
Abstract
Rotavirus (RV) is a common double-stranded RNA virus that causes diarrheal disease in young children. The prevalent species, Rotavirus A (RVA), is responsible for over 90% of human RV infections. With significant morbidity and mortality, this pathogen poses a serious global health challenge, particularly in underdeveloped countries. This study presents an immunoinformatics approach for designing an mRNA vaccine based on a multi-peptide construct to elicit robust immune responses against RVA. The VP4 was analyzed from 40 sequences using phylogenetic analysis. Prediction of cytotoxic (CTL) and helper T cell (HTL) epitopes was performed and validated. The 17 high-conservancy CTL/HTL epitopes were selected for vaccine construction. The mRNA vaccine based on multi-peptide was engineered with human beta-defensin 3 (hBD3) adjuvant and linkers to enhance immunogenicity. The designed mRNA vaccine product exhibited favorable physicochemical properties and was predicted to be a probable antigen, non-allergenic, and non-toxic. 2D and 3D structure validation demonstrated the quality of the model. Molecular docking with Toll-like receptor 2/3 (TLR2/3) indicated favorable interaction, and peptide docking with MHC-I/II alleles showed strong binding affinities and have significant Residue-Residue interactions. Simulation of immune responses revealed potent B-cell and T-cell activities, macrophage responses, and significant cytokine synthesis. Molecular dynamics simulation (MDS) confirmed the structural stability of the TLR3-vaccine complex, and MHC-peptide in 200ns and STQFTDFVSLNSLRF peptide have shown good interaction with MHC molecule. In addition, the MM/GBSA analysis yielded a binding free energy of − 89.77 kcal/mol, indicating a strong and stable interaction between the vaccine construct and the target receptor. Codon optimization and mRNA secondary structure prediction were carried out for efficient translation. Additionally, population coverage analysis indicated the vaccine’s effectiveness worldwide with 100% value. Overall, this study showcases a promising immunoinformatics approach for designing an mRNA vaccine based on a multi-peptide construct targeting RVA. The findings support the potential of this vaccine design to elicit robust and widespread immune responses against RVA infection, paving the way for future vaccine development strategies and this study needs experimental validation.
Similar content being viewed by others
Introduction
Rotaviruses (RVs) are a double-stranded RNA virus in the Reoviridae family. RVs are the most common cause of diarrheal disease among young children1. By the age of five, all children have been infected at least once, and adults are seldom afflicted by this virus2. RV has nine species the most prevalent RV is Rotavirus A (RVA), which causes more than 90% of RV infections in people3. The genome of RV comprised a total of approximately 18.6 Kbp, encompassing the encoding of 12 proteins, both structural and non-structural. Notably, among these proteins, VP1, VP2, VP3, VP4, VP6, and VP7 act as structural proteins, with VP4 and VP7 being particularly crucial as they reside on the virus’s surface4.
The icosahedral protein capsid of the mature RV virion is around 100 nm in diameter (VP4 Spikes included). Three concentric layers of protein make up the capsid. Together, the proteins VP7 and VP4 make up the outer capsid layer3,5,6. In the 1950 s and 1960 s, RV was first identified in the rectal swabs of monkeys by using electron microscopy7 and also, in humans discovered in 19738. The World Health Organization (WHO) estimates that each year, RV results in 450,000 deaths, over 2 million hospital admissions, and 25 million outpatient cases. More than 90% of these deaths take place in underdeveloped nations in Asia and Africa9,10,11.
In 1998, the US government approved the use of the Wyeth-produced RotaShield vaccination against RV. According to research carried out in the United States, Finland, and Venezuela, it was detected through clinical trials that showed 80 to 100% potential to inhibit RVA infection and there were no significant side reactions12. Indications for Rotarix include the prevention of RV gastroenteritis caused by G1 and non-G1 varieties (G3, G4, and G9) when provided as a 2-dose series in babies and kids. Rotarix is a monovalent, human, live attenuated RV vaccination containing one RV strain with G1P specificity13. Recent advances in in-silico reverse vaccinology and immune-informatics have revolutionized the early-stage development of vaccines by enabling rapid identification of immunogenic targets from pathogen genomes. These computational approaches allow for the rational design of multi-epitope vaccine candidates with enhanced safety, facilitating rapid, cost-effective, and efficacy profiles. Notably, the integration of immune simulations and structural modeling facilitates the prediction of immunogenicity and population coverage, supporting the translation of computationally designed vaccines into therapeutic applications against emerging and re-emerging viral pathogens14,15,16. In extensive clinical trials, RotaTeq provided 98% protection against severe cases of rotavirus gastroenteritis characterized by symptoms such as fever, vomiting, diarrhea, and behavioral changes and offered 74% protection against rotavirus-related gastroenteritis of any severity, including inflammation of the stomach and intestines, during the first rotavirus season17. Two clinical trials showed that Rotarix provided between 85% and 96% protection against severe rotavirus gastroenteritis over the course of two rotavirus seasons18.
In recent years, mRNA vaccine technology has emerged as a transformative platform in the field of vaccinology due to its high potency, rapid development timeline, and safety profile. Unlike live attenuated vaccines, mRNA vaccines are non-infectious and do not integrate into the host genome, reducing safety concerns. Current licensed Rotavirus vaccines, such as Rotarix and RotaTeq, although effective, have shown reduced efficacy in low-income countries and raise concerns related to cold chain storage19manufacturing complexity, and rare side effects like intussusception. Moreover, these live vaccines may not provide broad protection against emerging or region-specific genotypes. mRNA-based vaccines offer a scalable and adaptable alternative, capable of encoding highly conserved and immunogenic epitopes such as those from the VP4 protein20. This approach facilitates the development of multi-peptide vaccines tailored for global population coverage and robust immune activation, particularly in settings where traditional vaccines are less effective or less accessible21.
In addition, mRNA vaccines are a functional field that showed the efficient way to progress vaccines against viruses22. Researchers have developed novel mRNA-based vaccine candidates targeting the viral spike protein VP8 to combat Rotavirus-induced severe childhood diarrhea. The LS-P2-VP8 candidate demonstrated the ability to self-assemble into 60-mer nanoparticles, inducing superior humoral responses against prevalent P genotypes in guinea pigs. This promising next-generation RV vaccine shows the potential to provide widespread protection and warrants further clinical investigation23. In purpose of the in silico design of vaccines was done against SARS-CoV-2, MERS-CoV, Rotavirus, and other viruses that will help scientists enhance and avoid new variants and infections24,25,26,27.
Infectious diseases have plagued humanity throughout history, causing immense suffering and claiming countless lives. This unpredictable microscopic pathogen has threated worldwide, particularly among the youngest and most vulnerable members of society28. The significance of advancing vaccine design through the utilization of immunoinformatics approaches and mRNA vaccine design has been underscored. These approaches have proven to be instrumental in the creation of potent vaccines and the identification of immunogenic peptides. Consequently, we employed bioinformatics techniques to develop an innovative mRNA vaccine that is based on a multi-peptide formulation with immunogenic properties. This formulation has the potential to induce adaptive immune responses against RVA.
Method
Mining the protein and analysis
Utilizing the Uniprot database, the VP4 protein was extracted. We used the 40 reviewed (Swiss-prot) sequences to prepare for analyses of conserved sequences and phylogenetic trees using MEGA11 software (https://www.megasoftware.net/)29 to identify the relationship between the outer capsid protein (VP4) in RVA. Additionally, the neighborhood-joining tree method was utilized to visualize the phylogenetic tree, and the alignment of the sequences was performed using the MUSCLE algorithm’s default features. The process of utilizing immunoinformatics approaches is illustrated in Fig. 1. Finally, we conducted an assessment of the surface antigenicity of VP4 and VP7 using the Vaxijen tool.

The workflow of the study demonstrated the immunoinformatics approaches.
Prediction and evaluation of the MHC I and MHC II epitopes
The significance of MHC I and MHC II in adaptive immune responses through CD8 + T Cytotoxic cells and CD4 + T Helper cells has underscored the importance of predicting MHC I and MHC II epitopes. The IEDB web server was employed to anticipate CTL and HTL epitopes, utilizing the default parameters as recommended by IEDB. The NetMHCpan EL 4.1 method was utilized for MHC I prediction, while the IEDB recommend 2.22 method was employed for MHC II prediction. Subsequently, a selection was made of the 7-allele HLA reference for MHC II and the frequently occurring MHC alleles for MHC I. The IL4Pred30 platform was implemented in silico to discover IL4 (https://webs.iiitd.edu.in/raghava/il4pred/scan.php). In addition, for the validation of MHC-II epitopes to detect the IFN-gamma-inducing epitope, we used the IFNepitope tool (https://webs.iiitd.edu.in/raghava/ifnepitope/algo.php). In IFNepitope31we used the SVM model to predict the IFN-gamma inducing. To validate the epitopes, Protparam (https://web.expasy.org/protparam/) was employed to ensure the physicochemical properties. Finally, the conservancy of the epitopes was assessed through epitope conservancy analysis (http://tools.iedb.org/conservancy/). Additionally, the conservancy analysis utilized a set of 40 sequences from RVA. The epitope sequences with the highest minimum identity matching at identity < = 100% were selected.
