment in several pathways, including those for response to axon injury (GO:0,048,678), regulation of actin filament organization (GO:0,110,053), and regulation of protein depolymerization (GO:1,901,879) within the BP category. Additionally, there was significant enrichment observed for the intrinsic component of the Golgi membrane (GO:0,031,228), actin filament bundle (GO:0,032,432), and brush border (GO:0,005,903) in the CC category. In the MF category, enrichment was noted for phosphatidylinositol-4,5-bisphosphate binding (GO:0,005,546), GTPase binding (GO:0,051,020), and phosphatidylinositol bisphosphate binding (GO:1,902,936) (Fig. 6B, Table 8).

Hub gene interaction analysis. (A) Gene co-expression network mapping. The types of proteins represented by nodes, and the types of interactions represented by edges. (B) Gene Ontology analysis for co-expressed genes. (C) KEGG analysis of co-expressed genes.
The KEGG pathway analysis demonstrated considerable enrichment in a selection of pathways, including glycosaminoglycan biosynthesis—keratan sulfate (hsa00533), glycosphingolipid biosynthesis—lacto and neolacto series (hsa00601), Mannose type O-glycan biosynthesis (hsa00515), and other related pathways (Fig. 6C, Table 9).
Hub gene diagnostic line plot and diagnostic value
The calibration line was employed to measure the predictive potential, and the diagnostic plotting model was used to further validate the hub genes (Fig. 7A).The high accuracy of the cLBP model was demonstrated by the calibration line, which revealed minimal discrepancies between the actual and predicted cLBP risks (Fig. 7B). The model’s precision was also verified using the ROC curve analysis (Fig. 7C), which demonstrated the diagnostic significance of the hub genes. The AUC values for AVIL, ST3GAL6, PADI4, BICDL2, and ARHGAP24 were all above 0.6, suggesting their capability to serve as biomarkers for cLBP (Fig. 7D-H).

Chronic LBP diagnostic column chart model was established, and its accuracy was confirmed. (A) The column chart facilitated the prediction of chronic LBP cases. (B) Calibration curves were used to evaluate the forecasting capacity of the linear sketch model. (C) ROC curve evaluations were deployed to ascertain the clinical efficacy of the linear sketch model. (D) ROC curve for AVIL performance. (E) The ROC curve for ST3GAL6 performance. (F) ROC curve for PADI4 performance. (G) ROC curve of BICDL2 performance. (H) ROC curve for ARHGAP24 performance.
Analysis of immune cell infiltration
Chronic LBP might be significantly influenced by the presence of immune cells, suggesting a role for immune cell infiltration in its development. To explore this, we compared immune cell infiltration between the groups, revealing significant deviations in the proportions of five immune cell types between chronic LBP and acute LBP groups (Fig. 8A, Table 10).

Comparing immune infiltration in chronic LBP versus acute LBP groups. (A) The predicted immune cell infiltration percentage. (B) Heatmap showing the changes in immune infiltration levels between the groups. (C) Scatter plot of AVIL and CD56bright natural killer cell level correlation. (D) Scatter plot of PADI4 and effector memory CD8 T cell level correlation. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05.
Figure 8B illustrates a notable disparity in the extent of immune cell infiltration between the groups. Additionally, there was a significant correlation detected between AVIL expression and CD56bright NK cell levels (R = −0.544, p < 0.001) (Fig. 8C); effector memory CD8 T cell levels were associated with PADI4 (R = −0.541, p < 0.001) (Fig. 8D).
Pathways related to key genes
Differences between the chronic LBP and acute LBP groups in the 50 hallmark pathways were studied using the gene set variation analysis. No upregulated hallmark pathways were found in cLBP patients, while 15 significantly downregulated pathways were identified, including HALLMARK_ADIPOGENESIS, HALLMARK_ALLOGRAFT_REJECTION, HALLMARK_DNA_REPAIR, HALLMARK_E2F_TARGETS, HALLMARK_ESTROGEN_RESPONSE_EARLY, HALLMARK_G2M_CHECKPOINT, HALLMARK_IL2_STAT5_SIGNALING, HALLMARK_MTORC1_SIGNALING, HALLMARK_MYC_TARGETS_V1, HALLMARK_MYC_TARGETS_V2, HALLMARK_OXIDATIVE_PHOSPHORYLATION, HALLMARK_PEROXISOME, HALLMARK_PI3K_AKT_MTOR_SIGNALING, HALLMARK_REACTIVE_OXYGEN_SPECIES_PATHWAY, and HALLMARK_UNFOLDED_PROTEIN_RESPONSE (Fig. 9A, Table 11).

