
Abstract
Tuberous sclerosis complex (TSC) is an autosomal dominant genetic disorder characterized by the development of benign tumors and lesions in multiple organ systems. The syndrome arises from heterozygous mutations in either TSC1 or TSC2. In this study, we identified a family with a TSC1 c.363 + 668G > C mutation exhibiting diverse clinical phenotypes. The proband and affected family members exhibited multifocal nodular pneumocyte hyperplasia (MMPH), renal hamartomas, bone marrow hyperplasia, and pulmonary lymphangioleiomyomatosis (LAM), with genetic co-segregation analysis confirming the association between the mutation and the clinical phenotype. Genetic co-segregation analysis demonstrated that the TSC1 c.363 + 668G > C mutation was consistently associated with the observed clinical features in this family. Using first-generation Sanger sequencing, we identified a heterozygous splicing variation located in intron 5 of TSC1 (NM_000368.5). In vitro cell and family Minigene results show that TSC1 c.363 + 668G > C mutation can lead to abnormal retention of 92 bp intron sequence in different positions, which may be related to the alternative splicing phenomenon that the same gene produces different splicing variants in different tissues or development stages. The 3D protein structure analysis using Chimera revealed that the mutation site was located at the 363rd base, within the intron between the 121st and 122nd amino acids. This mutation resulted in the insertion of a 92-base sequence, causing a frameshift that led to premature termination of the TSC1 protein after the translation of 26 amino acids. Additionally, the 121st amino acid was altered from lysine to asparagine, significantly shortening the mutated TSC1 protein. These findings provide critical experimental evidence supporting the potential pathogenic mechanism of the TSC1 c.363 + 668G > C mutation. Future research should focus on validating this splicing abnormality in patient-derived cells or tissues and investigating its impact on protein expression and functional activity to better understand its role in disease progression.
Clinical trial number: K2024-09-144.
Similar content being viewed by others

Introduction
Tuberous sclerosis complex (TSC) is a rare autosomal dominant genetic disorder that affects multiple organ systems across all races and ethnicities1. The global incidence is estimated at approximately 1 in 6,000 to 10,000 individuals2. TSC is primarily caused by heterozygous mutations in the TSC1 (located on chromosome 9q34) or TSC2 (on chromosome 16p13) genes3, which encode the proteins hamartin and tuberin, respectively. The mutation profiles of TSC1 and TSC2 genes differ significantly. TSC1 mutations predominantly involve truncating nonsense mutations, as well as small insertions or deletions, with relatively few functionally confirmed missense mutations4,5. In contrast, TSC2 mutations are more frequently observed as missense mutations and large genomic rearrangements2. To date, over 1,800 point mutations associated with TSC have been identified. Specifically, 449 mutations in the TSC1 gene (https://www.hgmd.cf.ac.uk/ac/gene.php? gene=TSC1) and 1,432 mutations in the TSC2 gene (https://www.hgmd.cf.ac.uk/ac/gene.php? gene=TSC2) have been cataloged in the HGMD database. These pathogenic mutations often result in constitutive activation of the mammalian target of rapamycin (mTOR) pathway. This dysregulation disrupts normal cellular growth and proliferation, contributing to the development of TSC-related pathological features6. Patients with TSC mutations exhibit significant individual differences in clinical characteristics, often manifesting a wide range of multisystem involvement affecting the brain, lungs, kidneys, heart, and skin. For example, approximately two-thirds of TSC patients develop cardiac rhabdomyomas, which can appear as single or multiple lesions. In the kidneys, renal angiomyolipomas and renal cysts are common tumors. Skin manifestations frequently include hypopigmented macules, facial angiofibromas, shagreen patches, and periungual fibromas of the fingers and toes7. Additionally, neurological and mental disorders such as seizures, autism spectrum disorder, and cognitive impairment may occur.
Notably, TSC gene mutations have a strong association with lung diseases. Common clinical presentations include lymphangioleiomyomatosis (LAM), multifocal micronodular pneumocyte hyperplasia (MMPH), and chylous effusions8. The primary objective of this study is to report a novel splicing mutation in the TSC1 gene identified during clinical evaluation and to characterize the diverse clinical manifestations of tuberous sclerosis observed in the affected family. Furthermore, we conducted a preliminary investigation into the pathogenic mechanisms underlying this splicing mutation, providing new insights into its role in disease development.
Materials and methods
Research subjects
An analysis of a Han family from Fujian, China, revealed that the proband (III1) is a 44-year-old woman diagnosed with tuberous sclerosis complex (TSC1). Among her relatives, four other individuals (III3, II2, II6, and I1) were identified as carrying the TSC1 gene mutation, while nine family members (III2, III4, III5, II1, II3, II4, II5, II7, and I2) were confirmed to be healthy without the mutation. All affected individuals in this family met the clinical diagnostic criteria for TSC established in 2012, underscoring the phenotypic diversity and genetic basis of the disease9.
Relevant medical history and clinical data were collected from members of this family, including chest high-resolution computed tomography (HRCT), enhanced CT scans, magnetic resonance imaging (MRI), PET-CT, laboratory test results, and histopathological findings. A genetic pedigree of the family was constructed, and the correlation between the identified clinical phenotypes and genetic mutations was analyzed to provide insights into the pathogenic mechanisms underlying TSC.
DNA extraction and genetic mutation screening
Molecular genetic techniques were employed to detect mutations in the TSC1 and TSC2 genes. Genomic DNA was extracted from blood samples using a commercial kit (Jinyu Inspection Company, Guangzhou, China) through a column-based method. The concentration and purity of the DNA were determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) to ensure that the quality met the requirements for subsequent experiments. Specific primers were designed to target all exons and flanking introns of the TSC1 and TSC2 genes. These regions were then amplified using high-fidelity DNA polymerase. After PCR product purification, sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and analyzed by capillary electrophoresis on an ABI Prism3730 DNA Analyzer (Applied Biosystems). Sequencing data were analyzed with Sequencher software (Gene Codes Corporation, Ann Arbor, MI, USA) and compared to the human genome reference sequence (GRCh37/hg19) to identify potential mutation sites. The sequences for the forward (F-end) and reverse (R-end) primers were as follows:
F-end: tgtaaaacgacggccagtATTCTCACCAATCCATTAATTAGAT;
R-end: caggaaacagctatgaccACTCAGGGTTATTTGTTTAATCTGT.
