
Abstract
Rare genetic variants that affect host defense against SARS-CoV-2 may contribute to COVID-19 progression, helping to explain severe or fatal cases in young and middle-aged patients. This study aimed to identify rare genetic variants potentially implicated in life-threatening COVID-19 in a cohort of Brazilian patients aged 18 to 60, with no prior history of health issues, who required intensive care unit admission (n = 161). Whole genome sequencing was performed, followed by a prioritization approach for rare variants in loci previously associated with severe COVID-19. A total of 104 rare and potentially deleterious variants were identified in 79 genes. Ultra-rare variants in MUC5AC, IFNA10, ZNF778, and PTOV1 were the most frequently observed. We report 17 novel variants, including those likely pathogenic or indicating strong loss-of-function (LoF) intolerance. Patients carrying prioritized rare variants had a significantly higher incidence of acute respiratory distress syndrome (ARDS) (p = 0.027, OR = 2.59). Additionally, patients with variants in highly LoF-intolerant genes had a fourfold higher risk of death (p = 0.0084, OR = 4.04). To date, this is the first genomic analysis of previously healthy young and middle-aged Latin American patients with severe COVID-19. Our findings highlight the importance of identifying population-specific genetic risk factors.
Similar content being viewed by others


Introduction
Coronavirus disease 2019 (COVID-19) is a respiratory and systemic disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), responsible for one of the largest pandemics in history. As of April 2024, the disease had affected more than 704 million people, accounting for approximately 7 million deaths worldwide1. Despite worldwide advances in vaccination and the consequent decrease in fatal cases, new variants of concern with greater potential for vaccine escape and spread have emerged, prompting health authorities to remain vigilant for possible new waves of infection and hospitalizations2.
Most SARS-CoV-2 infections are mild, typically presenting with fever and cough, and recovery usually occurs within 2 to 3 weeks. Nonetheless, some patients may progress to severe complications, including acute respiratory distress syndrome (ARDS), septic shock, coagulation disorders, and multiple organ failure3. Several risk factors for severe COVID-19 have been described, for example, advanced age, male sex, smoking, and pre-existing conditions such as hypertension, diabetes, and cardiovascular, renal, or respiratory diseases4,5. Nonetheless, these factors do not fully explain why some previously healthy young individuals require hospitalization and ventilatory support due to COVID-196,7.
Differences in a host’s genetic profile, determined by common and rare genetic variants that influence susceptibility and clinical outcomes of COVID-19, may be decisive in explaining the variability in disease severity among patients8. The genome-wide association study (GWAS) meta-analysis conducted by the COVID-19 Host Genetics Initiative (HGI), including 219,692 cases and over 3 million controls, identified 51 distinct significant loci associated with critical illness, hospitalization, and susceptibility to SARS-CoV-2 infection9. However, this classical GWAS approach has not been applied to detect rare genetic variants that may influence the host response to SARS-CoV-2, particularly in patients exhibiting extreme COVID-19 phenotypes, such as fatal outcomes in previously healthy young and middle-aged individuals.
The COVID Human Genetic Effort (COVIDHGE) has played an important role in uncovering rare genetic factors associated with severe COVID-19. Initial studies identified inborn errors of immunity (IEIs) affecting type I interferon (IFN-I) pathways in 23 patients with severe disease, involving mutations in genes such as TLR3 and IRF710. Furthermore, COVIDHGE demonstrated that some individuals without IEIs carry neutralizing autoantibodies against IFN-I, which act as a “phenocopy” of these genetic defects, impairing the antiviral response11. The consortium also reported that approximately 1% of men under 60 developed critical pneumonia due to rare variants in the TLR7 gene on the X chromosome, a crucial viral sensor for the immune response12. Subsequent research revealed rare autosomal inborn errors of type I IFN-dependent immunity to influenza viruses that also underlie critical COVID-19 cases, particularly in individuals under 60 years of age13.
Genetic variants with the most significant influence on COVID-19 severity are likely rare. Nonetheless, not all rare variants necessarily exert a significant effect on disease severity14. Therefore, we hypothesized that an in-depth screening of loci previously associated with COVID-19 and gene- and variant-level prioritization could uncover rare variants with significant effects on disease severity and mortality.
Prior studies investigating rare variants associated with COVID-19 severity and mortality have identified genes involved in viral invasion mechanisms and molecules involved in inflammatory signaling15,16,17. Nevertheless, these studies have primarily focused on identifying genetic factors influencing the risk of death in COVID-19 patients from predominantly European populations. Host genetic factors for COVID-19 remain underexplored in Latin American populations. Due to the limited availability of genomic data from non-European and admixed individuals, studies targeting this gap are crucial. Genomic information can be integrated into clinical decision-making, influencing patient management and prognosis protocols. In this study, we conducted whole genome sequencing (WGS) on young and middle-aged Brazilian adults without pre-existing health conditions to identify rare genetic variants potentially implicated in life-threatening COVID-19.