Connections between the 50 hallmark pathways and hub genes. (A) Comparisons between the chronic LBP and acute LBP groups. (B) Hub gene correlations with hallmark signaling pathways. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05.
The analysis of the correlation between the 50 hallmark pathways and the five hub genes was also conducted. AVIL and ST3GAL6 were associated with.
HALLMARK_ADIPOGENESIS and HALLMARK_ALLOGRAFT_REJECTION (Fig. 9B).
GSEA
Those genes that exhibited similar expression patterns to AVIL were predominantly enriched in certain pathways, including KEGG_FC_GAMMA_R_MEDIATED_PHAGOCYTOSIS, KEGG_FC_EPSILON_RI_SIGNALING_PATHWAY, KEGG_LONG_TERM_POTENTIATION, KEGG_NEUROTROPHIN_SIGNALING_PATHWAY, and KEGG_GNRH_SIGNALING_PATHWAY (Fig. 10A). The genes that exhibited similar expression patterns to ST3GAL6 were predominantly enriched in certain pathways, including KEGG_DORSO_VENTRAL_AXIS_FORMATION, KEGG_FC_EPSILON_RI_SIGNALING_PATHWAY, KEGG_MELANOMA, KEGG_LONG_TERM_POTENTIATION, and KEGG_LEUKOCYTE_TRANSENDOTHELIAL_MIGRATION (Fig. 10B). The genes that exhibited similar expression patterns to PADI4 were predominantly enriched in certain pathways, including KEGG_REGULATION_OF_ACTIN_CYTOSKELETON, KEGG_NEUROTROPHIN_SIGNALING_PATHWAY, KEGG_STARCH_AND_SUCROSE_METABOLISM, KEGG_LEUKOCYTE_TRANSENDOTHELIAL_MIGRATION, and KEGG_FC_GAMMA_R_MEDIATED_PHAGOCYTOSIS (Fig. 10C). The genes that exhibited similar expression patterns to BICDL2 were predominantly enriched in certain pathways, including KEGG_NEUROTROPHIN_SIGNALING_PATHWAY, KEGG_GNRH_SIGNALING_PATHWAY, KEGG_NOD_LIKE_RECEPTOR_SIGNALING_PATHWAY, KEGG_B_CELL_RECEPTOR_SIGNALING_PATHWAY, and KEGG_LEISHMANIA_INFECTION (Fig. 10D). The genes that exhibited similar expression patterns to ARHGAP24 were predominantly enriched in certain pathways, including KEGG_FC_EPSILON_RI_SIGNALING_PATHWAY, KEGG_LONG_TERM_POTENTIATION, KEGG_TYPE_II_DIABETES_MELLITUS, KEGG_LEISHMANIA_INFECTION, and KEGG_B_CELL_RECEPTOR_SIGNALING_PATHWAY (Fig. 10E).

Hub gene underwent single-gene GSEA enrichment analysis. (A) AVIL underwent single-gene GSEA enrichment analysis. (B) ST3GAL6 gene underwent single-gene GSEA enrichment analysis. (C) PADI4 underwent single-gene GSEA enrichment analysis. (D) BICDL2 underwent single-gene GSEA enrichment analysis. (E) ARHGAP24 underwent single-gene GSEA enrichment analysis.
Hub mRNA-related interaction network
In order to investigate the interactions between RBPs and mRNAs, we utilized the StarBase online database to identify the top five hub mRNAs, resulting in the acquisition of mRNA/RBP pairs. With the relationships delineated by the dataset available online, an RBP-mRNA interaction map was generated, comprising 45 nodes, 40 RNA binding proteins, five mRNAs, and 69 edges (Table 12, Fig. 11A).

Hub gene interaction network. (A) RBP-mRNA regulatory network. Green-colored circles represent RBP orange-colored circles represent mRNA. (B) Drug sensitivity analysis of hub genes. Orange-colored circles represent hub genes and blue-colored circles represent drugs or molecular compounds.