Bioinformatics tools, such as BAMtools and GATK, were then employed to predict the pathogenicity of the newly discovered missense mutation. Additionally, known mutations were assessed for pathogenicity by referencing databases like the Human Gene Mutation Database (HGMD) and specialized TSC1/TSC2 mutation databases, which provide curated information on previously reported mutations and their clinical significance.
RT-qPCR
Total RNA was isolated from HEK293T cells using the TRIPURE ISOLATION REAGENT (11667165001, Roche, Swit) according to the manufacturer’s protocol. First-strand cDNA synthesis was performed with the SYBR Green qPCR Master (25325100, Roche) in a 20 µL reaction containing 5× All-in-One RT Mix 4 µL, RNA template 1 µg, primer mix 1 µL each of forward and reverse primers, and nuclease-free water. The reverse-transcription program was 37 °C for 10 min, 55 °C for 15 min, and 95 °C for 3 min. cDNA was diluted 1:20 in nuclease-free water, and quantitative PCR was carried out on a Bio-Rad CFX96™ system.Primer sequences are provided in Table 1.
In vitro minigene detection
Genomic DNA from the patient was used as a template to amplify the gene fragment (approximately 500 bp) containing the target mutation and its flanking sequence by PCR. The wild-type sequence was also amplified as a control. After purification, the PCR products were cloned into the pSPL3 exon capture vector (Invitrogen, Carlsbad, CA, USA) using KpnI and BamHI restriction enzymes. Following sequencing, the recombinant plasmids were transfected into HEK293T cells (ATCC, Manassas, VA, USA). After 48 h of transfection, total RNA was extracted using TRIzol reagent (Invitrogen) and treated with DNase I (Thermo Fisher Scientific) to remove any genomic DNA contamination. The RNA was then reverse transcribed into cDNA using SuperScript III reverse transcriptase (Invitrogen). PCR amplification of the cDNA was performed with carrier-specific primers, and the products were analyzed by agarose gel electrophoresis. The target band was excised from the gel, and Sanger sequencing was performed to determine the splicing pattern. Additionally, real-time quantitative PCR (qPCR) was used to assess the relative abundance of different splicing products.
Family minigene detection
RNA template, 5×All-In-One RT Mix, and DEPC water were initially dissolved on ice. A standard 20 µL reaction system was prepared containing the following components: 4 µL 5× All-In-One RT Mix, 1 µg RNA template, 1 µL Primer Mix, and DEPC water to adjust the final volume to 20 µL. The mixture was vortexed thoroughly and briefly centrifuged to collect residual solution from the tube walls. Thermal cycling conditions consisted of sequential incubations at 37℃ for 10 min, 55℃ for 15 min, and finally 95℃ for 3 min. Post-reaction processing included brief centrifugation followed by long-term storage at −20℃. Reverse-transcribed RNA samples were subsequently subjected to PCR amplification under standardized conditions, with resultant products prepared for sequencing. The specific primer sequences used were:
TSC1 PCR F: 5’-CAAGCACCTCTTTTGCCTTC-3’;
TSC1 PCR R: 5’-CATGGTCCTTGGATCCAGTC-3’.
MPLA detection
First, remove the MLPA probe and buffer, and allow them to melt at room temperature. Then, briefly centrifuge. Shake the hybridization premix to mix evenly, followed by a brief centrifugation for later use. Next, add 3.0 µL of the hybridization premix to the denatured DNA, and gently pipette up and down to ensure thorough mixing. Place the PCR tube in the PCR instrument, setting the reaction volume to 8 µL. Prepare the ligase-65 ligation reaction system and incubate the mixture at 60 °C for 16 h. Afterward, cool the mixture to 54 °C for approximately 1 min. Open the hot lid of the PCR instrument and add 32 µL of the ligase-65 mix. Place the PCR tube back into the PCR instrument, setting the reaction volume to 40 µL. On ice, prepare the PCR mix (PM) and add 10 µL of PM to the tubes containing the reaction products at room temperature. Gently pipette and mix the contents, then place the PCR tubes in the PCR instrument and set the reaction volume to 50 µL. Finally, analyze the experimental data using GeneMapper software.
Histological and immunohistochemical examination of lung specimens
Following the manufacturer’s instructions, lung tissue from patient III1 was first fixed with 10% formaldehyde. The tissue was then embedded in paraffin, and paraffin-embedded sections were stained with hematoxylin-eosin (HE) for histopathological analysis. Immunohistochemical staining was performed using Ki67 (RMA-0542, Fuzhou Mai-xin Biotech, China), CK7 (Mab-0828, Fuzhou Mai-Xin Biotechnology, China), and TIF1 (Mab-0599, Fuzhou Mai-Xin Biotechnology, China) antibodies.
Informative prediction of protein structure of TSC1 c.363 + 668G > C
The TSC1 protein sequence was downloaded from the AlphaFold Protein Structure Database (https://alphafold.com/entry/Q92574). This sequence was then visualized using Chimera software10, which allows for the detailed analysis of protein structures.
Ethics and consent
This study was approved by the Ethics Committee of Fujian Provincial Hospital (K2024-09–144). Prior to participation, all recruited family members were informed about the study’s purpose, procedures, and potential risks, and they voluntarily signed an informed consent form, ensuring their understanding and agreement to participate. Confirming that all experiments were performed in accordance with relevant guidelines and regulations.