Materials and methods
Subjects and clinical data
This retrospective cross-sectional study included 161 unrelated patients admitted to intensive care units (ICUs) in Brazil, recruited from August 2020 to September 2021. This cohort is referred here as COVID-19-BR. In this study, severe COVID-19 was defined strictly based on ICU admission, which was the primary inclusion criterion. To select individuals with extreme phenotypes, participants had to be 18 to 60 years old, have no history of chronic health conditions (e.g., obesity, cancer, diabetes, hypertension, or HIV/AIDS), and have a confirmed SARS-CoV-2 infection. The absence of chronic conditions was determined based on medical history documented at the time of hospital admission, either directly from the patient or, when not possible, from a close family member or caregiver. SARS-CoV-2 infection was primarily confirmed by molecular testing (RT-qPCR). In some cases, serological tests (IgG/IgM by immunochromatographic test or ELISA) were also considered, particularly in the early phases of the pandemic when RT-qPCR testing was not always readily available. However, serology alone was not used as the sole diagnostic criterion for acute infection. Patients were recruited from referral hospitals for COVID-19 from the following Brazilian states, which represent all regions of the country: Pernambuco, Bahia, Pará, Mato Grosso, Rio de Janeiro, and Rio Grande do Sul (Fig. 1). Patients’ electronic medical records were accessed to collect information on sex, self-reported race/ethnicity, SARS-CoV-2 test results, and clinical and laboratory findings upon hospital admission. None of the individuals had been previously vaccinated against SARS-CoV-2.

Distribution of patients in the COVID-19-BR cohort by Brazilian states.
The study proposal was submitted to the Research Ethics Committee of the different institutes involved in the study and it received approval: Aggeu Magalhães Institute/Fiocruz, Pernambuco (CAAE 36403820.2.0000.5190); Universidade Federal de Mato Grosso (CAEE 32361020.0.0000.5541); Oswaldo Cruz Institute, Nacional Infectology Institute Evandro Chagas/Fiocruz and Universidade Federal Fluminense, Rio de Janeiro (CAAE 68118417.6.0000.5248; 32169120.1.0000.5262 and 0623520.5.0000.5243); Nossa Senhora da Conceição Hospital, Rio Grande do Sul (CAAE 68118417.6.3003.5530); Universidade Federal do Pará (CAAE 33470020.0.1001.0018). The study adhered to the tenets of the Declaration of Helsinki for research involving human subjects. All patients or their legal representatives signed an informed consent form. In cases where patients were intubated or unable to provide consent due to their medical condition, consent was obtained from the individual responsible for the patient’s hospitalization, in accordance with ethical guidelines and institutional protocols.
DNA extraction and whole genome sequencing
Patients’ genomic DNA was extracted from whole blood using a ReliaPrep Blood gDNA Miniprep System (Promega®) commercial kit. The concentration and purity of DNA samples were assessed using a Qubit® DNA assay kit, with the aid of a Qubit® 2.0 fluorometer (Life Technologies). Sequencing libraries were prepared according to the Illumina DNA PCR-Free Library Prep protocol and quantified using a ProNex NGS Library Quant kit. Genomic sequencing was performed on a NovaSeq 6000® system (Illumina).
For the initial genomic data processing, we employed an established workflow that implements the Genome Analysis Toolkit (GATK) with best practices for calling small germline variants (see https://github.com/snakemake-workflows/dna-seq-gatk-variant-calling version 2.1.1). For each sample, paired-end reads were quality-checked and trimmed using Trimmomatic (parameters: LEADING:3, TRAILING:3, SLIDINGWINDOW:4:15, MINLEN:36). The remaining sequenced reads were aligned to the reference human genome (GRCh38, ENSEMBL release 98) with BWA, sorted using samtools and deduplicated using Picard. Reads were realigned in regions with identified indels to improve accuracy. Base recalibration (base quality score recalibration [BQSR]) was used to identify systematic errors in sequencing data and to recalibrate the quality scores to reflect the actual probability of error.
The next steps of the pipeline identified genetic variants (base substitutions and short indels) employing a joint call with all samples. Subsequently, hard filters were applied to reduce false positives (for Single Nucleotide Variant (SNV): quality by depth [QD] < 2.0 || Fisher strand bias [FS] > 60.0 || mapping quality [MQ] < 40.0 || mapping quality rank sum test [MQRankSum] < − 12.5 || read position rank sum test [ReadPosRankSum] < − 8.0; and for indels: QD < 2.0 || FS > 200.0 || ReadPosRankSum < − 20.0).
Ancestry inference
To conduct principal component analysis (PCA), the full dataset of unrelated individuals of the 1000 Genomes Project (phase 3), which comprises subjects with European (EUR), African (AFR), East Asian (EAS), South Asian (SAS), and Native American (AMR) ancestries, was used as a reference. This 1000 Genomes panel was merged with the genetic data of the Brazilian cohort, using autosomal variants with minor allele frequency (MAF) > 0.1 that were common to both datasets. Subsequently, the merged data were pruned using PLINK 1.9 software with a window size of 50 markers, a step size of 5, and a variance inflation factor threshold of 1.5, leaving 95,844 markers to calculate principal components with PLINK18.