Engineering the construct of a mRNA vaccine based on multi-peptide
After the prediction of MHC-I and MHC-II epitopes for the VP4 protein, we used the various linkers to attach the MHC I and MHC II epitopes and complete the construct of the mRNA vaccine based on multi-peptide. The CTL epitopes linked with Gly-Pro-Gly-Pro-Gly (GPGPG) linkers and also linked to HTL epitopes used this. The Ala-Ala-Tyr (AAY) sequence is linked to HTL epitopes. In this study, we used human beta-defensin 3 (hBD3) as an adjuvant, attaching it to both the N- and C-termini of the vaccine construct via an EAAAK linker. This design enhances dendritic cell activation, thereby potentially boosting innate immune responses32. The adjuvant was added at the N-terminal of CTL and HTL epitopes with EAAAK linkers. The role of hBD3 in triggering cellular immune responses that contribute to antigen-specific adaptive immunity remains unclear. In this study, we demonstrate that hBD3, a member of the hBD family, can promote maturation and induce a T helper type 1 (Th1) response in human Langerhans cell-like dendritic cells (DCs).
Assessment of multi-peptide construct
The validation and characterization of multi-peptide vaccines were carried out with different web servers. The antigenicity of multi-peptide was performed, utilizing the VaxiJen33 based on the virus model and the ANTIGENpro server that is used (http://scratch.proteomics.ics.uci.edu/). The toxicity and allergenicity of multi-peptide were implemented by Toxinpred34AllerTop35and Algpred36 servers. The physicochemical characterizations were utilized in the ProtParam tool (https://web.expasy.org/protparam/) to probe the half-life, theoretical PI, molecular weight, and grand average of hydropathy (GRAVY). Moreover, the solubility of multi-peptide was calculated in Protein-Sol37 and SOLpro38. Protein-Sol is the server that predicted the solubility of proteins via data from the E.coli expression system and SOLpro is the two-stage SVM architecture that predicted the solubility with an accuracy of over 74%.
Prediction of secondary structure
With the aid of the SOPMA server39the 2D structure of multi-peptide (Helix, Sheet, Turn, and Coil) was predicted. In addition, the PSIPRED server (http://expasy.org/tool/gor4.html) was carried out to anticipate the 2D structure and the disorder region in the multi-peptide.
3D modeling and validation of tertiary structure
The Robetta server was utilized to model the multi-peptide that consists of 273 amino acids. The Robetta server is a 3D structure prediction service that was evaluated through CAMEO40 using RoseTTAFold, comparative modeling, and ab initio method to predict the 3D structure. In this case, we used the RoseTTAFold method to predict the 3D structure. The validation of the tertiary structure of multi-peptide was carried out with the different features of the SAVES v6.0 server (https://saves.mbi.ucla.edu/) such as PROCHECK for Ramachandran plot, ERRAT41. The ProsA-web server was performed to validate the structure42. This web server based on X-ray, NMR spectroscopy, and theoretical calculations that the service showed the quality of tertiary structure.
Prediction of B-cell conformational and linear epitopes
The Ellipro server (http://tools.iedb.org/ellipro/) predicts antibody epitopes for B-cells using linear and conformational epitopes. The default setting of the ElliPro server uses a minimum score threshold of 0.5. Given its foundation on the geometric features of protein structures, ElliPro holds potential for broader applications in predicting various protein–protein interactions43.
Molecular Docking of vaccine and TLR
In this investigation, we employed the ClusPro server, a highly sophisticated protein-protein docking web server (https://cluspro.bu.edu/)44. This tool was utilized to determine the docking of mRNA vaccine product with the Toll-like receptor 2/3 (TLR2/3), thereby facilitating the identification of potential interactions and their subsequent impact on the immune response. The structures of TLR3 and TLR2 (PDB IDs: 6NIG and 2A0Z, respectively) were carefully prepared using UCSF Chimera v1.16, a widely used and robust tool for molecular visualization and structural analysis. The higher scores for the molecular docking were selected for further analysis to confirm the interaction between TLR and vaccine. To discern the various forms of interactions, such as hydrogen bonds and salt bridges, the analysis of the molecular docking was meticulously filtered using the PDBsum tool45a tool specifically designed for the detection of protein-protein interactions. Additionally, to confirm the molecular docking results of the TLR3–vaccine complex, the Residue Interaction Network Generator (RING) tool was utilized to identify and visualize specific intermolecular interactions46. The molecular docking study between hBD-3 [PDB ID: 1kj6] and TLR3 was conducted to demonstrate the interaction and potential agonistic role of hBD-3 on TLR3. Specifically, the docking focused on the C-terminal domain of TLR3, which plays a critical role in ligand binding and signaling activity through its dimerization site. This interaction is pivotal for TLR3’s activation and function47.
Docking with corresponding epitopes and MHC alleles
The docking between corresponding epitopes of CTL, HTL, and MHC molecules [HLA-A*24:02.
, HLA-A*68:01, HLA-DRB1*04:01] was prepared. The HDOCK (http://hdock.phys.hust.edu.cn/) was performed to dock the peptide and the MHC molecules. The analysis of the peptide and MHC molecules was implemented by the PDBsum tool to detect the interaction of the peptide and MHC molecules. Finally, the peptide-MHC docking high score calculated the binding affinity, using the PRODIGY tool (https://wenmr.science.uu.nl/prodigy/) that was employed to anticipate the binding affinities of peptide and MHC molecules. The binding affinity is calculated by the below formula48:
Molecular dynamics simulation
To prove the structural stability and flexibility of TLR3-vaccine and MHC-peptide complex, we carried out Molecular dynamics (MD) simulation using the GROMACS (GROningenMAchine for Chemical Simulations) version 2019.0649. To construct the docked complex’s topology, the pdb2gmx module, and OPLS-AA force field were utilized, and the system contained TIP3P solvent and neutralized the system with 0.15 concentration of Na+/Cl−. Then the complex was centered in a cubic box. The energy was subsequently reduced by using the steepest descent integrator for 5000 nsteps and restricting the system parameter to 1000 KJ.mol−1.nm−1. Next, the two steps of equilibration were administered using 100 ps of NVT and NPT. The Brendsen pressure coupling method was employed with a base pressure of 1 bar at 310 oK. At last, MD simulation was carried out for the TLR3-vaccine and MHC-peptide complex for 200 ns. The MD trajectory data were then evaluated, as well as the TLR3-vaccine complex root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), buried surface area (BSA), solvent accessible surface area (SASA), radius of gyration (Rg), free energy landscape (FEL), and definition secondary structure of protein (DSSP). Furthermore, an examination of the molecular dynamic trajectory of the MHC-peptide was undertaken utilizing various analytical methods, including RMSD, RMSF, Rg, and SASA. In addition, the BSA was calculated by the formula:
Calculation of binding energy
HawkDock web server was used to calculate the binding free energy (ΔG)50. To calculate the binding free energy (ΔG) using the Molecular Mechanics/Generalized Born Surface Area (MM-GBSA) method, we extracted representative snapshots of the TLR3–vaccine complex from the molecular dynamics simulation at 10, 50, 100, 150, and 200 ns time intervals. To calculate binding energy and binding affinity, the MM-GBSA technique makes use of gas-phase energy (MM), polar electrostatic solvation energy (GB), and non-polar (SA) solvation energy51. At the final, we average the ΔG during the selected time steps to show the binding during the simulation. Furthermore, the formula for the calculation of binding energy is shown below.
mRNA vaccine development
Codon optimization, mRNA construction, 2D structure, and in Silico cloning
The codon optimization of the peptide vaccine design is required for efficient synthesis in human cells. As a result, the JCAT (Java codon adaption tool) software was implemented (http://www.jcat.de/). Then the evaluation of the optimized sequence’s quality was done and the Codon Adaption Index (CAI)52. The mRNA vaccine included the main part that the structure consists of m7GCap, 5’UTR (Untranslated regions, signal peptide (tPA), multi-peptide, stop codon, 3’UTR, and polyadenylation (PolA). In addition, the signal peptide is used to secrete the multi-peptide53the stop codon (TGA), and PolyA that consist of 100 and 250 Adenines54. The mFold tool (http://www.unafold.org/mfold/applications/rna-folding-form.php) to predict the secondary structure of the mRNA vaccine. The final sequence was inserted into the pET28a (+) vector and its predicted viability was assessed using SnapGene software (https://snapgene.com).
Prediction of the mRNA vaccine 3D structure and molecular dynamics simulation
To predict the 3D structure of the mRNA vaccine utilized the Xiao Lab server (http://biophy.hust.edu.cn/new/3dRNA). In this web server, we chose the RNAfold option for the prediction of the 2D structure of the mRNA structure55. In addition, we selected the best model for preparing for the MD simulation. MD simulations of the mRNA vaccine construct were performed using GROMACS 2019.v, as developed by Van Der Spoel et al. (2005). The CHARMM force field and SPC/E water model were employed for the simulation. A cubic simulation box with dimensions of 31.79 × 31.79 × 31.79 ų was constructed, and the system was neutralized by adding Na⁺ and Cl⁻ ions at a physiological concentration of 0.15 M. The solvated system was then subjected to energy minimization using 1000 steps of the steepest descent algorithm. Once the temperature was set at 310 K and the pressure at 1 bar, a production simulation was executed for a duration of 1000ps, allowing for an assessment of the dynamics of the mRNA structure. Consequently, profiles for RMSD, RMSF, and SASA were generated for the mRNA vaccine construct.
Population coverage
To build a vaccination that is effective for every individual in the globe, the coverage of expected epitopes throughout the world was examined using the IEDB population coverage web server (http://tools.iedb.org/population/). We integrated the MHC I and MHC II in this investigation. The world map generated by MapChart (https://www.mapchart.net/).
Immune simulation
The immune responses (innate immune and adaptive immune) against the product of the mRNA vaccine were probed by C-Immsim web server56 (https://kraken.iac.rm.cnr.it/C-IMMSIM). In this investigation, the cell-mediated response and humeral response were focused on, and also the cytokine responses. The option of time steps at 1, 84, 140, 180, and 240 (each time step is 8 h) with 5 injections. The simulation steps were set at 1050 to perceive the immune system for about 350 days with injection days at 1, 28, 47, 60, and 80 and the other features were default values.