The drug sensitivity analysis, focusing on the five hub genes, revealed a significant correlation with PADI4. This finding suggests that PADI4 might be a key determinant of a response to a specific pharmaceutical. The drug-gene interaction network (Fig. 11B) revealed that three drugs or molecular compounds (CHEMBL1910970, STREPTONIGRIN, CHEMBL1910971) had different degrees of regulatory effects on PADI4.
Validation of hub gene expression patterns
All five hub genes showed substantial expression disparities between the two cohorts, with the cLBP group having increased expression levels compared to those of the control group (Fig. 12A). The hub genes were correlated (Fig. 12B).

Verification of hub gene expression levels. (A) Hub gene expression boxplot. (B) A heatmap illustrating the correlation between hub genes. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05.
Discussion
This study examined the gene expression patterns linked to pyroptosis in people with cLBP, seeking to understand the mechanisms behind them. Pyroptosis, characterized by inflammation, has been associated with numerous chronic inflammatory diseases27. By comparing the DEGs related to pyroptosis between the study groups, we aimed to identify key molecular pathways and potential therapeutic targets. This study may help advance the diagnostics and therapeutics of cLBP.
First, we identified 883 DEGs (397 upregulated and 486 downregulated genes) that distinguished chronic LBP from acute LBP groups. GO results showed that the biological functions of pyroptosis-associated DEGs were enriched in the positive regulation of NF-kappaB transcription factor activity, MyD88-dependent toll-like receptor signaling pathway. Previous studies have made similar findings. Intervertebral disc degeneration (IDD) is an important cause of chronic low back pain,Deng, Z.et al.showed that the BRD9 expression is upregulated in IDD and promotes IDD progression through the NOX1/ROS/NF-κB axis by inducing matrix degradation and pyroptosis28. Neuropathic pain(NP) is also a cause of chronic low back pain, and evidence suggests a causal relationship between neuropathic pain and immune-mediated inflammatory disorders. Wang, Y. H.et al. established a rat spinal nerve ligation(SNL)model to explore the potential association between melatonin and pyrotosis. It was shown that SNL significantly up-regulated inflammatory cytokines associated with over-activation of NLRP3 components and NF-κB signaling as well as pronounced pyroptosis activation, triggering severe abnormal pain. Melatonin prevents focal death through the NF-κB/NLRP3 inflammasome signaling pathway and has potent analgesic and anti-inflammatory effects in SNL models29. Kurt, S. et al. found that MyD88-dependent toll-like receptor plays an important role in RIPK1-driven necroptosis, pyroptosis, and inflammatory responses in an LPS-induced inflammatory pain model30.
Then, we employed WGCNA analysis and discovered a significant positive correlation with pyroptosis, which included 48 pyroptosis-related DEGs.Subsequently, based on three machine learning algorithms, we identified five key pyroptosis-related DEGs: AVIL, PADI4, ARHGAP24, ST3GAL6, and BICDL2. These hub genes are prime candidates for additional functional assessment and may potentially function as diagnostic indicators or targets for treatment. Alterations in the AVIL gene have been linked to steroid-resistant nephrotic syndrome, which is a common cause of chronic kidney disease. These mutations impair the AVIL actin-bundling ability, disrupting the associated processes, which are crucial for preserving the structural stability of podocytes in the kidney27. In cLBP, AVIL effects on actin filament organization and cellular migration could be pivotal. The gene’s involvement in pathways such as NF-kappaB transcription factor function and toll-like receptor signaling suggests that it may influence inflammatory responses and cellular stress mechanisms, which are critical in the pathophysiology of cLBP. Therefore, AVIL and its associated pathways could be therapeutic targets in cLBP.
The PADI4 gene, which encodes peptidyl arginine deiminase 4, contributes to inflammation and immune responses. Research has shown that in knockout mice lacking the PADI4 gene, the severity of arthritis induced by recombinant human glucose-6-phosphate isomerase (GPI) is significantly diminished. The various immunological changes observed in the GPI-immunized Padi4 knockout mice correlate with a decrease, which includes a notable reduction in serum IL-6 levels, suppression of Th17 cell differentiation, and a diminished production of anti-GPI antibodies31. The differentiation of NB4 cells induced by ATRA seems to involve PADI4, which might also be responsible for the increased expression of inflammatory cytokines32. By catalyzing the citrullination of proteins, PADI4 participates in the regulation of macrophage differentiation and inflammatory responses, and could potentially have a substantial effect on the development of autoimmune diseases, such as rheumatoid arthritis33. PADI4 plays a role in the immune response by triggering the NF-kappaB signaling pathway and adjusting the levels of inflammatory factors. PADI4 participates in the citrullination of proteins, which may be related to NAD + nucleosidase activity, thereby participating in the activation of inflammation. Thus, PADI4 might be a new target for managing cLBP.