The ADMIXTURE software19 was used to estimate individual ancestry of the COVID-19-BR sample. This analysis was conducted under an unsupervised mode, using the EUR, AFR, and AMR samples of the 1000 Genomes Project and the same pruning approach described above. K = 3 was assumed based on the main continental parental groups (Europeans, Africans, and Native Americans) that contributed to the formation of the Brazilian population20.
Variants prioritization
To investigate rare variants in loci previously associated with COVID-19 severity, we utilized a list of genomic regions identified by the COVID-19 Host Genetics Initiative (HGI) using the filtered dataset “COVID19_HGI_A2_ALL_leave_23andme_20220403_1e-5.tsv”. Specifically, we collected data from the GWAS meta-analysis, round 7 (very severe respiratory confirmed COVID-19 versus population controls). The total population analyzed included 18,152 cases and 1,145,546 controls. We considered only significant variants (p < 5 × 10−8) identified by the HGI and grouped these variants into loci, including a 50 kb extension in each of their flanking regions (Supplementary Table S1).
VCFtools21 was used to select in our samples variants in loci previously associated with COVID-19 by the HGI consortium. The variant effects were predicted with the Ensembl Variant Effect Predictor and the Ensembl GRCh38.p14 reference database. Variant prioritization was conducted as follows: (a) functional impact predicted as moderate to high according to the Variant Effect Predictor (Ensembl) algorithm22including non-synonymous or splice-site variants. Synonymous, intronic, and non-coding variants were excluded from the analysis; (b) MAF ≤ 0.01 in the entire dataset (ALL) from the 1000 Genomes Project and the Genome Aggregation Database (gnomAD); (c) Combined Annotation-Dependent Depletion (CADD) score > 1523; and Gene Damage Index (GDI) score < 13.8424. For variants with a presumably disruptive impact on the protein, such as splice-site, stop-gained, frameshift, stop-lost, and start-lost variants, which could be analyzed by the GDI score but not the CADD score, additional annotations were performed considering the probability of being a loss-of-function (LoF) intolerant (pLI) gene25 and the LofTool26 metrics. It is important to note that neither the pLI threshold nor the LofTool score was used as a filtering criterion during the initial variant prioritization process, but rather as complementary annotations to further characterize the identified genes and assess their potential intolerance to loss-of-function variants.
ClinVar (ncbi.nlm.nih.gov/clinvar/), AlphaMissense27 and the American College of Medical Genetics (ACMG) guidelines were used to assess the pathogenic potential of the prioritized variants. ClinVar was utilized to identify variants previously associated with clinical phenotypes in humans, while AlphaMissense provided predictive classification for missense variants. The pathogenicity assessment followed the ACMG guidelines and was performed using the Franklin software by Genoox28. Variants classified as pathogenic or likely pathogenic by at least one of these sources were included in the analysis, whereas those described as likely benign or benign were excluded.
To verify whether the variants identified in the COVID-19-BR cohort were exclusive to severe cases, we applied an additional filtering step using our internal database, which contains 39 genomes from unvaccinated Brazilian patients with mild COVID-19 symptoms, collected during the same period as the severe cases. Supplementary Figure S1 provides the characterization of these individuals.
Brazilian reference populations
The frequencies of candidate variants for severe COVID-19 identified in the COVID-19-BR cohort were investigated using two reference databases of the Brazilian population: SABE29which includes 1,171 unrelated individuals from the city of São Paulo and 61,174,462 variants, and the Variant Browser of the “DNA do Brasil” Project (http://www.dnabr.science), which has a sample size of 2,723 individuals with WGS data.
Statistical analysis
Associations between the occurrence of candidate variants for severe COVID-19 and clinical/laboratory parameters were assessed using the Mann-Whitney U test for continuous variables or the chi-square test for categorical variables. The COVID-19-BR cohort was stratified into the following two subgroups: (1) patients carrying prioritized variants and (2) patients not carrying these variants. Additionally, to investigate a potential association between candidate genetic variants and individual global ancestry, we performed a stratified analysis based on global ancestry components (EUR, AFR, AMR), categorizing individuals as above or below the median for each ancestry group. P < 0.05 was considered statistically significant.