Results
Phylogenetic tree and alignment
To identify a representative and broadly conserved vaccine target, we retrieved the VP4 protein sequences of RVA from the UniProt database. Specifically, 40 reviewed (Swiss-Prot) VP4 sequences were selected to ensure high-quality, manually annotated entries, avoiding potential annotation errors common in unreviewed sequences. These 40 sequences were chosen to represent geographically diverse and clinically relevant human RVA strains, thus allowing for a meaningful conservation and evolutionary analysis. Multiple sequence alignment was carried out using the MUSCLE algorithm with default parameters to evaluate conserved regions across the selected strains and phylogenetic tree was demonstrated in Fig. S1. To explore the evolutionary relationships among these VP4 sequences, a phylogenetic tree was constructed using the Neighbor-Joining method in MEGA11 software. The purpose of this analysis was to assess the degree of conservation across strains and to ensure that the selected vaccine strain is representative of broader circulating variants. Following phylogenetic analysis, a single representative strain (UniProt ID: sp|P36308) was selected for downstream immunoinformatics analyses. This strain was chosen based on its central positioning in the phylogenetic tree, indicating high similarity to multiple other strains, and its prevalence in both clinical and epidemiological data, making it a rational choice for the design of a broadly protective vaccine. Subsequently, we evaluated the surface antigenicity of both VP4 and VP7 proteins using the VaxiJen v2.0 server with a default threshold of 0.4 to ensure selection of antigenic proteins. In addition, the prediction of protective antigenicity using the VaxiJen server revealed that VP4 had a higher score (0.5279) compared to VP7 (0.4957), indicating that VP4 is a more likely antigen candidate. The threshold for the viral model was set at 0.4, further confirming that VP4 is more probable as a protective antigen than VP7.
Cytotoxic/Helper T cell epitopes and assessment
The MHC I/II is the major part of the adaptive immune system that has a connection with cytotoxic and helper T cells via CD4 + and CD8 +57. To predict the CTL and HTL epitopes, we carried out the IEDB web server that filtered via conservancy. The 7 CTL and 10 HTL epitopes were picked and a percentile rank between 0.01 and 1 (high affinity and intermediate affinity) was selected with a high conservancy that is shown in Tables 1 and 2. Most of the epitopes are good binders. Furthermore, analysis of MHC Class II epitopes revealed that nine of the predicted HTL epitopes have the potential to induce IL-4 production, a key cytokine in activating humoral immunity. Over the past decade, numerous computational approaches have been developed to predict MHC Class II binding peptides, with the goal of identifying HTL epitopes that can effectively mimic the activity of natural T-helper cells. It is crucial to acknowledge the diversity in cytokine responses associated with MHC II binding. This study endeavors to create a predictive model specifically tailored to identify MHC Class II binders that induce Interleukin-4 (IL-4) production, recognizing that not all MHC II binders elicit the same cytokine responses30. Via analysis of IFNepitope tool all MHC-II epitopes are IFN-gamma inducing. At last, the physicochemical properties were employed and most of them have a stable nature.
Construction and evaluation of multi-peptide
A total of seven CTL epitopes and ten HTL epitopes were selected for the construction of a novel multi-peptide mRNA vaccine. To enhance the immunogenicity of the vaccine construct, human β-defensin 3 was incorporated as an adjuvant at both the N- and C-termini. The adjuvant was connected to the CTL and HTL epitopes using the rigid EAAAK linker, which helps maintain structural stability and ensures efficient epitope presentation. The CTL epitopes were attached with a GPGPG linker, and the HTL epitopes were linked by AAY. The evaluation of multi-peptide construction was carried out, and the physicochemical assessment showed that the vaccine is stable with a 22.47 value and the molecular weight is about 41167.99 Da. The computational analyses have confirmed that the designed mRNA based on multi-peptide vaccine candidate exhibits favorable characteristics, including strong antigenicity, non-toxicity, and non-allergenicity, as outlined in Table 3. These in silico predictions suggest the potential of the vaccine to elicit a robust immune response with minimal adverse effects. However, while these computational results are promising, they must be validated through experimental studies to fully establish the vaccine’s efficacy and safety in real-world conditions.
2D/3D structure and validation
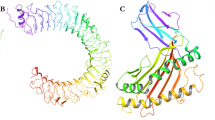
The SOPMA estimation of the secondary structure showed a 39.30% α-helix, 17.38% extended strand, and 43.32% random coil and the PSIPRED results showed the disorder region and secondary structure in Fig. 2A, B. The multi-peptide was modeled by the Robetta server (Fig. 2C) and validated with different methods in SAVES v.6.0 that included the ERRAT, and PROCHECK. Also, the ERRAT score showed a 96.94 overall quality factor that good high-resolution structures have values around 95%. The analysis of the Ramachandran plot has shown in the Fig. 3A. In addition, the percent of the residues in most favoured, additional allowed, generously allowed, and disallowed regions are 83.5%, 13.1%, 2.5%, and 0.9%, respectively. The Z-score of ProSA-web (Fig. 3B) showed a −5.91 score and all validations determined that the model has high quality for tertiary structure. At last, the ERRAT score (96.94) showed that significant quality structure of the vaccine (Fig. 3C).

(A) The secondary structure of multi-peptide that used PSIPRED server. (B) The detailed percent of secondary structure (Alpha helix, strand, Beta turn, and coil) (C) The 3D model of the final vaccine construct.

The validation of vaccine 3D structure. (A) The Ramachandran plot (B) Z-score of 3D structure with 5.91 scores followed by ProSA-web. (C) The ERRAT plot showed an overall quality factor of 96.94 score.
The conformational/linear B-lymphocyte epitopes
The Ellipro tool was used to anticipate the B-cell epitopes. All predicted B-cell epitope scores were between 0.594 and 0.885. All epitopes are shown in Table S1 and then the conformational and linear epitopes are demonstrated in Fig. S2.
TLR2/3 and multi-peptide Docking
Protein-protein docking was used by Cluspro 2.0 to determine how the vaccine complex interacted with TLR3. The 10 poses were predicted and the highest score was selected for the rest of the interaction analysis. After selecting the model 1 complex (−975.9 Kj/mol), the model that nominated for further study. The protein-protein interaction was carried out by the PDBsum server that demonstrated the hydrogen bond and salt bridge in Fig. 4A. Result displayed the 3 salt bridges, 7 hydrogen bonds, and the 23–25 residues of TLR3-vaccine have interaction as shown in Table S2. For the molecular docking analysis between TLR2 and the vaccine, the highest-scoring pose, with a binding energy of −1182.3 Kj/mol, was selected for further investigation of the protein-protein interactions (Fig. 4B). The results revealed strong interactions between TLR2 and the vaccine, characterized by the formation of 38 hydrogen bonds, 4 salt bridges, and 335 non-bonded contacts (Table S2). Finally, the interaction of TLR3 and hBD3 was done that have − 789.1 (kj/mol). The analysis of interaction showed that have 14 hydrogen bonds, 1 salt bridge, and 137 non-bonded contacts. This interaction between hBD3 proved that can be an agonist of the TLR3. The interaction of hBD3 and TLR3 is demonstrated in Fig. S3. In addition, to validate the interactions within the TLR3–vaccine complex, we employed the RING tool. The analysis confirmed several significant intermolecular interactions, including hydrogen bonds, π–π stacking, ionic interactions, van der Waals forces, and π–hydrogen bonds, as illustrated in Fig. S4.

The result of molecular docking. (A) The TLR3 and multi-peptide vaccine docking (Blue: TLR3, Cyan: multi-peptide vaccine). (B) The TLR2 and multi-peptide vaccine result (Red: TLR2, Cyan: multi-peptide vaccine). The interaction analysis of molecular docking showed the residue’s contact, ion bridge, and hydrogen bond. The number of residues that have interaction (Chain A: TLR2/3, Chain B: multi-peptide vaccine).
MHC alleles and peptide Docking
Among 17 lymphocyte epitopes, we selected 3 corresponding epitopes for analyzing the MHC I/II alleles and epitopes that docked with HDock that selected high scores as demonstrated in Table 4. The molecular docking outcomes from the HDock server, however, in terms of binding affinity are shown in Table 4. The epitopes SFDDISAAV showed the strongest binding free energy with its corresponding MHC I allele (HLA-A*68:01) with − 8.4 (kcal mol−1). Then, in Fig. 5 The peptide-MHC I/II demonstrated that included the interaction of peptide and MHC I/II with specific residue chain A/B.

Visualization of MHC alleles and MHC I/II peptide docking. (A) The SFDDISAAV epitope and HLA-A*68:01 (Cyan) (B) The KYGGRIWTF epitope and HLA-A*24:02 allele (Green) (C) The STQFTDFVSLNSLRF epitope and HLA-DRB1*04:01 allele (Purple). The interaction of the peptide and MHC molecules is visualized by the PDBsum tool.