ARHGAP24 belongs to the Rho GTPase-activating protein (RhoGAP) family, which is essential for controlling a range of cellular processes. Studies have revealed that ARHGAP24 possesses the ability to suppress cellular multiplication and the advancement of cellular cycling34. When ARHGAP24 is overexpressed, it notably suppress cellular multiplication and causes cell cycle arrest at the G0-G1 phase35. Furthermore, an increase in ARHGAP24 levels substantially reduces the ability of cells to invade and initiates cell death in renal cancer cells. In bladder cancer, ARHGAP24 has linked with the presence of immune cell infiltration, suggesting its potential involvement in immune regulation36. Elevated levels of ARHGAP24 have been observed to substantially decrease the concentrations of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6, consequently alleviating the intensity of acute pneumonia by dampening inflammatory reactions in the pulmonary system37. DEGs are predominantly involved in the biological process of regulating the production of cytokines, which is essential for the immune reaction. The ARHGAP24 may influence the occurrence and progression of cLBP through the regulation of cytokines.
The pathways in which key genes were significantly enriched, as identified by our study, include the positive modulation of NF-kappaB transcription factor activity, the MyD88-dependent cascade of toll-like receptor signaling, and several other related pathways. The NF-kappaB signaling cascade is of paramount importance in coordinating the body’s immune responses, managing inflammatory reactions, and ensuring the ongoing survival of cells. It acts as a pivotal regulator of inflammatory and apoptotic cell signaling, and upon its activation, it causes an uptick in the transcription of genes involved in these pathways38. This pathway is subject to stringent regulation through various mechanisms, such as phosphorylation and protein–protein interactions, which are essential for modulating its activity and ensuring precise control over gene expression39,40.
In our study, another critical pathway identified was the MyD88-dependent toll-like receptor signaling pathway. It contributes to the natural immune defense, facilitating the initiation of NF-kappaB and several other transcription factors when encountering PAMPs (pathogen-related molecular signatures)39. This signaling cascade is crucial for initiating immune responses It has also been linked to various inflammatory conditions.
In our analysis,the percentage of CD56bright NK cells was significantly reduced in patients with chronic low back pain.CD56bright NK cells play an important role in immune regulation,especially in inflammatory responses and immune surveillance. Studies have shown that CD56bright NK cells are able to modulate the immune response through the secretion of cytokines such as IFN-γ,so their reduction may be related to the onset and persistence of chronic inflammation41. Dominique Josephin et al. had an interesting finding that in patients with cLBP pain intensity was positively correlated with anxiety and depression scores,and depression scores were positively correlated with CD3-CD56bright natural killer cell counts were significantly negatively correlated42. In the chronic pain patient population,patients with opioid use or higher NRS scores had relatively low CD56bright NK cell counts41,43. We speculate that a decrease in CD56bright NK cells may exacerbate the pain intensity in patients with chronic low back pain through multiple pathways.
In our study, the percentage of Effector Memory CD8 + T cells was significantly reduced in patients with chronic low back pain. Previous studies have shown that changes in Effector Memory CD8 + T cells in pain depend on the specific pathological setting and type of pain. In chronic pain induced by autoimmune neuropathy and nerve injury, Effector Memory CD8 + T cells are usually increased and play an important role in the development and maintenance of pain. In long-term chronic pain or immunosuppressed states, Effector Memory CD8 + T cells may be reduced or depleted44. In studies of neuropathic pain (NP), Effector Memory CD8 + T cells have been suggested to be possibly involved in the regulation of neuroinflammation,and in some cases the number of Effector Memory CD8 + T cells decreases45.
Our research indicates that the identified pyroptosis-related DEGs are involved in key pathways, underscoring their possible contribution to the development of cLBP. The significant enrichment of genes in the pathway may affect NF-kappaB transcription factor function, potentially leading to inflammatory dysregulation, which is observed in cLBP. Additionally, the presence of MyD88-dependent toll-like receptor signaling suggests that innate immune responses could affect disease progression. These pathways establish a potential connection between inflammation, immune system activation, and chronic pain, which may drive cLBP.