Results
Characteristics of patients with life-threatening or fatal COVID-19
This study analyzed 161 unrelated patients, including those with life-threatening COVID-19 and individuals who did not survive the disease. The median age of the patients was 44 years (interquartile range: 37–53), with the majority being male (n = 112; 69.6%) (Supplementary Fig. S2A). The median time from symptom onset to admission was 10 days, and the median hospital stay was 13 days (Supplementary Fig. S2B; Supplementary Fig. S3A). The patients’ main symptoms upon hospitalization (Supplementary Fig. S3B) included dry cough (70.2%) and dyspnea (68.9%), followed by fever ≥ 38 °C (44.7%) and oxygen saturation below 95% (40.9%). Other frequently reported symptoms were asthenia and muscle pain, both present in 28.6% of cases. Headache was observed in 22.4% of patients. Alterations in smell and/or taste occurred in 16.77%, followed by general malaise (13.4%) and diarrhea (11.8%). As shown in Supplementary Fig. S3C, all patients required ICU admission. Of these, 93.8% required oxygen support, including nasal catheter, Venturi mask, or orotracheal intubation. Ventilatory support was required in 71.4% of cases, with orotracheal intubation alone performed in 45.3%. Additionally, 27.9% of patients required vasopressor therapy. ARDS was diagnosed in 32.9% of patients. Other complications included renal failure (8.0%), need for dialysis/hemodialysis (7.4%), shock (6.8%), and sepsis (4.9%). In total, 33 patients (20.5%) died.
Laboratory results (Supplementary Table S2) showed abnormalities that were consistent with the clinical severity observed in the cohort. The majority of patients had neutrophilia (median 8,228 cells/mm³; IQR: 5,967 to 10,557) and lymphopenia (median 869 cells/mm³; IQR: 565 to 1,188). In addition, elevated C-reactive protein levels were detected, with a median of 21.9 mg/L (IQR: 7.25 to 93.5), providing evidence of systemic inflammation.
Whole genome sequencing and ancestry analysis of COVID-19-BR patients
The median read count for the genomes of the 161 patients was 759.6 million, with a median read depth of 69.1× (Supplementary Fig. S4). Supplementary Table S3 presents a statistical summary of the SNVs and short indels identified through WGS in the COVID-19-BR cohort. The principal component analysis (PCA) of the genomic data revealed that the COVID-19-BR sample formed a heterogeneous group, distributed mainly among European (EUR), African (AFR), and Native American (AMR) reference populations (Fig. 2A). The median global ancestry results were 0.60 (IQR: 0.45 to 0.77) EUR, 0.23 (IQR: 0.11 to 0.35) AFR, and 0.10 (IQR: 0.06 to 0.16) AMR (Fig. 2B).

Ancestry analyses of the COVID-19-BR cases. (A) Principal component analysis of 161 patients from the COVID-19-BR cohort and samples from the 1000 Genomes Project. (B) Individual ancestry bar plot of COVID-19-BR using unsupervised ADMIXTURE analysis. Abbreviations – AFR: African; AMR: Native American; EUR: European; SAS: South Asian; EAS: East Asian; IQR: interquartile range (first to third quartiles).
Variant prioritization
The entire genome of patients was sequenced to identify rare genetic variants potentially implicated in COVID-19 severity. We initially identified 242,855 variants in loci previously described as being associated with COVID-19 severity by the HGI consortium. Only non-synonymous and splice variants with predicted moderate to high functional impact were selected (n = 3,625). Considering that variants with a major effect on COVID-19 severity are rare, only variants with MAF ≤ 1% in the global datasets of the 1000 Genomes and gnomAD projects were selected (n = 1,498). Variants with a CADD score greater than 15 were prioritized due to their higher likelihood of being deleterious. Additionally, genes with a GDI score below 13.84—indicating lower mutation tolerance and an increased probability of harboring pathogenic variants—were also selected (n = 140).
Finally, we checked the occurrence of these variants in the group of 39 genomes from Brazilian patients with mild COVID-19 symptoms (see ‘Methods’). This analysis revealed that 104 variants, across 79 genes, were exclusively found in severe or fatal COVID-19 cases (Fig. 3; Supplementary Table S4).

Strategy for prioritizing functional variants in COVID-19-BR cases. The numbers shown at each stage represent the remaining variants after applying the corresponding filters. Abbreviations – 1KGP: 1000 Genomes Project, Phase 3; CADD: Combined Annotation-Dependent Depletion algorithm; GDI: Gene Damage Index; gnomAD: Genome Aggregation Database version 4.1; MAF: minor allele frequency.
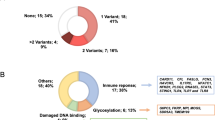
These variants were found in 89 patients (55.3%), with 35 of these patients carrying two or more variants (Fig. 4A). Most of the variants were found in a heterozygous state, with only two variants identified in a homozygous state. The majority of these variants were classified as missense, representing (41.5%) of the total (Fig. 4B). Twenty-six variants (24.5%) were classified as pathogenic or likely pathogenic based on data from ClinVar, AlphaMissense, or ACMG criteria (Table 1).

Frequency of patients and functional classification of variants in COVID-19-BR cases. (A) Distribution of patients by the number of rare variants potentially implicated in COVID-19 severity. (B) Distribution of rare variants by functional consequences.
Six variants in the MUC5AC gene, including three frameshift variants and three in-frame deletions, were identified in nine patients, all carrying at least one of these variants (Supplementary Table S4). Among these alterations, the rs1590143470 variant had a MAF below 0.0005 in reference populations, while the remaining variants were novel and not reported in reference population databases.