Molecular dynamics simulation
TLR3-Vaccine
The structural stability of the TLR3-vaccine complex was assessed using a 200ns molecular dynamics simulation. To determine the conformational stability of the complexes, a time plot analysis was performed using the RMSD method (Fig. 6A). The TLR3-vaccine’s mean and standard deviation (SD) RMSD were measured at approximately 0.836 nm and 0.17, respectively. The RMSD plot of the vaccine–TLR3 complex showed an increase up to 60 ns, followed by a slight decrease around 80 ns, after which it stabilized with minor fluctuations until the end of the simulation. The RMSD values reached up to 1.5 nm, which reflects the inherent flexibility and dynamic conformational changes of the multi-epitope vaccine construct particularly in the loop regions that accommodate the epitopes and linkers. Such elevated RMSD values are common in flexible, engineered peptide constructs and do not necessarily imply structural instability. Previous studies have reported similar fluctuations, attributing them to natural motion in highly flexible regions25,58,59,60. In addition, the observed fluctuations in the multi-peptide vaccine construct are largely attributed to the hydrophilic epitopes, which exhibit increased mobility due to their position on the outer regions of the VP4 protein. These hydrophilic epitopes are not only more dynamic but also possess high antigenicity and accessibility, enhancing their likelihood of being recognized by the immune system. Their prominent location on the protein’s surface allows for optimal exposure to immune cells, facilitating a more robust and efficient immune response. This structural flexibility, combined with their surface accessibility, further supports the immune system’s capacity to detect and target the epitopes effectively, reinforcing the design’s immunogenic potential61. To find the mobility, the TLR3-vaccine complex’s residue flexibility was evaluated. (Fig. 6B). TLR3 and the vaccine have average RMSFs of 0.28 and 0.48 nm. The maximum RMSF for the vaccine is 0.93 (Residue: Ser318), and the RMSF for the vaccine is 0.92 (Residue: Gly54). Both residues in the vaccine are located in the loop, which is a flexible region of the vaccine structure. The RMSF of TLR3 demonstrated rigidity at the N-terminal and smoothness at the C-terminal. To assess the structural compactness, Fig. 6C showed the Rg profile of the TLR3-vaccine complex. The tertiary structure’s exceptional compactness was demonstrated by the average Rg of 4.03 that was estimated for each complex and the stable Rg plot of these complexes throughout the simulation, which was consistent with the RMSD values. The Rg plot decreased to 60ns and then got stable. Changes in the complex volume were carried out via SASA analysis. The purpose of SASA analysis yielded a mean SASA of 493.14 nm (Fig. 6D). The H-bond analysis shown in Fig. 6E showed the stable binding between vaccine and receptor showed that the maximum/minimum number of the H-bond is 11/4. At last, the transition phase of the complex was implemented through the FEL. The generation of FEL (Fig. 6F) involved the plotting of the first two eigenvectors that originated from the resultant trajectory. This was done to comprehensively analyze and understand the progression from the initial condition to the metastable condition. Furthermore, a study on the low energy state of the TLR3-vaccine complex system was carried out, with an emphasis on analyzing potential structural alterations that transpired after the construct’s binding to the receptor. The system continuously demonstrated an exceptional degree of translation confirmation during the entire experiment, guaranteeing the accuracy and validity of the results. Interestingly, the simulation revealed several distinct metastable states, each characterized by different energy levels. These included a stable low-energy state (red), a moderate-energy state (green), and a high-energy state (blue). The identification and classification of these states provided valuable insights into the conformational transitions and structural dynamics of the complex throughout the simulation. Analyzing these energy states helped elucidate the nature of structural rearrangements within the system. The DSSP analysis showed the secondary structure of the TLR3 and vaccine during the simulation that showed stable secondary structure in TLR3 and in vaccine residues 1 to 50 changes through the simulation, and other residues have stable secondary structure in 200ns simulation that demonstrated in Fig. 6G and H. At last, the BSA analysis was implemented as demonstrated in Fig. S5. Typically, higher BSA values suggest stronger interactions or more significant contact between the two structures, which is essential for understanding the binding affinity and stability of the complex. From the dataset, it looks like each residue involved in the interface has a corresponding BSA value, indicating how much of each residue is buried during an interaction. The ΔiG (interaction Gibbs free energy) is another critical factor. Negative values typically indicate favorable interactions, meaning the complex formation is energetically favorable. By plotting the BSA values across the residues, we visualized the regions of strong interaction (higher BSA) and weaker interaction (lower BSA) between TLR3 and the vaccine.

(A) Alpha carbon: Root mean square deviation (RMSD) (B) Root mean square fluctuation (C) Radius of Gyration (D) Solvent- Accessible surface area (SASA) (E) Hydrogen bond (H-bond) (F) Free energy landscape (FEL). The definition of the secondary structure of the protein (DSSP) was used to analyze the 2D structure of the complex. (G) The TLR3 secondary structure (H) The multi-peptide vaccine secondary structure.
MHC-epitope
Molecular dynamics simulation was employed to examine the three complexes over a 200ns timescale. The purpose of this simulation was to examine the variety of MHC molecules and their corresponding CTL and HTL peptides, as well as to validate the stable binding of epitopes. The HLA-DRB1*04:01 and peptide complexes reached stability after 10ns, according to the Root RMSD analysis, whereas the HLA-A*24:02-peptide complex increased until 60ns, at which point it approximated stability. However, it took 25ns for the HLA-A*68:01-peptide complex to stabilize (Fig. 7A). The HLA-DRB1*04*01 and the STQFTDFVSLNSLRF peptide had the lowest value, with an average RMSD of about 0.3 nm and a standard deviation of 0.028, according to an analysis of the RMSD profiles of the three complexes. In contrast to the other two complexes, the HLA-A*24:02 and KYGGRIWTF peptides demonstrated a significant conformational shift, with a mean RMSD of 1.17 and a standard deviation of 0.37. Additionally, the analysis of the Rg profile corroborated the findings of the RMSD analysis. As seen in Fig. 7B, the Rg plot of HLA-A*24:02 and KYGGRIWTF peptides showed lower compactness, while the HLA-DRB1*04:01 and peptide complexes showed high compactness. Proteins that have higher SASA values typically have more surface area exposed to solvent. According to Fig. 7C, the HLA-A*68:01-SFDDISAAV complex showed a high SASA value in this situation, whereas the HLA-DRB1*04*01-STQFTDFVSLNSLRF peptide complex showed a relatively lower SASA value. The HLA-DRB1*04*01 and STQFTDFVSLNSLRF peptide complexes showed more stable and robust interactions than the other complexes, according to an analysis of the hydrogen bond interactions between the three complexes (Fig. 7D). As seen in Fig. 7E, RMSF profile showed that the majority of the residues within the complexes stayed stable during the simulation. In particular, the least fluctuating peptide was STQFTDFVSLNSLRF, and the most fluctuating peptide was SFDDISAAV (Fig. 7F).

The Molecular dynamics simulation of MHC-peptide. (A) RMSD (B) Rg (C) SASA (D) H-bond number (E) RMSF for MHC molecules (F) RMSF for peptide. (Peptide 1: STQFTDFVSLNSLRF, Peptide 2: KYGGRIWTF, Peptide 3: SFDDISAAV).
MM-GBSA calculation
The binding free energy was calculated by the Hawkdock server and was carried out via 5 complexes during the simulation that extracted the snapshot from GROMACS. The average binding free energy is about − 89.77 which shows the stable binding between TLR3 and vaccine (Table 5). The plot of residues and ΔG shown in Fig. S6.
mRNA vaccine development
mRNA codon optimization, mRNA 2D structure, and in Silico cloning
The mRNA vaccine’s translation in its hosts was improved by using the JCAT Codon Optimization tool. There are 1505 nucleotides in the entire mRNA vaccination. The vaccine’s successful expression within human cells was shown by the GC content percentage of 66.57 and the CAI of roughly 0.95. The web server of mFold estimated the 2D structure of the mRNA vaccine. For the input of this server, we used the whole sequences of the mRNA vaccine used, which the improved sequence is in Table S3. Finally, Fig. 8A displays the general structure of mRNA, whereas Fig. 8B depicts the 2D structure of the mRNA vaccine. The thermodynamics of folds calculation revealed that the free energy is approximately ΔG = −455.70 kcal/mol, and the detail 5’ of the mRNA vaccine is shown in Table S4. The in silico cloning process was carried out using SnapGene software. The open reading frame (ORF) of the multi-peptide vaccine, comprising 1134 base pairs, was engineered by adding the sequences CTCGA and CATATG at the N-terminal and C-terminal, respectively, to enable cleavage by the restriction enzymes XhoI and NdeI. This detailed procedure is provided in Fig. S7.
3D structure of mRNA and MD simulation
The best 3D model out of the top 5 was carefully selected and showcased in Fig. 9A. To gain a deeper understanding of molecular dynamics, a comprehensive simulation was conducted using the GROMACS software. The trajectory analysis, spanning a period of 100ns, was meticulously performed and thoroughly examined. This simulation specifically focused on the stability of the structure of an mRNA vaccine. The RMSD profile, a crucial parameter in assessing the structural changes, exhibited an upward trend in its value at the onset of the simulation, only to stabilize after 20ns. The average RMSD value was found to be approximately 3.14 nm, with a standard deviation of 0.61 (Fig. 9B). Furthermore, the RMSF profile shed light on the nucleotides’ behavior, revealing minimal fluctuations for the majority of them (Fig. 9C). Lastly, the analysis of the SASA demonstrated a gradual decrease throughout the 10ns, indicative of a relatively low surface area (Fig. 9D).

(A) The general structure of mRNA vaccine that consists of 5’UTR, Kozak sequence, signal peptide, and multi-peptide. 3’UTR, and PolyA. In addition, the epitopes and linkers were demonstrated. (B) The secondary structure of mRNA vaccine and the 5’ of mRNA structure.

The 3D structure of mRNA vaccine and molecular dynamics simulation analysis. (A) The tertiary structure and detailed position of mRNA vaccine. (B) The RMSD (C) RMSF (D) SASA.