Results
Clinical phenotype
The clinical manifestations of four patients with TSC1 gene mutations (III1, II2, III3, and II6) and nine remaining healthy relatives in the family were evaluated. All affected individuals exhibited skin manifestations in the form of shark skin spots. The family diagram, shown in Fig. 1a illustrates the relationships: circles represent women, squares represent men, and individuals with TSC1 mutations are marked in green. The proband is indicated by an arrow, and slant marks denote deceased family members. In this family, the proband (III1) primarily presented with pulmonary nodules, exhibiting the MMPH phenotype. The other affected patients (III3, II2, II6) displayed characteristics of lymphatic and renal nodules. The proband’s grandfather (I1), upon reviewing his medical history, was found to have had both MMPH and shark skin spots, and he is deceased.

(a) The family diagram of the TSC1 c.363 + 668G > C mutation reported in this study. Circles represent women, and squares represent men. Patients with the TSC1 gene mutation are marked in green. The proband is indicated by an arrow. Diagonal lines indicate death. In this family, four surviving patients show considerable clinical phenotypic heterogeneity. The proband’s condition is primarily characterized by pulmonary nodules (such as MMPH), while others exhibit lymphatic nodules (such as LAM). (b) The first-generation Sanger sequencing map of the TSC1 c.363 + 668G > C mutation. When the G→C mutation occurs, the originally unimodal signal becomes bimodal. (c) The first-generation Sanger sequencing map of the TSC1 wild type. In the absence of mutation, the signal corresponding to this base is unimodal.
The proband (III1) is a 44-year-old woman who does not exhibit any clinically significant symptoms, such as seizures or vision loss. Brain MRI revealed slight hyperintensity on both T1 and T2 sequences in the left frontal lobe, along with a mildly elevated T2-FLAIR signal. These findings were suggestive of a demyelinating lesion or ischemic focus (Fig. 2b-c). During a routine physical examination, a chest computed tomography (CT) scan revealed a ground-glass opacity in the upper lobe of the left lung, measuring approximately 3.7 cm × 1.6 cm, with truncation of the corresponding bronchi. Additionally, multiple ground-glass nodules were noted in both lungs, the largest being approximately 0.5 cm in diameter (Fig. 2d-g).

a. Shark leather spots on the back of the proband’s trunk. b, c. Brain MRI revealed slight hyperintensity on both T1 and T2 sequences in the left frontal lobe, along with a mildly elevated T2-FLAIR signal. These findings were suggestive of a demyelinating lesion or ischemic focus. d-g. Chest CT scan of the proband’s lung window showed multiple ground-glass nodules distributed across both lungs, ranging in size from 0.2 to 0.5 cm. Additionally, a mixed ground-glass density shadow was observed in the upper lobe of the left lung, with the corresponding bronchus appearing truncated. h, i. Chest CT scan of the proband’s father showed normal lung structures with no nodules or air sac lesions identified. j-l. Chest CT scan of the proband’s cousin revealed uniformly distributed air sacs of varying sizes throughout both lungs.
To further investigate the nature of the lung shadow, the patient underwent thoracoscopic surgery to remove the left upper lobe. We can notice that nodular hyperplasia of MMPH multifocal alveolar epithelial cells at ×50, ×100, ×200 and ×400 magnification have shown that alveolar epithelial cells proliferate, with most cells showing atypical features. The volume of both the cells and their nuclei increases, and pseudoinclusions or nucleoli are visible in some nuclei (Fig. 3a-d). Pathological examination confirmed the presence of multicentric lung adenocarcinoma (Fig. 3f), with the largest nodule located in the tongue segment, which was classified as invasive adenocarcinoma (90% adherent type, 7% acinar type, and 3% solid type) (Fig. 3g).

(a) Nodular hyperplasia of MMPH multifocal alveolar epithelial cells at ×50 magnification. The lung shows scattered, multifocal, well-defined small nodules. (b) MMPH multifocal nodular hyperplasia of alveolar epithelial cells at ×100 magnification. The alveolar cavity narrows or collapses, with proliferation of alveolar epithelial cells. (c) MMPH multifocal nodular hyperplasia of alveolar epithelial cells at ×200 magnification. (d) MMPH multifocal nodular hyperplasia of alveolar epithelial cells at ×400 magnification. Alveolar epithelial cells proliferate, with most cells showing atypical features. The volume of both the cells and their nuclei increases, and pseudoinclusions or nucleoli are visible in some nuclei. (e) AAH at ×200 magnification. Small, localized lesions (usually ≤ 0.5 cm) show proliferated alveolar cells, which are round, cuboidal, or low columnar in shape, with round or oval nuclei. These cells display mild to moderate dysplasia and line the alveolar walls, occasionally extending into the respiratory bronchioles. Often, gaps between cells are present, and the cells do not form continuous clusters. (f) AIS adenocarcinoma in situ at ×200 magnification. It is composed of alveolar type II epithelium or Clara cells, with cancer cells growing strictly along the original alveolar wall. There is no evidence of other growth patterns, such as interstitial infiltration, vascular or pleural infiltration, or airway spread. (g) MIA at ×200 magnification. Part of the alveolar cavity is narrowed and collapsed, with homogeneous, gentle proliferation of alveolar epithelial cells along the alveolar wall. This forms a non-mucinous adenocarcinoma with an infiltration focus measuring less than 5 mm. (h) Bone marrow biopsy lesion map. Bone marrow biopsy confirmed active bone marrow hyperplasia, predominantly erythroid hyperplasia, with young red blood cells being the most common.