Furthermore, in the IFNA10 gene, a LoF variant (rs145785282) that introduces a premature stop codon, was identified in seven patients. This variant is very rare, with a MAF of 0.008 in the 1KGP and 0.009 in gnomAD. The third gene with the highest recurrence of variants was ZNF778, with three SNVs identified in six patients. Moreover, the missense variant rs563641001 (MAF = 0.009) in the PTOV1 gene was found in five patients. Variants in the ATG4D, HSD17B14, PRSS50, and RAB25 genes were observed in four patients each, while SNVs in the C4B, C6orf15, DNAJC28, DXO, and HRC genes were identified in three patients.
The variants rs45534831 (located in the DXO gene) and rs147316998 (located in the PRSS50 gene) were the only ones identified in a homozygous state, each found in a single patient. Both were classified as likely pathogenic based on the AlphaMissense prediction, reinforcing their possible role in the predisposition to severe COVID-19.
We identified 17 novel variants, with no frequency recorded in the main public databases. Among these, the following three missense variants were classified as likely pathogenic: NM_004381.5:c.1787 C > A in the ATF6B gene, NM_000258.3:c.250G > A in the MYL3 gene, and NM_020126.5:c.245 A > T in the SPHK2 gene. Furthermore, other variants showed high functional relevance, such as NM_001136.5:c.798_802del in the AGER gene, NM_003024.3:c.2304 + 1G > A in the ITSN1 gene, and two variants in the SCAF1 gene (NM_021228.3:c.619_620insGC and NM_021228.3:c.1783_1794dup), all showing a pLI > 0.9, which indicates high gene intolerance to LoF mutations.
In total, 17 variants in genes with pLI > 0.9 were identified, including: DPP9, ILF3, AGER, ITSN1, SNRNP70, SCAF1, AP2A1, KAT7, MYH14, GON4L, RXFP4, and LMNA. Among the 15 patients carrying these variants, 7 died (46.7%). Given this frequency, we compared it to the overall cohort mortality rate of 20.5% and found a statistically significant association (p = 0.0084, OR = 4.04, 95% CI = 1.35–12.13), indicating that patients with variants in high-pLI genes had a higher risk of death (Supplementary Table S5).
Candidate genetic variants to severe COVID-19 and clinical/laboratory parameters
To investigate the association between candidate genetic variants and clinical/laboratory parameters of severe COVID-19, we stratified the COVID-19-BR cohort into two subgroups: patients carrying prioritized variants (n = 89) and those without these variants (n = 72). Our analysis did not involve testing each of the 104 prioritized variants individually for association with clinical outcomes. Instead, we created a single binary variable indicating whether each patient carried at least one of the 104 previously prioritized rare variants. This variable (“carrier of ≥ 1 prioritized variant: Yes/No”) was then used to assess potential associations with clinical and laboratory features. These groups were compared in terms of demographic, clinical, and laboratory characteristics to identify potential differences that might influence the outcomes.
Importantly, there were no significant differences between the groups regarding sex distribution (p = 0.472) or age (p = 0.605), indicating that these factors, known to be potential confounders, were well balanced between the subgroups (Supplementary Table S6). Additionally, to investigate the potential association between candidate genetic variants and patients’ ancestry, we performed a stratified analysis based on global ancestry (EUR, AFR, AMR). Individuals were categorized based on the median of each ancestry group. This analysis did not reveal any significant associations between the identified variants and a specific ancestral group, suggesting that these variants are not strongly influenced by ancestry in our cohort (Supplementary Table S6).
Patients with variants potentially implicated in severe COVID-19 had a significantly higher incidence of acute respiratory distress syndrome (ARDS) compared to those without such variants (40.4% vs. 23.6%, p = 0.027, OR = 2.59, 95% CI: 1.11–6.05). Other clinical and laboratory parameters did not differ significantly between the groups (Table 2).
Discussion
Investigations into host genetic factors involved in COVID-19 are essential for advancing our understanding of the disease’s clinical progression, improving healthcare outcomes, and reducing mortality rates. These studies are expected to play a critical role in genomic and precision medicine. This is particularly important for underrepresented populations in global genomic studies and databases. In this study, WGS was performed in 161 young and middle-aged Brazilian adults with life-threatening or fatal COVID-19, aiming to identify rare genetic factors that may explain individual predisposition to disease severity. To date, this is the first genomic analysis of previously healthy young and middle-aged Latin American patients with severe COVID-19.
Genomic studies focusing on rare variants in Brazilian patients with COVID-19 remain limited. Secolin et al. (2021) reported three rare and four ultra-rare variants in four COVID-19-related genes—SLC6A20, LZTFL1, XCR1, and FURIN—that were exclusively identified in the Brazilian dataset and are predicted to affect protein function30. Other studies involving Brazilian patients and rare variants have predominantly focused on children with SARS-CoV-2–related Multisystem Inflammatory Syndrome (MIS-C)31,32,33.
A recent meta-analysis by the HGI (data release 7) identified several candidate genes that define the main biological pathways (virus entry, mucus defense, and role of interferons) involved in COVID-19 susceptibility and severity9. Their approach has provided useful information to identify specific host pathways and molecules that are important in COVID-19 pathogenesis; however, common variants have low effect sizes and explain only a very small fraction of clinical variability34.