Population coverage
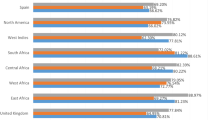
Using MHC-I/II combined, the IEDB analysis method anticipated the coverage of the global population. The total MHC-I/II epitope coverage of the global population is 100%, (Fig. S8A) with an average coverage of 86.09%. With a total population coverage of 100% for the class of chosen epitopes, Europe and North America had the greatest vaccination coverage. Additionally, Fig. S8B demonstrates that South Africa and Central America have the least coverage, whereas West Africa, South Asia, South America, Oceania, Northeast Asia, North Africa, Central Africa, and East Asia all have coverage levels above 90%. The final representation of the population coverage map is shown (Fig. S8).
Assessment of immune response
The immune responses elicited by the mRNA vaccine, based on a multi-peptide strategy, are detailed in Fig. 10. The vaccine was administered in five doses, spaced over different time intervals. The immune simulation results reveal that the immune responses generated during the fourth and fifth administrations were significantly stronger than the initial primary response. Notably, these later responses demonstrated a marked enhancement in immunoglobulin activity, especially between the third and fifth doses, indicating an improvement in the overall immune efficiency of the vaccine (Fig. 10A). Additionally, the analysis of B-cell isotype formation was carried out, with predictions suggesting a diversified immune profile in response to the multi-peptide vaccine. This isotype variation, shown in Fig. 10B, underscores the capacity of the vaccine to promote a broad immune recognition, enhancing its potential to protect against various viral strains. Further investigation of the T-cell responses (both helper T lymphocytes, HTL, and cytotoxic T lymphocytes, CTL) demonstrated a marked increase in both populations, suggesting a balanced and robust cellular immune response. These results are depicted in Fig. 10C and D, where the elevated levels of HTL and CTL populations highlight the ability of the vaccine to induce both humoral and cell-mediated immunity. This dual mechanism of action is crucial for the vaccine’s effectiveness against viral infections. Moreover, macrophage activation was observed to increase significantly, as shown in Fig. 10E. This heightened macrophage activity suggests a more efficient immune clearance and pathogen elimination process, reinforcing the overall protective capacity of the vaccine. Another pivotal finding was the synthesis of several key cytokines, with IFN-gamma levels reaching a peak concentration of over 500,000 ng/ml. The elevated levels of IFN-gamma, as illustrated in Fig. 10F, are indicative of a strong Th1-driven immune response, which plays a critical role in promoting viral clearance and enhancing the effectiveness of the immune system in combating infections. In conclusion, these results highlight the promising potential of the multi-peptide mRNA vaccine to elicit a potent and well-rounded immune response, not only through antibody production but also via a robust cellular immune response. The observed immune activation, including macrophage engagement, cytokine release, and T-cell responses, positions this vaccine as a promising candidate for the prevention of RVA infection.

The computational immune response of multi-peptide vaccine that injected 5 doses. (A) The relation of antigen concentration and antibody responses showed the efficiency of vaccines that have high concentration of IgG. (B) indicates the equivalent number of antibody-producing plasma cells. (C-E) The HTL, CTL, and macrophage responses. (F) The releasing cytokines.
Discussion
Every year, RVA is a prevalent illness that affects children. Concerns about RV are raised by the vaccinations’ yearly decline in effectiveness (Rotarix and RotaTeq)62. While other scientists used this treatment, we concentrated on immunoinformatics, which is crucial to the development of vaccines. The present SARS-CoV-2 pandemic has demonstrated the advantages of mRNA vaccine therapy, as demonstrated by Pfizer and Moderna vaccines63,64. The development of mRNA vaccines represents a promising approach to improving the efficacy of vaccines such as Rotarix and RotaTeq, which are currently the primary vaccines used for the prevention of RVA infection65,66. Although these vaccines have proven effective in reducing the incidence of severe RV infections, they still present certain limitations, including suboptimal efficacy in some populations, particularly in low-resource settings and among malnourished or immunocompromised children. These issues highlight the need for the development of more effective and universally applicable vaccines. The mRNA vaccine platform offers several advantages over traditional vaccines like Rotarix and RotaTeq. Firstly, mRNA vaccines can be designed to target a wide array of epitopes, including those from multiple strains of the virus, potentially increasing the breadth of immune protection. By encoding several peptides from the RVA virus in a single mRNA construct, such a vaccine could stimulate both humoral and cellular immunity more effectively67. This multi-peptide approach could also enable the development of vaccines that can target antigenic variation in the virus, a challenge for traditional vaccines that rely on specific viral strains. Moreover, mRNA vaccines can be produced rapidly and at scale. In the case of Rotavirus vaccines, this speed of production is critical, particularly in the face of emerging viral strains68. Since mRNA vaccines do not require the production of live virus or its proteins, they could be more easily updated and adapted to new variants, providing a more flexible solution compared to traditional inactivated or live-attenuated vaccines69. In addition, mRNA vaccines are designed to deliver the genetic information necessary for the host’s cells to produce the viral antigen directly, leading to a more robust immune response. This mechanism has the potential to overcome some of the limitations seen with current vaccines, such as poor immunogenicity in certain demographics or lower efficacy due to incomplete immune activation70. The ability to stimulate a more comprehensive immune response, including the activation of T-cells, B-cells, and the production of high levels of cytokines such as IFN-gamma, further enhances the potential for mRNA vaccines to provide long-lasting immunity. Ultimately, the need for an mRNA-based vaccine for Rotavirus arises from the desire to improve upon the efficacy of current vaccines like Rotarix and RotaTeq, particularly in populations with compromised immune systems or in regions where vaccine efficacy is less than ideal. With the ability to target multiple viral strains, rapidly respond to new variants, and produce a more potent immune response, mRNA vaccines offer a promising solution to overcome the existing limitations of Rotavirus vaccines. In this study, a novel immunoinformatics method was used to create an mRNA vaccine against RVA, the most frequent cause of diarrheal illness in young children worldwide71. During the early phases of vaccine development, immunoinformatics techniques provide several benefits, including the ability to quickly identify and assess possible vaccine candidates before conducting experiments to validate them72. VP4 proteins are essential to rotavirus biology, especially in facilitating the virus’s entry into host cells73,74,75. The previous study showed that all VP4 proteins are capable of inducing strong neutralizing antibody responses in both guinea pigs and rabbits when combined with an aluminum adjuvant. Moreover, a bivalent VP4-based vaccine (P[8] + P[6]-VP4) can elicit similarly high levels of neutralizing antibodies across multiple rotavirus genotypes, showing no significant difference in effectiveness compared to trivalent vaccines. Due to the significant potency of VP4, we chose the VP4 protein as the target for designing the mRNA vaccine. The findings of the investigation show that this strategy has the potential to produce an mRNA vaccine based on multiple peptides that can effectively and broadly elicit immune responses against RVA infection. From a pool of 40 strains, we selected a single strain for detailed analysis of the VP4 sequences. The phylogenetic analysis and alignment of the RVA VP4 protein sequences is one of the study’s main components to predict the conserved epitopes76. The researchers were able to concentrate on areas that the immune system is more likely to recognize across various RVA strains by identifying conserved sequences. This strategy raises the possibility of creating a vaccine that is more broadly effective, even against various strains of RVA. To induce efficient cellular and humoral immune responses that are most conservancy in other variants and IL4 inducing for HTL epitopes, it is imperative to predict CTL and HTL epitopes77. Epitope prediction using the IEDB web server is a commonly used and approved technique that has proven successful in several vaccine design studies. For the multi-peptide vaccine to effectively protect against RVA, a balanced and comprehensive immune response is guaranteed by the selection of seven CTL and ten HTL epitopes. The vaccine’s immunogenicity is increased through the engineering of the multi-peptide construct using the hBD3 adjuvant and suitable linkers. A promising option in this context is hBD3, which is known to stimulate dendritic cells and has anti-microbial properties. Adjuvants are essential for enhancing the immune response78,79,80,81. EAAAK is a peptide linker that forms a rigid α-helix and has a closed-packed backbone due to intramolecular hydrogen bonding82. GPGPG is a glycine-rich linker that offers high accessibility and freedom of activity in addition to improving construct solubility83. In this study, AAY also referred to as proteasomal cleavage sites in mammalian cells that employed as a linker to improve the stability of proteins84. The hBD3 adjuvant was selected due to its superior protein folding and flexibility, which enhance its potential as a vaccine candidate. Additionally, hBD3 is known for its immunomodulatory properties, as well as its antimicrobial and antiviral effects. Vaccines utilizing beta-defensins or other defensins as adjuvants have been shown to stimulate the primary innate antiviral immune response both in vivo and in vitro, while also facilitating various immunomodulatory actions against several viruses85,86,87.The appropriate linkers are employed to join the epitopes, ensuring optimal presentation and processing that facilitates immune cell recognition. An analysis of the multi-peptide vaccine’s physicochemical characteristics, antigenicity, allergenicity, and toxicity offers crucial information about both its efficacy and safety. The vaccine’s stability, non-toxicity, and non-allergenicity are indicated by the results, which are positive characteristics for future development. The validation of the tertiary structure through 3D modeling and various validation tools establishes the reliability of the vaccine construct. The high-quality tertiary structure suggests that the vaccine is structurally suitable for interaction with immune receptors and subsequent immune activation. The identification of B-cell epitopes is also a critical aspect of vaccine design, as B-cell responses contribute to antibody production and long-term immunity. The prediction of conformational and linear B-cell epitopes further supports the potential of the vaccine to elicit robust humoral responses. The protein-protein docking results obtained through Cluspro 2.0 provide critical insights into the interaction dynamics between the vaccine complex and TLR3, with the highest-scoring model (Model 1, with a binding energy of −975.9 kJ/mol) selected for further analysis. This model revealed key interaction features, including the formation of 3 salt bridges and 7 hydrogen bonds, as well as specific contacts between residues 23–25 of TLR3 and the vaccine. The relatively high binding affinity of the vaccine-TLR3 complex highlights its potential for robust activation of TLR3-mediated immune pathways, suggesting an effective stimulation of the innate immune response. When analyzing the docking between the vaccine and TLR2, the selected pose demonstrated a significantly higher binding energy of −1182.3 kJ/mol, suggesting an even stronger interaction compared to TLR3. This interaction was characterized by the formation of 38 hydrogen bonds, 4 salt bridges, and a substantial number of 335 non-bonded contacts, indicating a highly stable and potentially efficacious interaction. The extensive binding interactions between the vaccine and TLR2 may suggest a broad immune activation profile, likely involving the activation of both TLR2 and TLR3 pathways. This multi-receptor engagement could amplify the vaccine’s immune-stimulating potential by activating both antiviral and antimicrobial immune responses. TLRs play a crucial role in regulating viral pathogenesis by activating specific pathways that trigger antiviral interferon (IFN) and inflammatory responses88. However, many viruses have developed sophisticated strategies to evade TLR-mediated innate immunity, positioning TLRs as promising targets for therapeutic interventions89. hBD-3, a natural antimicrobial peptide, promotes the upregulation of costimulatory molecules like CD80, CD86, and CD40 on monocytes and myeloid dendritic cells through a mechanism reliant on Toll-like receptors (TLRs). This activation process is specifically driven by hBD-3’s engagement with TLR2, initiating a signaling pathway dependent on the adaptor protein myeloid differentiating factor 88 (MyD88) and culminating in the phosphorylation of IL-1 receptor-associated kinase-1 (IRAK-1)90. Moreover, the docking analysis of hBD3 with TLR3 further supports its potential as an agonist of the receptor, with a binding energy of −789.1 kJ/mol. TLR3 plays a crucial role in the innate immune response by recognizing double-stranded RNA (dsRNA), a molecular pattern commonly associated with viral infections. Upon activation, TLR3 initiates a signaling cascade via the TRIF (TIR-domain-containing adapter-inducing interferon-β) pathway, leading to the production of type I interferons and pro-inflammatory cytokines. This signaling not only facilitates antiviral defense mechanisms but also enhances the activation and maturation of dendritic cells and subsequent adaptive immune responses91,92.