The tumor invaded the lung membrane, leading to the rupture of elastic fibers, but did not extend beyond the membrane. The remaining nodules were consistent with in situ adenocarcinoma, some accompanied by atypical adenomatous hyperplasia (Fig. 3e). Most of these nodules ranged in size from 0.2 to 0.6 cm. Histological and molecular pathological examinations suggest that these nodules represent multifocal nodular alveolar epithelial hyperplasia (Fig. 3a-d). The total abdominal color Doppler ultrasound revealed multiple renal cysts and hamartomas in the patient’s left kidney. Whole-body PET-CT scans showed multiple osteogenic lesions in the cervical, thoracic, lumbar, and sacral vertebrae, as well as in the bilateral ilium and ribs (Fig. 4). Cardiac color Doppler ultrasound suggested the presence of cardiac rhabdomyoma. Bone marrow biopsy confirmed active bone marrow hyperplasia, predominantly erythroid hyperplasia, with young red blood cells being the most common (Fig. 3h). On the back of the patient’s trunk, multiple shark skin spots were observed (Fig. 2a). MRI of the patient’s head did not show any lesions consistent with cortical dysplasia, subependymal nodules, or giant cell astrocytoma. All of the above clinical manifestations meet the diagnostic criteria for Tuberous Sclerosis Complex (TSC).

PET-CT images showed multiple osteogenic lesions in cervical and thoracic vertebrae.
The proband’s father (II2), an 86-year-old male, had a tumor removed from his right kidney 20 years ago. Postoperative pathology confirmed renal cancer and revealed multiple renal cysts. Similarly, polymorphic shark skin spots were observed on his back. However, his chest CT scan did not reveal any significant findings (Fig. 2h, i) were not present, meeting only the primary and secondary diagnostic criteria for TSC.
The proband’s cousin (III3) is a 45-year-old woman who reported coughing and shortness of breath after exercise. Chest CT showed diffuse changes in the air sacs of both lungs, consistent with LAM (Fig. 2j-l). Similar to the previous two patients, shark skin spots were observed on her waist. No other TSC-related manifestations were identified. Based on these clinical findings, she meets the diagnostic criteria for TSC.
The proband’s uncle (II6) is an 80-year-old male. During a physical examination, diffuse changes in the air sacs of both lungs were noted, again suggesting LAM. Shark skin spots were also visible on his back, similar to the other affected family members.
Mutation analysis of TSC1 and TSC2 germ lines
The coding region and intron-exon junctions of TSC1 and TSC2 genes were sequenced by Sanger sequencing. Additionally, the 23 exons of TSC1 and 42 exons of TSC2 were analyzed using the MLPA method to exclude large fragment deletions or repeat mutations. In patient 1, a heterozygous splicing variation in intron 5 of the TSC1 c.363 + 668G > A (NM_000368.5) was identified. This mutation has previously been reported in a family with multifocal small nodular pulmonary cell hyperplasia. In our study, we also detected the same point mutation (c.363 + 668G > A) in intron 5 of TSC1, which results in a non-synonymous amino acid substitution (p.Gln3Glu). This mutation disrupts normal splicing, leading to a loss of function of the TSC1 protein11. Moreover, the novel mutation c.363 + 668G > C identified in this study has not been reported in the gnomAD or GGMD databases. According to the guidelines of the American College of Medical Genetics and Genomics (ACMG), this mutation is classified as pathogenic (PVS1 + PM2 + PM4 + PP1 + PP3). Further analysis showed that all affected family members (III1, III3, II2, and II6) carried the TSC1 c.363 + 668G > C mutation, while no mutation was detected in any of the healthy relatives. Additionally, the TSC1 gene mutation exhibited consistent genetic co-segregation within the family.
RT-qPCR
To assess TSC1 gene expression levels in the study cohort, we conducted quantitative PCR (qPCR) analysis on four biological samples comprising affected family members and demographically-matched healthy controls. Primer localization details are provided in Fig. 5e. Analysis of amplification data revealed significantly reduced TSC1 expression in affected individuals compared with controls, as demonstrated in Figs. 5a-d. Notably, affected subjects exhibited varying degrees of TSC1 mRNA reduction. This expression pattern strongly suggests mutation-mediated transcriptional dysregulation, potentially explaining the observed phenotypic manifestations. Mechanistically, these findings align with the functional inactivation hypothesis of TSC1, providing experimental validation for the pathogenic potential of the identified mutations in this pedigree.

a-d. Based on RT-qPCR analysis using four distinct primer sets targeting the TSC1 gene, the results reveal that patient expression levels are variably reduced compared with those of normal family members. e. T The figure illustrates the positions of the primers used in the RT qPCR experiments. f. After amplification with TSC1 PCR F/R primer in the family, the PCR amplification result is shown in the figure. g. The agarose gel electrophoresis results confirming the family minigene are presented.
In vitro splicing analysis
To evaluate the effect of the c.363 + 668G > C variation in the TSC1 gene on mRNA splicing, we performed in vitro splicing analysis. HEK293T cells were transiently transfected with plasmids containing wild-type and mutant sequences, and cell growth was observed after 24 h of culture (Fig. 6a). Total RNA was then extracted, and the target fragment (Fig. 6b) was amplified by reverse transcription PCR (RT-PCR). Agarose gel electrophoresis of the RT-PCR products revealed that the c.363 + 668G > C mutation generated additional large fragments compared to the wild-type. To confirm the specific sequence of the abnormal splicing products, we sequenced the PCR products. Sequencing results showed that, unlike the wild-type, which displayed a single peak signal (Fig. 1c), the mutation resulted in a double peak at the same base site (Fig. 1b). Further sequence analysis identified an additional 92 bp intron sequence between c.363 and c.364: GATGGCAGTACCATT GGATTCATCTGCTACAGCA CCATGCAAATT GATTTTTGTGTCTGC CAAGAA GGGTAACTC TTTTATTATC CCTAGAG (Fig. 6c). This abnormal retention of the 92 bp sequence causes a frame-shift, leading to premature translation termination and the functional inactivation of the TSC1 protein.