Rare variants play unique roles in the genetics of complex diseases, as they could have a greater impact on gene function and expression as well as greater population specificity35. For this reason, we investigated rare variants in loci whose common variants have already been associated with COVID-19. Despite significant efforts to understand the biological mechanisms underlying COVID-19, the wide clinical variability between individuals remains a fundamental scientific challenge. This variability has direct implications on the identification of high-risk patients, clinical decision-making, and the development of personalized treatments.
A recent study investigated the presence of rare genetic variants in 44 patients from a Spanish cohort with very severe or fatal COVID-19 under the age of 6517. They found variants in genes related to immune response, carbohydrate metabolism, and DNA repair processes; however, most of their patients were of European descent (86%), and the inclusion criteria were not restricted to previously healthy individuals. On the other hand, our cohort included, for the first time, previously healthy patients with severe COVID-19 from all regions of Brazil. Our ancestry analyzes show a high genetic diversity20which highlights the complexity of the genetic composition of the Brazilian population. It is crucial to study diverse populations in order to identify specific genetic variations and better understand the genetic architecture of severe COVID-19.
The MUC5AC gene stood out as the most recurrent in our cohort, with nine patients presenting ultra-rare variants, including frameshift and inframe deletions. A previous study conducted in the Bulgarian population also reported a high prevalence of individuals carrying rare variants in this gene (rs36195734, rs200292517, and rs74811639), particularly among patients with critical COVID-1936. In contrast, our study identified other rare variants, including rs1590143470 and five additional novel variants. MUC5AC is one of the primary mucins produced in the airways, playing a crucial role in pathogen defense and being upregulated during respiratory infections37.These alterations may compromise the mucosal defense of the airways, thus increasing vulnerability to severe infections.
As previously suggested by other studies, the dysregulation of the type I interferon response plays an important role in COVID-19 severity, particularly in young (< 60 years) patients with severe or critical COVID-19 without comorbidities10,13,36,38. We identified the rs145785282 variant in the IFNA10 gene in seven patients, five of whom were severely affected with ARDS. This SNV was recently reported in a Brazilian child with multisystem inflammatory syndrome associated with SARS-CoV-231. Although this variant has not yet been directly implicated in COVID-19 susceptibility or severity, another variant in the same gene, rs28368148, has already been described as a risk factor for critical cases of COVID-19 (OR = 1.56; 95% CI = 1.38 to 1.77; p = 3.7 × 10⁻¹²)39. In our cohort, the rs28368148 variant was identified in five patients. However, as our goal was to identify variants exclusive to the severe COVID-19 group, this variant was excluded from the final list after applying the filtering step, since it was also present in one patient with mild COVID-19.
Seventeen new variants were identified in genes involved in different biological processes in COVID-19, such as mucosal immune response (MUC5AC, MUC21), extracellular matrix disassembly (ATF6B), regulation of molecular functions (TBC1D17, ARHGAP27, SPHK2, ITSN1, MYL3), and mitochondrial efficiency (SCAF1). We also identified a new variant in the C6orf15 gene, a functionally uncharacterized MHC gene, and a new variant in the AGER gene, linked to the requirement for mechanical ventilation and increased mortality40.
We identified candidate variants in genes that showed a high likelihood of intolerance to heterozygous LoF variation (pLI ≥ 0.9), as defined by Lek et al. (2016). This means that a single LoF variant can cause a severe clinical phenotype due to haploinsufficiency in genes such as DPP9, ILF3, AGER, ITSN1, SNRNP70, SCAF1, AP2A1, KAT7, MYH14, GON4L, RXFP4, LMNA, and GFY. Interestingly, a statistical comparison between the mortality rate of patients carrying variants in high-pLI genes and the overall cohort mortality rate showed a statistically significant association (p = 0.0084, OR = 4.04, 95% CI = 1.35–12.13), indicating that patients with variants in highly LoF-intolerant genes had a fourfold higher risk of death. In addition to allelic dosage, it is likely that the genetic effects of many rare variants contribute to the overall polygenic effect in severe COVID-1941. In this context, 52% of patients carried more than one candidate variant.
Our findings revealed that patients carrying variants potentially implicated in the severity of COVID-19 had a significantly higher incidence of ARDS compared to patients not carrying these variants. These results point to an involvement between rare high-impact genetic factors and the progression to more critical stages of the disease, which is in line with previous studies that associated specific genetic variants with exacerbated immune and inflammatory dysfunction in severe cases17,36,41.
Among the prioritized variants, 20 were classified as likely pathogenic by AlphaMissense, with a score higher than 0.6, indicating a high probability of causing functional disorders. While experimental validation remains the gold standard for confirming pathogenicity, these predictive analyses represent a valuable preliminary step in identifying candidates for further investigation27.