The interaction analysis revealed the formation of 14 hydrogen bonds, 1 salt bridge, and 137 non-bonded contacts, further strengthening the hypothesis that hBD3 can modulate TLR3 signaling. The high number of hydrogen bonds and specific contacts points to a strong and stable interaction, which could contribute to the adjuvant’s ability to enhance the immune response. The role of hBD3 as a TLR3 agonist is particularly significant as it suggests that this defensin not only has antimicrobial and antiviral properties but can also directly modulate immune signaling through TLR3, potentially amplifying vaccine efficacy. The other study suggested that hBD3 exacerbated MDA5 and suppressed TLR3 responses93,94. The hBD3 has a significant role in the activation of innate immunity as anti-bacteria and anti-viruses80,95. hBD3 can block viral attachment, entry, and fusion. It specifically reduces the surface expression of CXCR4 on peripheral blood mononuclear cells, without affecting CCR5 expression, in serum-free conditions96.
Additionally, our study demonstrated that hBD3 has the potential to bind and interact with TLR3, warranting further experimental evaluation to validate this interaction. The combination of these docking studies highlights the robust interaction between the vaccine and the key innate immune receptors TLR2 and TLR3, along with the adjuvant effect of hBD3 on TLR3. Together, these findings indicate that the vaccine formulation could lead to potent activation of the innate immune system, leveraging multiple receptors to generate a broad and strong immune response. The detailed mapping of hydrogen bonds, salt bridges, and non-bonded contacts underscores the potential stability and efficacy of these interactions, laying a foundation for further experimental validation and optimization of the vaccine candidate97,98. Additionally, peptide docking with MHC-I/II alleles reveals strong binding affinities, indicating efficient presentation of the epitopes to T cells99,100. An important source of information about the possible effectiveness of the mRNA vaccine is the stimulation of immune responses. A strong and varied immune response against RVA infection can be induced by the vaccine, as evidenced by the increased B-cell and T-cell activities, macrophage responses, and significant cytokine synthesis observed in the C-ImmSim tool101. To have a potent vaccine should induce the increase of IL-2, TNF-α, IFN-γ, IL-4, IL-6, and IL-10102. The molecular dynamics simulation confirms the structural stability of the TLR3-vaccine complex for 200 ns. The analysis of RMSD, RMSF, Rg, SASA, FEL, DSSP, and BSA shows that the vaccine complex maintains a stable and compact structure, supporting its potential as a viable vaccine candidate103. Chawla et al. described the MDS via several analyses The estimation of binding free energy through MM-GBSA calculation provides an additional measure of the strong interaction between the vaccine and TLR3, reinforcing the construct’s immunogenicity104. The codon optimization of the mRNA vaccine ensures efficient translation in human cells, which is critical for optimizing its expression and the stability of the mRNA vaccine structure as assessed by MD simulation. The mRNA vaccine was analyzed using RMSD, RMSF, and SASA. Population coverage analysis shows that the multi-peptide vaccine has the potential to be effective in a wide range of populations around the world, making it a promising candidate for global vaccination efforts. In general, this study demonstrates a comprehensive and promising immunoinformatics approach for developing an mRNA vaccine against RVA. The combination of computational methods, molecular docking, immune simulation, and structural analysis offers a robust framework for vaccine development. However, it is important to note that experimental validation, including in vitro and in vivo studies, is necessary to confirm the efficacy, safety, and practicality of the vaccine candidate. If successful, this mRNA vaccine design could represent a significant advancement in the combat against RVA and contribute to global efforts to improve child health and reduce RVA-related morbidity and mortality. While immunoinformatics has revolutionized vaccine development by accelerating the identification of promising antigenic targets and epitopes, it is not without limitations. One of the primary concerns is the reliance on predictive algorithms, which, although increasingly accurate, are not infallible. These tools often utilize training datasets that may not fully represent the antigenic diversity of all pathogens or host genetic variations across global populations105,106. Consequently, predicted epitopes may not consistently elicit strong or protective immune responses in vivo. Furthermore, the accuracy of tools used for predicting MHC binding, allergenicity, antigenicity, and population coverage can vary significantly depending on the algorithms and databases used107. This introduces a degree of uncertainty in the extrapolation of computational findings to real biological systems. Another limitation lies in the structural modeling and molecular docking processes, which often simplify complex biological interactions. For instance, docking between vaccine constructs and immune receptors (such as TLRs) may not fully capture the dynamic nature of these interactions under physiological conditions. Even with molecular dynamics simulations, the timescale and environmental factors modeled may not accurately replicate the in vivo scenario. Additionally, immune simulation tools, though useful for preliminary assessments, cannot fully emulate the intricacies of the human immune system, including tolerance, memory response, and cytokine signaling networks. In terms of mRNA vaccine design, several challenges persist. Firstly, the delivery of mRNA into host cells requires effective and safe delivery systems such as lipid nanoparticles (LNPs)108which may vary in efficiency and biocompatibility. Stability of mRNA constructs is also a major concern, as mRNA is inherently unstable and susceptible to degradation by nucleases. Although modifications like nucleoside analogs and optimized UTRs are used to enhance stability and translation efficiency, these require careful optimization and may not be universally effective. Moreover, mRNA vaccines can elicit innate immune responses that lead to inflammation or adverse effects if not properly designed. The translation efficiency, immunogenicity, and duration of expression also depend on multiple host-related factors, making consistent outcomes across populations challenging. Finally, experimental validation remains a key bottleneck. While in silico methods can guide rational design, laboratory-based studies (in vitro and in vivo) are essential to confirm safety, immunogenicity, and efficacy yet such validation is often limited by time, cost, and infrastructure constraints.
Conclusion
The current study utilized computational approaches to design an mRNA-based vaccine targeting the VP4 protein of Rotavirus A, incorporating multi-peptide and conserved epitopes. The designed vaccine exhibits promising immunological properties, capable of stimulating both the innate and adaptive immune systems, and shows broad population coverage. It also demonstrated favorable physicochemical characteristics, structural stability, and strong binding affinity to MHC molecules and TLR receptors. As a result, this vaccine candidate holds potential as an effective strategy for combating RVA outbreaks. However, experimental validation is essential to confirm its efficacy, requiring further in vitro and in vivo studies, including clinical trials, to evaluate its immunogenicity and safety.
Data availability
All data generated during this study are included in this published article.
References
Dennehy, P. H. & Rotavirus Infection A disease of the past?? Infect. Dis. Clin. North. Am. 29, 617–635 (2015).
Bernstein, D. I. Rotavirus overview. Pediatr. Infect. Dis. J. 28, S50–S53 (2009).
Lekana-Douki, S. E. et al. Molecular epidemiology of enteric viruses and genotyping of rotavirus A, adenovirus and astrovirus among children under 5 years old in Gabon. Int. J. Infect. Dis. IJID Off Publ Int. Soc. Infect. Dis. 34, 90–95 (2015).
Hoxie, I. & Dennehy, J. J. Rotavirus A Genome Segments Show Distinct Segregation and Codon Usage Patterns. Viruses 13, (2021).
on Taxonomy of Viruses, I. C. & & King, A. M. Q. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses (Elsevier Science, 2011).
de Schoondermark-van, E. et al. Rabbit colony infected with a bovine-like G6P[11] rotavirus strain. Vet. Microbiol. 166, 154–164 (2013).
ADAMS, W. R. & KRAFT, L. M. Epizootic diarrhea of infant mice: indentification of the etiologic agent. Science 141, 359–360 (1963).
Bishop, R. F., Davidson, G. P., Holmes, I. H. & Ruck, B. J. Virus particles in epithelial cells of duodenal mucosa from children with acute non-bacterial gastroenteritis. Lancet (London England). 2, 1281–1283 (1973).