(a) The growth of wild-type and mutant cells for Minigene after transient transfection for 24 h. (b) After collecting the wild-type and mutant cells for precipitation, RNA was extracted and reverse-transcribed. The target gene was then amplified by PCR. (c) The amplified product was recovered and sequenced. After the c.363 + 668G > C mutation occurred, a 92 bp intron sequence was inserted between c.363 and c.364.
Family minigene detection
To investigate the mRNA splicing impact of the TSC1 c.363 + 668G > C mutation, we performed a human minigene splicing assay. The genomic region spanning the mutation site was amplified using specific primers (TSC1 PCR F/R), with amplification efficiency verified by agarose gel electrophoresis (Fig. 5f-g). Sanger sequencing revealed distinct splicing patterns: (1) Proband samples exhibited a 92-bp insertion between nucleotides 699–700 of the coding sequence (CDS) compared to healthy controls; (2) Minigene constructs containing the mutant allele showed a 92-bp retention between nucleotides 363–367 in the CDS region relative to wild-type counterparts. This aberrant splicing pattern suggests activation of a cryptic splice site in mutant samples, resulting in partial intron retention.
Informative prediction of protein structure of TSC1 c.363 + 668G > C
The protein structure diagram of TSC1 was obtained (Fig. 7a). The mutation site identified in this study occurred at the 363rd base, which is located between the 121 st and 122nd amino acids (Fig. 7b), resulting in the insertion of 92 base pairs. This insertion causes the TSC1 protein to continue translation for 26 additional amino acids before terminating prematurely (Fig. 7c). Additionally, the amino acid at position 121 was altered from lysine to asparagine (Fig. 7d). Chimera software was used to compare the structures of the TSC1 c.363 + 668G > C mutant protein and the wild-type TSC1 protein (Fig. 7e). The comparison revealed that the mutated TSC1 protein was significantly truncated.

Prediction and analysis of the 3D structure of the TSC1 protein. (a) The protein structure diagram of TSC1. (b) The local amino acid structure diagram of lysine 121 (121 LYS). The mutation occurs at the 363rd base, resulting in the insertion of a 92-base sequence into the intron between the 121 st and 122nd amino acids. (c) The predicted protein structure diagram of the TSC1 c.363 + 668G > C mutation. The red area in the diagram highlights the mutation site and the altered amino acid sequence structure. As a result, translation of the TSC1 protein continues for 26 additional amino acids before terminating prematurely. (d) The local amino acid structure of asparagine 121 (121 ASN) after the mutation. The 121 st amino acid is altered from lysine to asparagine. (e) Comparison of the structures of the TSC1 c.363 + 668G > C mutant protein and the wild-type TSC1 protein. The mutated TSC1 protein is significantly truncated.
Discussion
Studies have shown that TSC patients with the same mutation can present with diverse clinical phenotypes12,13. However, reports on varying manifestations of tuberous sclerosis caused by splicing mutations in the same TSC1 gene within a single family remain relatively limited. Notably, the TSC family reported in this study exhibits a wide range of clinical manifestations. Patient III1 presents with multifocal micronodular pulmonary cell hyperplasia (MMPH) associated with lung cancer, skin shark dermoid, cardiac rhabdomyosarcoma, multiple osteopetrotic changes, and multiple renal cysts and hamartomas. Patient II2 shows renal carcinoma, renal cysts, and shark-like skin spots. Meanwhile, patients III3 and II6 present with pulmonary LAM and skin shark dermoid spots.
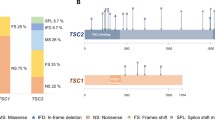
Among patients clinically diagnosed with TSC, TSC2 mutations are found in 70% of cases, while TSC1 mutations account for 20%13,14,15. The remaining 10% of cases, as reported in previous studies, involve low-level somatic mosaicism or intron splicing variations that affect either TSC1 or TSC22. Most TSC1 mutations are either short truncations or insertion/deletion (indel) mutations, which lead to loss-of-function. Only a few missense mutations with confirmed functional consequences have been identified, and these are primarily located within five specific regions of the gene5,16.
Tetsuaki Shoji et al.11, reported another mutation of the same allele, c.363 + 668G > A, found in intron 5 of TSC1, which was functionally validated. In this study, patients with the TSC1 gene mutation and MMPH phenotype exhibited MMPH, melanotic macula, seizures, cerebral cortical dysplasia, cerebral cortical nodules, and subependymal nodules. RT-PCR analysis confirmed the presence of the c.363 + 668G > A mutation in all patients, leading to an abnormal splicing process. This splicing abnormality causes a frameshift and results in the loss of function of TSC1. Specifically, the point mutation introduces a new 6 bp branch site sequence, “AATAGT”, which disrupts the normal splicing mechanism. The sequence “AG” upstream of the insertion is now recognized as the splicing acceptor site, while the two “GT” sequences downstream are treated as the splicing donor site, leading to abnormal splicing. Regardless of whether the inserted sequence length is 68–92 bp, the insertion is not a multiple of three, further contributing to the frameshift and loss of TSC1 function. In addition, the TSC1 gene plays a crucial role in tumor suppression through its regulation of the phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin pathway. Immunohistochemistry confirmed that when the TSC1 c.363 + 668G > A mutation occurs, TSC1 gene products are unable to inhibit mTOR complex 1, specifically phosphorylated p70S6K and 4E-BP1, leading to the disruption of the hamartoma protein/tuber protein complex. The family with the TSC1 mutation identified in this study carries a heterozygous c.363 + 668G > C mutation in intron 5. In vitro analysis revealed that a 92 bp intronic sequence between c.363 and c.364 may cause splicing abnormalities.