Some limitations should be considered. Although 89 patients (55.28%) had prioritized variants in this study, 72 did not present these variants. This may be attributed to the fact that we evaluated only significant regions identified by the HGI analyses, mainly focused on genes related to viral entry, airway mucosal defense, and response to type I interferon. These results suggest that, in patients not carrying these candidate variants, the severity of COVID-19 may be explained by loci that were not analyzed in this study or by a set of rare genetic variants with small effects that, even so, contribute to more severe clinical presentations. Additionally, while functional validation is essential for determining the true impact of genetic variants, experimentally assessing every identified variant is not feasible—particularly in studies involving rare variants across multiple genes. Our study follows a widely accepted framework for variant prioritization based on predictive annotations, facilitating the identification of potentially impactful genetic factors associated with severe COVID-19.
Our findings suggest the potential involvement of rare genetic variants in the severity of COVID-19, especially in the host immune response, as shown by ARDS, a characteristic of the most severe forms of the disease, which predisposes to an exacerbated inflammatory condition and increased demand for oxygen support. The novel variants identified in our cohort, in conjunction with the heterogeneity observed among patients, highlight the complexity of the genetic architecture of COVID-19, especially in diverse populations. Rare variants may contribute to increased susceptibility to life-threatening COVID-19; however, further studies are needed to confirm the definitive role of the variants described by this study in disease severity.
Data availability
The raw data generated from the sequencing experiments are available in the NCBI database under controlled access mode (BioProject accession number PRJNA1059130).
Change history
15 September 2025
The original online version of this Article was revised: In the original version of this Article, the last five paragraphs of the “Variant prioritization” subsection, the “Candidate genetic variants to severe COVID-19 and clinical/laboratory parameters” subsection and Table 2 were incorrectly placed at the end of the Discussion section. This has now been corrected.
References
Worldometer COVID - Coronavirus Statistics - Worldometer. (2024). https://www.worldometers.info/coronavirus/
Callaway, E. Will there be a COVID winter wave? What scientists say. Nature 610, 239–241 (2022).
Guan, W. et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl. J. Med. 382, 1708–1720 (2020).
O’Driscoll, M. et al. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 590, 140–145 (2021).
Yang, J. et al. Prevalence of comorbidities and its effects in coronavirus disease 2019 patients: A systematic review and meta-analysis. Int. J. Infect. Dis. 94, 91–95 (2020).
Zhang, Q. et al. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature 603, 587–598 (2022).
Carapito, R. et al. Identification of driver genes for critical forms of COVID-19 in a deeply phenotyped young patient cohort. Sci. Transl Med. 14, 1–20 (2022).
van der Made, C. I., Netea, M. G., van der Veerdonk, F. L. & Hoischen, A. Clinical implications of host genetic variation and susceptibility to severe or critical COVID-19. Genome Med. 14, 96 (2022).
Kanai, M. et al. A second update on mapping the human genetic architecture of COVID-19. Nat. 2023 6217977. 621, E7–E26 (2023).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370, 6515 (2020).
Bastard, P. et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370, 6515 (2020).
Asano, T. et al. X-linked recessive TLR7 deficiency in ~ 1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 6, 65 (2021).
Matuozzo, D. et al. Rare predicted loss-of-function variants of type I IFN immunity genes are associated with life-threatening COVID-19. Genome Med. 15, 1–25 (2023).
Ganna, A. et al. Quantifying the impact of rare and Ultra-rare coding variation across the phenotypic spectrum. Am. J. Hum. Genet. 102, 1204–1211 (2018).
Butler-Laporte, G. et al. Exome-wide association study to identify rare variants influencing COVID-19 outcomes: results from the host genetics initiative. PLOS Genet. 18, e1010367 (2022).
Horowitz, J. E. et al. Genome-wide analysis provides genetic evidence that ACE2 influences COVID-19 risk and yields risk scores associated with severe disease. Nat. Genet. 54, 382–392 (2022). (2022).
López-Rodríguez, R. et al. Presence of rare potential pathogenic variants in subjects under 65 years old with very severe or fatal COVID-19. Sci. Rep. |. 12, 10369 (2023).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based Estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
S. G. Kehdy, F. et al. Origin and dynamics of admixture in Brazilians and its effect on the pattern of deleterious mutations. Proc. Natl. Acad. Sci. U S A. 112, 8696–8701 (2015).
Danecek, P. et al. The variant call format and vcftools. Bioinforma Appl. NOTE. 27, 2156–2158 (2011).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 1–14 (2016).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
Itan, Y. et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc. Natl. Acad. Sci. U S A. 112, 13615–13620 (2015).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Fadista, J., Oskolkov, N., Hansson, O. & Groop, L. LoFtool: a gene intolerance score based on loss-of-function variants in 60 706 individuals. Bioinformatics 33, 471–474 (2017).
Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with alphamissense. (2023). https://doi.org/10.1126/science.adg7492
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424 (2015).
Naslavsky, M. S. et al. Whole-genome sequencing of 1,171 elderly admixed individuals from Brazil. Nat. Commun. 13, 1–11 (2022). (2022).