Conclusions
Our research has uncovered notable disparities immunological cell permeation among individuals experiencing chronic LBP in contrast to those with acute LBP, highlighting the possible role of immune cells in the progression of chronic LBP. Notably, our findings revealed that AVIL was inversely related to CD56bright NK cells, while PADI4 was inversely associated with effector memory CD8 T cells. This concurrence aligns with other studies that emphasize the function of immune cells in persistent pain states. For example, research has shown that NK cells and their subsets are pivotal in regulating pain and inflammation. CD56bright NK cells, in particular, are recognized for their high cytokine production capabilities, which can significantly impact inflammatory responses and the perception of pain. This suggests that the modulation of immune cell activity, particularly NK cells, could be a key factor in the pathogenesis of cLBP, and further investigation into these correlations may provide valuable insights into the mechanisms driving back pain42.
Furthermore, CD8 T cells are involved in chronic pain syndromes by interacting with other immune cells and cytokines42. The observed negative correlations suggest that these immune cells might be downregulated or functionally altered in cLBP, potentially contributing to the persistence of pain.
Moreover, our findings are supported by studies that have shown altered immune cell profiles in chronic pain conditions, including NK cells and T cells, which can contribute to both triggering and sustaining chronic pain conditions46. Patients with cLBP have been shown to have reduced levels of CD56bright NK cells and altered T-cell subsets, compared to those healthy controls47. Targeting specific immune cells or their pathways may help manage chronic pain. For instance, therapies aimed at modulating NK cell activity or restoring the balance of T-cell subsets could potentially alleviate pain and improve patient outcomes. In our study, we identified DEGs between the chronic LBP and acute LBP groups and constructed a network of co-expressed genes to detect modules significantly associated with pyroptosis. The GO and KEGG enrichment analyses revealed substantial alterations in BP, MF, CC, and MP associated with the condition. Additionally, we mapped out a PPI network for pivotal genes, assessed their potential as diagnostic indicators using ROC curves, and compared the levels of immunological cell penetration between the two groups. Furthermore, we developed an RBP-mRNA network to shed light on the possible functions of key mRNA-RBP interactions in cLBP. These findings may help inform cLBP diagnostics and therapeutics.
In summary, the study’s findings on biomarkers and therapeutic targets for cLBP have significant clinical implications, offering promising avenues for improved diagnosis and treatment. The identification of specific biomarkers AVIL, PADI4, ARHGAP24, ST3GAL6, and BICDL2, can enhance diagnostic precision by enabling the stratification of cLBP subtypes based on underlying pathophysiology. This could lead to personalized treatment plans, moving away from the current one-size-fits-all approach. Additionally, the discovery of novel therapeutic targets, PADI4 regulator CHEMBL1910970, STREPTONIGRIN, CHEMBL1910971, opens the door to the development of new pharmacological agents.
Limitations
This study had some limitations. First, this was not an experimental study. Although our bioinformatics analysis offers some insights into the links among pyroptosis, the identified DEG, and cLBP, experimental validation studies are required. Second, as the study was based on 98 patients, the results may not be broadly generalizable. Third, batch effects from technical variability in sample processing could further distort results. Fourth, this study did not include a clinical validation arm. Additionally, incorporating data from various sources might lead to batch effects, which could skew the results. Future studies should aim to address these limitations by incorporating larger sample sizes, clinical data, and experimental validation arms. Future studies should focus on validating hub genes and modulating immune cell activity to assess their therapeutic potential.
Data availability
The dataset(GSE177034) employed in this study are available via the GEO database (GeneExpressionOmnibus, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE177034). Genes related to pyroptosis were sourced from earlier investigations(https://doi.org/10.1038/nri3452, https://doi.org/10.1038/s41420-021-00451-x, https://doi.org/10.1016/j.jsb.2017.08.001, https://doi.org/10.1038/s41568-019-0123-y, https://doi.org/10.1038/s41419-019-1883-8, https://doi.org/10.1111/imr.12296).
References
Hartvigsen, J. et al. What low back pain is and why we need to pay attention. Lancet 391, 2356–2367 (2018).
Knezevic, N. N., Candido, K. D., Vlaeyen, J. W. S., Van Zundert, J. & Cohen, S. P. Low back pain. Lancet 398, 78–92 (2021).