Mutations in the TSC1 gene cause cortical nodules, subependymal nodules, and abnormal central white matter, as well as various systemic manifestations outside the nervous system. For example, ocular involvement may present as hamartomas of the optic network; when the macular area is affected, decreased vision or visual field defects can occur. Cardiac involvement often manifests as rhabdomyoma, the most common tumor in fetuses and newborns with TSC1 mutations. Although these tumors are usually benign, they may cause arrhythmia or heart failure; however, most spontaneously regress during childhood. Renal involvement typically appears as angiomyolipoma (AML), the most common renal lesion in patients with TSC mutations. AMLs consist of abnormal proliferations of blood vessels, smooth muscle, and adipose tissue, which may result in bleeding, pain, or renal insufficiency. Facial angiofibroma is a characteristic skin manifestation of TSC gene mutations, usually emerging in childhood as small, reddish or skin-colored nodules. Early cutaneous signs can also include depigmented, irregular white patches common in infancy. Additionally, skin involvement may present as “shark’s patches” and periungual fibromas17. Symptoms of TSC caused by TSC1 mutations tend to be milder and may be easily overlooked13. For example, Patient 1 in this study was diagnosed with TSC following a surgical biopsy, which revealed lung abnormalities detected during a routine physical examination. Genetic testing and additional extrapulmonary manifestations further confirmed the diagnosis. Interestingly, the lung involvement was identified as MMPH in combination with lung adenocarcinoma. Currently, there are few reports on lung involvement in TSC patients with multifocal micronodular pulmonary cell hyperplasia (MMPH) and lung adenocarcinoma. Furthermore, no studies have yet explored whether TSC1/2 mutations are associated with lung adenocarcinoma or the relationship between MMPH and lung adenocarcinoma. Given these uncertainties, it is crucial for family members of individuals with TSC1/2 mutations to undergo regular screenings for related conditions. Common screening methods for TSC include brain MRI, abdominal ultrasound, and cardiac ultrasound to detect lesions in the central nervous system, kidneys, heart, and other organs. If necessary, the diagnosis can be confirmed by testing for TSC1 or TSC2 mutation18,19.
MMPH is a multifocal nodular lung cell proliferation, which is a rare lung disease of tuberous sclerosis. Chest CT shows multiple and small ground glass nodules11. Histologically, MMPH is characterized by multi-center and well-defined nodular growth of type II lung cells20. Its pathogenic mechanism is mainly due to TSC1/2 gene mutation, which leads to the weakening or loss of TSC complex’s inhibitory function on mTOR1, and then the weakening of TSC complex’s inhibitory function on mTOR1, which leads to abnormal cell proliferation caused by abnormal activation of signal transduction in mTOR1 cells. MMPH is very similar to atypical adenomatous hyperplasia (AAH) in morphology, so it is difficult to distinguish MMPH from AAH in histology21.
Cancer is a gradual process driven by the activation of oncogenes and the inactivation of tumor suppressor genes, leading to the accumulation of genetic changes over time. Studies have shown that genetic abnormalities and other immunohistochemical or morphological features observed in AAH overlap with those seen in adenocarcinoma20, which is why AAH is considered a precancerous lesion of lung adenocarcinoma22. Furthermore, loss of heterozygosity (LOH) in TSC gene-related regions is commonly found in lung adenocarcinoma with multiple AAHs23, highlighting the critical role of LOH in the development of AAH. In conclusion, we speculate that there is a causal relationship between LOH in TSC gene-related regions and the onset of lung adenocarcinoma.
Reports indicate that the prevalence of LAM in TSC patients is between 1% and 3%3, with a higher incidence observed in women. In women with TSC, the prevalence of LAM ranges from 26 to 49%, and this is closely related to age. Specifically, the prevalence in women under 21 years old is 27%, while in women over 40 years old, it increases to as high as 81%. In contrast, multifocal micronodular pneumocyte hyperplasia (MMPH) is observed in both male and female TSC patients, with no significant gender difference24,25. It is estimated that the incidence of MMPH in TSC patients ranges from 40 to 60%26,27. However, due to the wide differential diagnosis for nodules and the fact that most TSC patients with nodules are presumed to have MMPH without histopathological confirmation, determining the exact incidence of MMPH in TSC remains challenging28. In this family, the lung manifestations related to TSC include both MMPH and LAM, suggesting that the newly discovered TSC1 c.363 + 668G > C mutation in this family may be associated with heterogeneous lung manifestations.
Combining the results from qPCR and minigene experiments, our findings indicate that the TSC1 mutation impairs gene function via two main mechanisms: reduced expression and abnormal mRNA splicing. Quantitative PCR analysis revealed that TSC1 mRNA levels in patients were significantly lower than those in the normal control group. This reduction supports the hypothesis that the mutation may lead to a partial or complete loss of TSC1 function, potentially through nonsense-mediated decay and other mechanisms.
Additionally, minigene analysis demonstrated that the c.363 + 668G > C mutation disrupted normal splicing. Although all samples produced the expected amplicon, PCR sequencing showed that the proband’s transcript contained an insertion of 92 base pairs compared to those of healthy controls and the proband’s parents. This abnormal splicing event may disrupt the open reading frame or result in dysfunctional protein products, thereby contributing to disease pathogenesis. We can draw the following conclusions. First, the TSC1 c.363 + 668G > C mutation results in abnormal splicing, causing aberrant retention of intronic sequences in the mRNA and potentially leading to premature termination of protein translation. Second, qPCR analysis revealed that TSC1 mRNA levels in patients were significantly lower than those in normal controls (Fig. 5a-d), suggesting that the mutation may downregulate TSC1 gene expression by affecting mRNA stability or transcription efficiency. Finally, the functional inactivation of the TSC1 gene may underlie the diverse clinical manifestations observed in this family. As a key regulator of the mTOR pathway, loss of TSC1 protein function can lead to abnormal activation of mTOR signaling, which in turn may contribute to multi-system tumors and developmental abnormalities. In addition, in vitro experiments have revealed different splicing patterns between cells and human minigene constructs, likely reflecting alternative splicing events. Alternative splicing refers to the process by which the same pre-mRNA selectively retains or excludes certain exons or introns through various splicing mechanisms, thereby generating multiple mRNA variants. This process allows a single gene to encode multiple protein isoforms with distinct functions, significantly increasing protein diversity. The same gene may produce different splice variants in various tissues or at different developmental stages. For example, troponin T isoforms in the heart and skeletal muscle perform distinct roles. This complex biological process may involve differential splicing mechanisms and the activation of cryptic splice sites, with multiple hidden donor or acceptor sites potentially present within the same sequence29.