Martella, V., Bányai, K., Matthijnssens, J., Buonavoglia, C. & Ciarlet, M. Zoonotic aspects of rotaviruses. Vet. Microbiol. 140, 246–255 (2010).
Tate, J. E. et al. 2008 estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: a systematic review and meta-analysis. Lancet Infect. Dis. 12, 136–141 (2012).
Parashar, U. D., Nelson, E. A. S. & Kang, G. Diagnosis, management, and prevention of rotavirus gastroenteritis in children. BMJ 347, f7204 (2013).
Hochwald, C. & Kivela, L. Rotavirus vaccine, live, oral, tetravalent (RotaShield). Pediatr. Nurs. 25, 203 (1999).
O’Ryan, M. Rotarix™ (RIX4414): an oral human rotavirus vaccine. Expert Rev. Vaccines. 6, 11–19 (2007).
Das, N. C. et al. Immune targeting of filarial glutaredoxin through a multi-epitope peptide-based vaccine: A reverse vaccinology approach. Int. Immunopharmacol. 133, 112120 (2024).
Kaur, A. et al. Rational design and computational evaluation of a multi-epitope vaccine for Monkeypox virus: insights into binding stability and immunological memory. Heliyon 10, e36154 (2024).
Choudhury, A., Sen Gupta, P. S., Panda, S. K., Rana, M. K. & Mukherjee, S. Designing AbhiSCoVac - A single potential vaccine for all ‘corona culprits’: immunoinformatics and immune simulation approaches. J. Mol. Liq. 351, 118633 (2022).
Yi, Y. et al. Effectiveness of Lanzhou lamb rotavirus vaccine and RotaTeq against hospitalized rotavirus infections among children during 2020–2023 in Guangdong province, china: A Test-Negative Case-Control study. Infect. Dis. Ther. 13, 2301–2317 (2024).
Sun, Z. W. et al. Association of rotavirus vaccines with reduction in rotavirus gastroenteritis in children younger than 5 years: A systematic review and Meta-analysis of randomized clinical trials and observational studies. JAMA Pediatr. 175, e210347 (2021).
Glass, R. I., Parashar, U., Patel, M., Gentsch, J. & Jiang, B. Rotavirus vaccines: successes and challenges. J. Infect. 68, S9–S18 (2014).
Cao, Z., Zhu, J., Wang, Z., Peng, Y. & Zeng, L. Comprehensive pan-cancer analysis reveals ENC1 as a promising prognostic biomarker for tumor microenvironment and therapeutic responses. Sci. Rep. 14, 25331 (2024).
Yin, H. et al. Design, synthesis and anticancer evaluation of novel oncolytic peptide-chlorambucil conjugates. Bioorg. Chem. 138, 106674 (2023).
Qin, S. et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Signal. Transduct. Target. Ther. 7, 166 (2022).
Roier, S. et al. Novel mRNA-based VP8* vaccines against rotavirus are highly Immunogenic in rodents. BioRxiv https://doi.org/10.1101/2023.03.29.534747 (2023).
Rahmani, A., Baee, M., Saleki, K., Moradi, S. & Nouri, H. R. Applying high throughput and comprehensive immunoinformatics approaches to design a trivalent subunit vaccine for induction of immune response against emerging human coronaviruses SARS-CoV, MERS-CoV and SARS-CoV-2. J. Biomol. Struct. Dyn. 40, 6097–6113 (2022).
Grewal, A. D. S. R. K., A., S. G. & Chawla F. C. V. K. A. R. S. L. C. M. T cell epitope based vaccine design while targeting outer capsid proteins of rotavirus strains infecting neonates: an immunoinformatics approach. J. Biomol. Struct. Dyn. 42, 4937–4955 (2024).
Li, X. F. et al. The association of post-embryo transfer SARS-CoV-2 infection with early pregnancy outcomes in in vitro fertilization: a prospective cohort study. Am. J. Obstet. Gynecol. 230, 436.e1-436.e12 (2024).
Lin, W. et al. Programmable macrophage vesicle based bionic Self-Adjuvanting vaccine for immunization against Monkeypox virus. Adv. Sci. (Weinheim Baden-Wurttemberg Ger. 12, e2408608 (2025).
Fang, W., Sun, W., Fang, W., Zhang, J. & Wang, C. Clinical features, treatment, and outcome of pembrolizumab induced cholangitis. Naunyn Schmiedebergs Arch. Pharmacol. 397, 7905–7912 (2024).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Dhanda, S. K., Gupta, S., Vir, P. & Raghava, G. P. S. Prediction of IL4 inducing peptides. Clin. Dev. Immunol. 263952 (2013). (2013).
Dhanda, S. K., Vir, P. & Raghava, G. P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct. 8, 30 (2013).
Semple, F. et al. Human beta-defensin 3 has immunosuppressive activity in vitro and in vivo. Eur. J. Immunol. 40, 1073–1078 (2010).
Doytchinova, I. A. & Flower, D. R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 8, 4 (2007).
Gupta, S. et al. Peptide toxicity prediction. Methods Mol. Biol. 1268, 143–157 (2015).
Dimitrov, I., Bangov, I., Flower, D. R. & Doytchinova, I. AllerTOP v.2–a server for in Silico prediction of allergens. J. Mol. Model. 20, 2278 (2014).
Saha, S. & Raghava, G. P. S. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 34, W202–W209 (2006).
Hebditch, M., Carballo-Amador, M. A., Charonis, S., Curtis, R. & Warwicker, J. Protein-Sol: a web tool for predicting protein solubility from sequence. Bioinformatics 33, 3098–3100 (2017).
Magnan, C. N., Randall, A. & Baldi, P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics 25, 2200–2207 (2009).
Elalouf, A. In-silico structural modeling of human immunodeficiency virus proteins. Biomed. Eng. Comput. Biol. 14, 11795972231154402 (2023).
Kim, D. E., Chivian, D. & Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531 (2004).
Colovos, C. & Yeates, T. O. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 2, 1511–1519 (1993).
Wiederstein, M. & Sippl, M. J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410 (2007).
Ponomarenko, J. et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 9, 514 (2008).
Kozakov, D. et al. The cluspro web server for protein-protein Docking. Nat. Protoc. 12, 255–278 (2017).
Laskowski, R. A. et al. Structural summaries of PDB entries. Protein Sci. 27, 129–134 (2018).
Del Conte, A. et al. RING 4.0: faster residue interaction networks with novel interaction types across over 35,000 different chemical structures. Nucleic Acids Res. gkae337 https://doi.org/10.1093/nar/gkae337 (2024).
Gosu, V., Son, S., Shin, D. & Song, K. D. Insights into the dynamic nature of the dsRNA-bound TLR3 complex. Sci. Rep. 9, 3652 (2019).
Xue, L. C., Rodrigues, J. P., Kastritis, P. L., Bonvin, A. M. & Vangone A. PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 32, 3676–3678 (2016).
Van Der Spoel, D. et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005).
Weng, G. et al. HawkDock: a web server to predict and analyze the protein–protein complex based on computational Docking and MM/GBSA. Nucleic Acids Res. 47, W322–W330 (2019).
Ylilauri, M. & Pentikäinen, O. T. MMGBSA as a tool to understand the binding affinities of Filamin–Peptide interactions. J. Chem. Inf. Model. 53, 2626–2633 (2013).
Grote, A. et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33, W526–W531 (2005).
von Heijne, G. The signal peptide. J. Membr. Biol. 115, 195–201 (1990).
Gergen, J. & Petsch, B. mRNA-Based vaccines and mode of action. Curr. Top. Microbiol. Immunol. 440, 1–30 (2022).
Xiong, Y., Zhang, Y., Wang, J. & Xiao, Y. Using 3dRNA/DNA for RNA and DNA 3D structure prediction and evaluation. Curr. Protoc. 3, e770 (2023).
Rapin, N., Lund, O. & Castiglione, F. Immune system simulation online. Bioinformatics 27, 2013–2014 (2011).
Saleki, K. et al. Engineering a novel Immunogenic chimera protein utilizing bacterial infections associated with atherosclerosis to induce a deviation in adaptive immune responses via immunoinformatics approaches. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 102, 105290 (2022).
Kaushik, V. et al. Immunoinformatics-Aided design and in vivo validation of a Peptide-Based multiepitope vaccine targeting canine circovirus. ACS Pharmacol. Transl Sci. 5, 679–691 (2022).
Akhtar, N. et al. Secreted Aspartyl Proteinases Targeted Multi-Epitope Vaccine Design for Candida dubliniensis Using Immunoinformatics. Vaccines 11, (2023).
Chawla, M. et al. Immunoinformatics-aided rational design of a multi-epitope vaccine targeting feline infectious peritonitis virus. Front Vet. Sci 10, 1280273 (2023).
Abbas, Abul, K., Andrew, H. & Lichtman, S. P. Cellular and Molecular Immunology. Elsevier (2022).
Cates, J. E., Amin, A. B., Tate, J. E., Lopman, B. & Parashar, U. Do rotavirus strains affect vaccine effectiveness?? A systematic review and Meta-analysis. Pediatr. Infect. Dis. J. 40, 1135–1143 (2021).
Noor, R. Developmental status of the potential vaccines for the mitigation of the COVID-19 pandemic and a focus on the effectiveness of the Pfizer-BioNTech and moderna mRNA vaccines. Curr. Clin. Microbiol. Rep. 8, 178–185 (2021).
Atefi, A. et al. Meningitis after COVID-19 vaccination, a systematic review of case reports and case series. BMC Infect. Dis. 24, 1138 (2024).
Esona, M. D., Gautam, R. & Rotavirus Clin. Lab. Med. 35, 363–391 (2015).