Alleva, J., Hudgins, T., Belous, J. & Kristin Origenes, A. Chronic low back pain. Dis. Mon. 62, 330–333 (2016).
Shirado, O. et al. Formulation of Japanese Orthopaedic Association (JOA) clinical practice guideline for the management of low back pain- the revised 2019 edition. J. Orthop. Sci. 27, 3–30 (2022).
Suntsov, V., Jovanovic, F., Knezevic, N. N., Knezevic, E. & Candido, K. D. Can implementation of genetics and pharmacogenomics improve treatment of chronic low back pain?. Pharmaceutics 12, 1–22 (2020).
Chen, S. et al. The role of the bone morphogenetic protein antagonist noggin in nucleus pulposus intervertebral disc cells. Int. J. Mol. Sci. 25, 11803 (2024).
Liao, Z. et al. Autophagic degradation of Gasdermin D Protects against nucleus pulposus cell pyroptosis and retards intervertebral disc degeneration in vivo. Oxid. Med. Cell Longev. 2021, 5584447 (2021).
Li, H. et al. IL-1β-mediated inflammatory responses in intervertebral disc degeneration: Mechanisms, signaling pathways, and therapeutic potential. Heliyon 9, e19951 (2023).
Zhang, R.-N., Sun, Z.-J. & Zhang, L. Pyroptosis in inflammatory bone diseases: Molecular insights and targeting strategies. FASEB J. 36, e22670 (2022).
Latz, E., Xiao, T. S. & Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411 (2013).
Ge, Y. et al. Pyroptosis and intervertebral disc degeneration: Mechanistic insights and therapeutic implications. J. Inflamm. Res. 15, 5857–5871 (2022).
Barrett, T. et al. NCBI GEO: Archive for functional genomics data sets - Update. Nucleic Acids Res. 41, 991–995 (2013).
Ditri, E. L. Z. Acute low back pain: Differentially somatosensory function and gene expression compared to healthy no-pain controls. Physiol. Behav. 176, 139–148 (2017).
Ye, Y., Dai, Q. & Qi, H. A novel defined pyroptosis-related gene signature for predicting the prognosis of ovarian cancer. Cell Death Discov. 7, 71 (2021).
Wang, B. & Yin, Q. AIM2 inflammasome activation and regulation: A structural perspective. J. Struct. Biol. 200, 279–282 (2017).
Karki, R. & Kanneganti, T. D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 19, 197–214 (2019).
Xia, X. et al. The role of pyroptosis in cancer: pro-cancer or pro-“host”?. Cell Death Dis. 10, 650 (2019).
Man, S. M. & Kanneganti, T. D. Regulation of inflammasome activation. Immunol. Rev. 265, 6–21 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 1–13 (2008).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol. 16, 284–287 (2012).
Blake, J. A. et al. Gene ontology consortium: Going forward. Nucleic Acids Res. 43, D1049–D1056 (2015).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Warde-Farley, D. et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38, 214–220 (2010).
Ru, B. et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 35, 4200–4202 (2019).
Ito, K. & Murphy, D. Tutorial: Application of ggplot2 to pharmacometric graphics. CPT Pharmacometrics Syst. Pharmacol. 2, 1–16 (2013).
Vasudevan, S. O., Behl, B. & Rathinam, V. A. Pyroptosis-induced inflammation and tissue damage. Semin. Immunol. 69, 101781 (2023).
Deng, Z. et al. BRD9 Inhibition Attenuates Matrix Degradation and Pyroptosis in Nucleus Pulposus by Modulating the NOX1/ROS/NF-κB axis. Inflammation 46, 1002–1021 (2023).
Wang, Y. H. et al. The Role of NF-κB/NLRP3 Inflammasome Signaling Pathway in Attenuating Pyroptosis by Melatonin Upon Spinal Nerve Ligation Models. Neurochem. Res. 47, 335–346 (2022).
Kurt, S. et al. Inhibition of RIPK1-driven necroptosis ameliorates inflammatory hyperalgesia caused by lipopolysaccharide: Involvement of TLR-, NLRP3-, and caspase-11-mediated signaling pathways. Cell Mol. Biol. 70, 52–58 (2025).