Analysis of the TSC1 protein structure revealed that the c.363 + 668G > C mutation results in a significant alteration, characterized by the insertion of 92 base pairs within the intronic region between the 121 st and 122nd amino acids. This mutation not only causes premature termination of TSC1 protein translation after 26 amino acids but also changes the 121 st amino acid from lysine (Lys) to asparagine (Asn). Using Chimera software to compare the mutant and wild-type protein structures, we observed a pronounced truncation in the mutant protein. These findings suggest that the truncation of the TSC1 protein may lead to a complete loss or a significant reduction of its function. TSC1 protein forms a complex with TSC2, and together they negatively regulate the mTOR signaling pathway. However, the truncated TSC1 protein may fail to bind TSC2 properly, leading to abnormal mTOR activation that promotes cell proliferation and tumor formation. Additionally, the substitution of lysine with asparagine at the 121 st amino acid could alter the local structure or function of TSC1. Lysine, a positively charged amino acid, typically participates in protein interactions and the formation of enzyme active sites, whereas asparagine is neutral; this substitution may disrupt critical functional regions of TSC1. The consequent loss of TSC1 function can result in constitutive activation of the mTOR pathway, which is associated with various pathological processes such as cell proliferation, metabolic disorders, and tumorigenesis. This aberrant mTOR activation may be a key factor underlying the development of multisystem tumors, including renal angiomyolipoma and pulmonary lymphangioleiomyomatosis, in patients with TSC1 mutations. Abnormal activation of the mTOR pathway may further alter the activity of downstream effectors, such as S6K and 4E-BP1, resulting in uncontrolled protein synthesis and cell growth. These changes could contribute to nervous system symptoms—including epilepsy and cognitive impairment—as well as tumor development in TSC patients. The protein truncation and functional loss induced by the TSC1 c.363 + 668G > C mutation likely form the molecular basis for the diverse clinical manifestations observed in this family. Moreover, variations in genetic background and environmental factors may account for differences in disease severity and organ involvement among patients.
In the future, we plan to verify the effects of the TSC1 c.363 + 668G > C mutation on protein expression and the mTOR pathway using patient-derived cells or tissues. For example, we will assess TSC1 protein levels by Western blot and analyze the activity of mTOR downstream molecules via immunohistochemistry. Based on the observed abnormal activation of the mTOR pathway, we also aim to explore the therapeutic potential of mTOR inhibitors (such as rapamycin) in patients with TSC1 mutations. In addition, strategies to restore TSC1 function—such as gene editing or protein stabilizers—warrant further investigation. Since the TSC1 c.363 + 668G > C mutation leads to protein truncation and amino acid substitution, resulting in loss of TSC1 function and subsequent abnormal mTOR pathway activation, this molecular mechanism may underlie the diverse clinical manifestations in affected patients. Future research should further validate the mutation’s impact on protein function and develop individualized mTOR-targeted treatment strategies to improve patient prognosis.
Limitations and future directions
The number of family members included in this study is small. In future studies, we plan to expand the sample size by including more TSC1 patients and families—particularly those with diverse clinical manifestations—to further validate the phenotypic diversity and pathogenic mechanisms of the TSC1 c.363 + 668G > C mutation. We also aim to investigate the relationship between the TSC1 c.363 + 668G > C mutation and lung adenocarcinoma, with a special focus on the potential role of MMPH as a precancerous lesion. Molecular pathology and genomic analyses will be employed to elucidate the mutation’s role in lung adenocarcinoma. Furthermore, patients harboring the TSC1 c.363 + 668G > C mutation will be followed for an extended period to assess disease progression, treatment efficacy, and prognosis, thereby informing more effective clinical management strategies. Additionally, we plan to establish an extensive database of patients with TSC1 gene mutations by integrating clinical, genetic, and molecular data to advance both research and clinical management of TSC1. This initiative will further our understanding of the genetic heterogeneity and phenotypic diversity of TSC1.
Conclusions
This study reports a tuberous sclerosis family exhibiting diverse clinical manifestations caused by a novel splicing mutation in the TSC1 gene. In vitro splicing analysis provided crucial experimental evidence supporting the potential pathogenic mechanism of the TSC1 c.363 + 668G > C mutation. Future research should aim to further validate this splicing abnormality in patient-derived cells or tissues and assess its impact on the expression and function of the TSC1 protein.
Data availability
The datasets generated and/or analysed during the current study are available in the manuscript, supplementary material files, the ClinVAR repository, and the accession number is RCV002274870.1 (https://www.ncbi.nlm.nih.gov/clinvar/RCV002274870.1/).
References
Crino, P. B., Nathanson, K. L. & Henske, E. P. The tuberous sclerosis complex. N Engl. J. Med. 355 (13), 1345–1356 (2006).
Henske, E. P., Jozwiak, S., Kingswood, J. C., Sampson, J. R. & Thiele, E. A. Tuberous sclerosis complex. Nat. Rev. Dis. Primers. 2, 16035 (2016).
Curatolo, P., Bombardieri, R. & Jozwiak, S. Tuberous sclerosis. Lancet 372 (9639), 657–668 (2008).
Morrison, P. J., Shepherd, C. H., Stewart, F. J. & Nevin, N. C. Prevalence of tuberous sclerosis in UK. Lancet 352 (9124), 318–319 (1998).
Mayer, K., Ballhausen, W. & Rott, H. D. Mutation screening of the entire coding regions of the TSC1 and the TSC2 gene with the protein Truncation test (PTT) identifies frequent splicing defects. Hum. Mutat. 14 (5), 401–411 (1999).