Secolin, R. et al. Genetic variability in COVID-19-related genes in the Brazilian population. Hum. Genome Var. 8, 1–9 (2021). (2021).
Barreto, T. M. M. et al. Rare genetic variants of NLRP12 in admixed Latino-American children with SARS-CoV-2–Related multisystem inflammatory syndrome. J. Infect. Dis. 230, 1400–1409 (2024).
Reis, B. C. S. D. et al. Rare genetic variants involved in multisystem inflammatory syndrome in children: a multicenter Brazilian cohort study. Front. Cell. Infect. Microbiol. 13, 1182257 (2023).
Santos-Rebouças, C. B. et al. Host genetic susceptibility underlying SARS-CoV-2-associated Multisystem Inflammatory Syndrome in Brazilian Children. Mol. Med. 28, 153 (2022).
Pairo-Castineira, E. et al. Genetic mechanisms of critical illness in COVID-19. Nature 591, 92–98 (2021).
Momozawa, Y. & Mizukami, K. Unique roles of rare variants in the genetics of complex diseases in humans. J. Hum. Genet. 66, 11–23 (2020). (2020).
Kamenarova, K. et al. Rare host variants in ciliary expressed genes contribute to COVID-19 severity in Bulgarian patients Veselina Koleva Acibadem City clinic, multidisciplinary hospital for active treatment ‘tokuda’ Anton Penev Acibadem City clinic, multidisciplinary hospital. (2024). https://doi.org/10.21203/rs.3.rs-4347522/v1
Chatterjee, M., van Putten, J. P. M. & Strijbis, K. Defensive properties of mucin glycoproteins during respiratory infections—relevance for sars-cov-2. MBio 11, 1–12 (2020).
van der Made, C. I., Netea, M. G., van der Veerdonk, F. L. & Hoischen, A. Clinical implications of host genetic variation and susceptibility to severe or critical COVID-19. Genome Med. 14, 1–22 (2022). (2022).
Initiative, T. C. & Ganna, A. A second update on mapping the human genetic architecture of COVID-19. medRxiv 2022.12.24.22283874 (2023). https://doi.org/10.1101/2022.12.24.22283874
Lim, A., Radujkovic, A., Weigand, M. A. & Merle, U. Soluble receptor for advanced glycation end products (sRAGE) as a biomarker of COVID-19 disease severity and indicator of the need for mechanical ventilation, ARDS and mortality. Ann. Intensive Care. 11, 1–13 (2021).
Khadzhieva, M. B. et al. COVID-19 severity: does the genetic landscape of rare variants matter? Front. Genet. 14, 1152768 (2023).
Acknowledgements
We thank the Núcleo de Bioinformática at the Aggeu Magalhães Institute for providing the computational infrastructure, technical support essential to the genomic analyses conducted in this study and Genomics Platform, P01-009, Technological Platforms Network, Oswaldo Cruz Foundation, Fiocruz.
Funding
This work was supported by the Fundação de Apoio à Fundação Oswaldo Cruz (FIOTEC), INOVA FIOCRUZ Program (project VPPCB-005-FIO-20-2-57-30, VPPCB-005-FIO-20-2-21); Foundation for the Support of Science and Technology of the State of Pernambuco (FACEPE, Brazil acronym in Portuguese) (grant numbers APQ-0422–2.02/19 and IBPG-1553-2.02/22); the Brazilian Coordination for the Improvement of Higher Education Personnel (CAPES, Brazil acronym in Portuguese), Finance Code 001; the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (grants 401235/2020-3; 302935/2021-5; 444181/2023-7); R.D.S. and L.R.S.V received scientific productivity scholarship grant from CNPq (309750/2020-2 and 311048/2022-6, respectively); R.F.C. has a scientific productivity scholarship grant from the FACEPE (BPP-0032-2.02/24).
Author information
Authors and Affiliations
Contributions
R.C.F. and L.R.S.V. contributed to the conception and design of the study. S.L.G.G., A.S.S., P.M., R.B.S.P., N.M.T., V.S.B., S.N., I. B. S., J. R. C., M.V.B.O.S., E.H. R., J.R.A., A.A.S, T.B., A.C.C., A.C.R.V., R.S., E.J.M.S., C.C.G., R.D.S., and A.C.A contributed to biological sample collection and acquisition of medical data. P.R.S.O., R.C.F., L.R.S.V., and T.L.C. contributed to sequencing data acquisition. P.R.S.O. contributed with ancestry analysis. G.D.R performed the bioinformatic and statistical analysis. G.D.R, P.R.S.O, R.C.F. contributed to the interpretation and analysis of the genetic data. G.D.R and P.R.S.O wrote the original draft. All authors were involved in the reviewing and editing of the manuscript and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rocha, G.D., Oliveira, P.R.S., de Oliveira Sá, M.V.B. et al. Rare genetic variants and severe COVID-19 in previously healthy admixed Latin American adults. Sci Rep 15, 23074 (2025). https://doi.org/10.1038/s41598-025-08416-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-08416-1