Seri, Y. et al. Peptidylarginine deiminase type 4 deficiency reduced arthritis severity in a glucose-6-phosphate isomerase-induced arthritis model. Sci. Rep. 5, 1–7 (2015).
Sun, X. et al. Roles of PADI4 in the expression of cytokines involved in inflammation and adhesion in differentiated NB4 cells treated with ATRA. Exp. Ther. Med. 25, 1–7 (2023).
Lai, N. S. et al. Increased peptidylarginine deiminases expression during the macrophage differentiation and participated inflammatory responses. Arthritis Res. Ther. 21, 1–14 (2019).
Yang, W. et al. ARHGAP24 represses β-catenin transactivation-induced invasiveness in hepatocellular carcinoma mainly by acting as a GTPase-independent scaffold. Theranostics 12, 6189–6206 (2022).
Wang, L. et al. ARHGAP24 inhibits cell proliferation and cell cycle progression and induces apoptosis of lung cancer via a STAT6-WWP2-p27 axis. Carcinogenesis 41, 711–721 (2020).
Yang, C. et al. Transcriptomic Analysis Identified ARHGAP Family as a Novel Biomarker Associated With Tumor-Promoting Immune Infiltration and Nanomechanical Characteristics in Bladder Cancer. Front. Cell Dev. Biol. 9, 1–20 (2021).
Liu, H., Wang, W., Shen, W., Wang, L. & Zuo, Y. ARHGAP24 ameliorates inflammatory response through inactivating Rac1/Akt/NF-κB pathway in acute pneumonia model of rat. Ann. Transl. Med. 8, 1289–1289 (2020).
Yin, X., Krikorian, P., Logan, T. & Csizmadia, V. Induction of RIP-2 kinase by proinflammatory cytokines is mediated via NF-κB signaling pathways and involves a novel feed-forward regulatory mechanism. Mol. Cell. Biochem. 333, 251–259 (2010).
Kumar, P., Schroder, E. A., Rajaram, M. V. S., Harris, E. N. & Ganesan, L. P. The Battle of LPS Clearance in Host Defense vs. Inflamm. Signal. Cells 13, 1–21 (2024).
Guldenpfennig, C., Teixeiro, E. & Daniels, M. NF-kB’s contribution to B cell fate decisions. Front. Immunol. 14, 1–14 (2023).
Diasso, P. D. K. et al. The effects of long-term opioid treatment on the immune system in chronic non-cancer pain patients: A systematic review. Eur. J. Pain (United Kingdom) 24, 481–496 (2020).
Dimmek, D. J. et al. Brain-derived neurotrophic factor and immune cells in osteoarthritis, chronic low back pain, and chronic widespread pain patients: Association with anxiety and depression. Med. 57, 327 (2021).
Yoon, J. J. et al. Cytotoxic activity and subset populations of peripheral blood natural killer cells in patients with chronic pain. Korean J. Pain 31, 43–49 (2018).
Yang, M. et al. Effector/memory CD8 + T cells synergize with co-stimulation competent macrophages to trigger autoimmune peripheral neuropathy. Brain. Behav. Immun. 71, 142–157 (2018).
Tian, S. et al. Identification of autophagy-related genes in neuropathic pain through bioinformatic analysis. Hereditas 160, 1–16 (2023).
Grace, P. M. et al. The neuroimmunology of chronic pain: From rodents to humans. J. Neurosci. 41, 855–865 (2021).
Parisien, M. et al. Acute inflammatory response via neutrophil activation protects against the development of chronic pain. Sci. Transl. Med. 14, 1–23 (2022).
Author information
Authors and Affiliations
Contributions
Tao Sun was responsible for the conception and design, and study supervision.Jintao Guo performed the analysis and was major contributors in writing the manuscript and submitting the manuscript. All authors read and approved the final manuscript. Jintao Guo and Tao Sun designed and directed the project; Jintao Guo performed the experiments; Jintao Guo analyzed data and wrote the article. Jintao Guo and Tao Sun designed and directed the project; Jintao Guo performed the analysis; Jintao Guo analyzed data and wrote the article,Tao Sun reviewed and modified the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Our study is based on open source data, so there are no ethical issues and other conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, J., Sun, T. Potential diagnostic and therapeutic gene in chronic low back pain through pyroptosis modulation: A silico study based on the dataset analysis. Sci Rep 15, 25767 (2025). https://doi.org/10.1038/s41598-025-06756-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-06756-6