Crino, P. B. Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol. 125 (3), 317–332 (2013).
Northrup, H., Koenig, M. K., Pearson, D. A. & Au, K. S. Tuberous Sclerosis Complex. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews((R)). Seattle (WA)1993.
Rebaine, Y., Nasser, M., Girerd, B., Leroux, C. & Cottin, V. Tuberous sclerosis complex for the pulmonologist. Eur. Respir Rev. 30(161), 200348 (2021).
Northrup, H., Krueger, D. A. & International Tuberous Sclerosis Complex Consensus G. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 iinternational tuberous sclerosis complex consensus conference. Pediatr. Neurol. 49 (4), 243–254 (2013).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25 (13), 1605–1612 (2004).
Shoji, T. et al. Familial multifocal micronodular pneumocyte hyperplasia with a novel splicing mutation in TSC1: three cases in one family. PLoS One. 14 (2), e0212370 (2019).
Niida, Y. et al. Mutational analysis of TSC1 and TSC2 in Japanese patients with tuberous sclerosis complex revealed higher incidence of TSC1 patients than previously reported. J. Hum. Genet. 58 (4), 216–225 (2013).
Dabora, S. L. et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 68 (1), 64–80 (2001).
Sancak, O. et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur. J. Hum. Genet. 13 (6), 731–741 (2005).
Jones, A. C. et al. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am. J. Hum. Genet. 64 (5), 1305–1315 (1999).
Hoogeveen-Westerveld, M. et al. Functional assessment of TSC1 missense variants identified in individuals with tuberous sclerosis complex. Hum. Mutat. 33 (3), 476–479 (2012).
von Ranke, F. M. et al. Tuberous sclerosis complex: State-of-the-Art review with a focus on pulmonary involvement. Lung 193 (5), 619–627 (2015).
Krueger, D. A., Northrup, H. & International Tuberous Sclerosis Complex Consensus G. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. ;49(4):255 – 65. (2013).
Northrup, H. et al. Updated international tuberous sclerosis complex diagnostic criteria and surveillance and management recommendations. Pediatr. Neurol. 123, 50–66 (2021).
Kitamura, H. et al. Atypical adenomatous hyperplasia and Bronchoalveolar lung carcinoma. Analysis by morphometry and the expressions of p53 and carcinoembryonic antigen. Am. J. Surg. Pathol. 20 (5), 553–562 (1996).
Guinee, D. et al. Multifocal micronodular pneumocyte hyperplasia: a distinctive pulmonary manifestation of tuberous sclerosis. Mod. Pathol. 8 (9), 902–906 (1995).
Takamochi, K. et al. Loss of heterozygosity on chromosomes 9q and 16p in atypical adenomatous hyperplasia concomitant with adenocarcinoma of the lung. Am. J. Pathol. 159 (5), 1941–1948 (2001).
Suzuki, K. et al. Loss of heterozygosity in the tuberous sclerosis gene associated regions in adenocarcinoma of the lung accompanied by multiple atypical adenomatous hyperplasia. Int. J. Cancer. 79 (4), 384–389 (1998).
Lim, K. H., Silverstone, E. J. & Yates, D. H. Multifocal micronodular pneumocyte hyperplasia in tuberous sclerosis complex: resolution with everolimus treatment. Am. J. Respir Crit. Care Med. 201 (10), e76 (2020).
Moss, J., DeCastro, R., Patronas, N. J. & Taveira-DaSilva, A. Meningiomas in Lymphangioleiomyomatosis. JAMA 286 (15), 1879–1881 (2001).
Tanaka, M., Hirata, H., Wataya-Kaneda, M., Yoshida, M. & Katayama, I. Lymphangioleiomyomatosis and multifocal micronodular pneumocyte hyperplasia in Japanese patients with tuberous sclerosis complex. Respir Investig. 54 (1), 8–13 (2016).
Muzykewicz, D. A. et al. Multifocal micronodular pneumocyte hyperplasia: computed tomographic appearance and follow-up in tuberous sclerosis complex. J. Comput. Assist. Tomogr. 36 (5), 518–522 (2012).
Gupta, N. & Henske, E. P. Pulmonary manifestations in tuberous sclerosis complex. Am. J. Med. Genet. C Semin Med. Genet. 178 (3), 326–337 (2018).
Ule, J. & Blencowe, B. J. Alternative splicing regulatory networks: functions, mechanisms, and evolution. Mol. Cell. 76 (2), 329–345 (2019).
Funding
This work was supported by the National Key Clinical Specialty (Grant No. 2023002), the Fujian Province Natural Science Fund Project (2022J01417, 2022J01409, 2023J011159, 2022J01996, 2024Y0033), Joint Funds for the innovation of science and Technology in Fujian province (2023Y9284), the Fujian Province Medical Innovation Foundation (2022CXA001), Startup Fund for scientific research, Fujian Medical University (2022QH2042, 2023QH2038), National famous and old Chinese medicine experts (Xuemei Zhang, Xiaohua Yan, Lv Shaoguang, Yichun Jin) inheritance studio construction project.
Author information
Authors and Affiliations
Contributions
BSX, JWL and LZ conceived and designed the study. ZLC, ZYX and XQL, collected the data. QC, HPY, JZ and JHZ conducted the data analysis and interpretation. XJY, YL and JZ wrote the manuscript. DDR, MZG, YL, and XJY made critical revisions to the important contents of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was reviewed and approved by the Ethics Committee of Fujian Provincial Hospital, Fuzhou, China, with the approval number: K2024-09-144. All participants and legal guardians have written informed consent in the study.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yao, Xj., Lin, Y., Zou, J. et al. Genetic analysis of a novel TSC1 splice mutation causing tuberous sclerosis without neurological phenotypes. Sci Rep 15, 24780 (2025). https://doi.org/10.1038/s41598-025-07906-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-07906-6