Morozova, O. V., Sashina, T. A., Fomina, S. G. & Novikova, N. A. Comparative characteristics of the VP7 and VP4 antigenic epitopes of the rotaviruses Circulating in Russia (Nizhny Novgorod) and the rotarix and RotaTeq vaccines. Arch. Virol. 160, 1693–1703 (2015).
Gote, V. et al. A comprehensive review of mRNA vaccines. Int J. Mol. Sci 24, 2700 (2023).
Rzymski, P., Szuster-Ciesielska, A., Dzieciątkowski, T., Gwenzi, W. & Fal, A. mRNA vaccines: the future of prevention of viral infections? J. Med. Virol. 95, e28572 (2023).
Chaudhary, N., Weissman, D. & Whitehead, K. A. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat. Rev. Drug Discov. 20, 817–838 (2021).
Leong, K. Y., Tham, S. K. & Poh, C. L. Revolutionizing immunization: a comprehensive review of mRNA vaccine technology and applications. Virol. J. 22, 71 (2025).
Samdan, A. et al. Hospital-based surveillance for rotavirus diarrhea in ulaanbaatar, mongolia, April 2009 through March 2016. Vaccine 36, 7883–7887 (2018).
Lata, K. S., Vaghasia, V., Bhairappanvar, S., Patel, S. & Das, J. Vaccine design against leptospirosis using an immunoinformatic approach. Methods Mol. Biol. 2131, 173–184 (2020).
Denisova, E. et al. Rotavirus capsid protein VP5* permeabilizes membranes. J. Virol. 73, 3147–3153 (1999).
Gilbert, J. M. & Greenberg, H. B. Cleavage of rhesus rotavirus VP4 after arginine 247 is essential for rotavirus-like particle-induced fusion from without. J. Virol. 72, 5323–5327 (1998).
Lizano, M., López, S. & Arias, C. F. The amino-terminal half of rotavirus SA114fM VP4 protein contains a hemagglutination domain and primes for neutralizing antibodies to the virus. J. Virol. 65, 1383–1391 (1991).
Mahmud, S. et al. Designing a multi-epitope vaccine candidate to combat MERS-CoV by employing an immunoinformatics approach. Sci. Rep. 11, 15431 (2021).
Fatoba, A. J. et al. Immunoinformatics prediction of overlapping CD8 + T-cell, IFN-γ and IL-4 inducer CD4 + T-cell and linear B-cell epitopes based vaccines against COVID-19 (SARS-CoV-2). Vaccine 39, 1111–1121 (2021).
Dong, R., Chu, Z., Yu, F. & Zha, Y. Contriving Multi-Epitope subunit of vaccine for COVID-19: immunoinformatics approaches. Front. Immunol. 11, 1784 (2020).
Haq, A. U. et al. Declaration of competing interest the authors confirms that they don’t have conflict of interest. Acknowledgement for improvement of writing used ChatGPT v.3.5. Funding Non Vaccines 9, (2021).
Ferris, L. K. et al. Human beta-defensin 3 induces maturation of human Langerhans cell-like dendritic cells: an antimicrobial peptide that functions as an endogenous adjuvant. J. Invest. Dermatol. 133, 460–468 (2013).
Dhople, V., Krukemeyer, A. & Ramamoorthy, A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim. Biophys. Acta. 1758, 1499–1512 (2006).
Ayyagari, V. S., C, T., Srirama, K. & V., K, A. P. & Design of a multi-epitope-based vaccine targeting M-protein of SARS-CoV2: an immunoinformatics approach. J. Biomol. Struct. Dyn. 40, 2963–2977 (2022).
Tarrahimofrad, H., Rahimnahal, S., Zamani, J., Jahangirian, E. & Aminzadeh, S. Designing a multi-epitope vaccine to provoke the robust immune response against influenza A H7N9. Sci. Rep. 11, 24485 (2021).
Obaidullah, A. J. et al. Immunoinformatics-guided design of a multi-epitope vaccine based on the structural proteins of severe acute respiratory syndrome coronavirus 2. RSC Adv. 11, 18103–18121 (2021).
Mohan, T., Mitra, D. & Rao, D. N. Nasal delivery of PLG microparticle encapsulated defensin peptides adjuvanted gp41 antigen confers strong and long-lasting immunoprotective response against HIV-1. Immunol. Res. 58, 139–153 (2014).
Mohan, T., Sharma, C., Bhat, A. A. & Rao, D. N. Modulation of HIV peptide antigen specific cellular immune response by synthetic α- and β-defensin peptides. Vaccine 31, 1707–1716 (2013).
Naz, A. et al. Designing Multi-Epitope vaccines to combat emerging coronavirus disease 2019 (COVID-19) by employing Immuno-Informatics approach. Front Immunol 11, 1663 (2020).
Mukherjee, S., Karmakar, S. & Babu, S. P. S. TLR2 and TLR4 mediated host immune responses in major infectious diseases: a review. Brazilian J. Infect. Dis. 20, 193–204 (2016).
Lester, S. N. & Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 426, 1246–1264 (2014).
Funderburg, N. et al. Human -defensin-3 activates professional antigen-presenting cells via Toll-like receptors 1 and 2. Proc. Natl. Acad. Sci. U S A. 104, 18631–18635 (2007).
Mukherjee, S., Huda, S. & Sinha Babu, S. P. Toll-like receptor polymorphism in host immune response to infectious diseases: A review. Scand. J. Immunol. 90, e12771 (2019).
Mukherjee, S. & Bayry, J. The Yin and Yang of TLR4 in COVID-19. Cytokine Growth Factor. Rev. 82, 70–85 (2025).
Semple, F. et al. Human β-Defensin 3 [corrected] exacerbates MDA5 but suppresses TLR3 responses to the viral molecular pattern mimic polyinosinic:polycytidylic acid. PLoS Genet. 11, e1005673 (2015).
Zhao, L. et al. Cadmium activates the innate immune system through the AIM2 inflammasome. Chem. Biol. Interact. 399, 111122 (2024).
Solanki, S. S., Singh, P., Kashyap, P., Sansi, M. S. & Ali, S. A. Promising role of defensins peptides as therapeutics to combat against viral infection. Microb. Pathog. 155, 104930 (2021).
Ding, J., Chou, Y. Y. & Chang, T. L. Defensins in viral infections. J. Innate Immun. 1, 413–420 (2009).
Semple, F. et al. Correction: Human β-D-3 exacerbates MDA5 but suppresses TLR3 responses to the viral molecular pattern mimic polyinosinic:polycytidylic acid. PLOS Genet. 12, 1 (2016).
Zhou, Y. et al. Regulatory roles of three MiRNAs on allergen mRNA expression in tyrophagus putrescentiae. Allergy 77, 469–482 (2022).
Al Tbeishat, H. Novel in Silico mRNA vaccine design exploiting proteins of M. tuberculosis that modulates host immune responses by inducing epigenetic modifications. Sci. Rep. 12, 4645 (2022).
Li, S. et al. Modified lentiviral globin gene therapy for pediatric β(0)/β(0) transfusion-dependent β-thalassemia: A single-center, single-arm pilot trial. Cell. Stem Cell. 31, 961–973e8 (2024).
Rapin, N., Lund, O., Bernaschi, M. & Castiglione, F. Computational immunology Meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One. 5, e9862 (2010).
Sun, Y. et al. Immune response after vaccination using inactivated vaccine for coronavirus disease 2019. Chin. Med. J. (Engl). 136, 1497–1499 (2023).
Aziz, S. et al. Contriving multi-epitope vaccine ensemble for Monkeypox disease using an immunoinformatics approach. Front Immunol 13, 1 (2022).
Das, N. C. et al. Designing of a novel multi-epitope peptide based vaccine against Brugia malayi: an in Silico approach. Infect. Genet. Evol. 87, 104633 (2021).
Aram, C., Alijanizadeh, P., Saleki, K. & Karami, L. Development of an ancestral DC and TLR4-inducing multi-epitope peptide vaccine against the Spike protein of SARS-CoV and SARS-CoV-2 using the advanced immunoinformatics approaches. Biochem. Biophys. Rep. 39, 101745 (2024).
Alishvandi, A. et al. Decoding virulence and resistance in Klebsiella pneumoniae: Pharmacological insights, immunological dynamics, and in Silico therapeutic strategies. Microb. Pathog. 107691 https://doi.org/10.1016/j.micpath.2025.107691 (2025).
Saleki, K. et al. Matrix metalloproteinase/Fas ligand (MMP/FasL) interaction dynamics in COVID-19: An < em > in silico study and neuroimmune perspective. Heliyon 10, (2024).
Firuzpour, F., Saleki, K., Aram, C. & Rezaei, N. Nanocarriers in glioblastoma treatment: a neuroimmunological perspective. Rev. Neurosci. https://doi.org/10.1515/revneuro-2024-0097 (2024).
Author information
Authors and Affiliations
Contributions
C.A. Conceptualization, Writing & Review, Methodology, Data analysis. L.K. Conceptualization, Project administration, Writing & Review, Software. M.M.R. Reviewing and Editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Aram, C., Karami, L. & Ranjbar, M.M. Development of a candidate mRNA vaccine based on Multi-Peptide targeting VP4 of rotavirus A: an immunoinformatics and molecular dynamics approach. Sci Rep 15, 22610 (2025). https://doi.org/10.1038/s41598-025-07433-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-07433-4
Keywords
This article is cited by
-
In silico design of a multi-epitope vaccine against NSCLC targeting T-cell exhaustion and metastasis: insights from RNA-seq and molecular dynamics simulation analysis
Network Modeling Analysis in Health Informatics and Bioinformatics (2026)
