
Abstract
Glycosylation is a post-translational modification essential for proper protein folding and function, with significant roles in diverse biological processes, including neurogenesis. MAN2A2 enzyme is required for proper N-glycan trimming/maturation in the N-glycosylation pathway. Whole-exome sequencing of a trio revealed two potentially causative variants in the MAN2A2 gene in a patient with autism spectrum disorder (ASD) and cognitive delay. The first variant, c.1679G > A (p.Arg560Gln), was inherited from the unaffected father. It is located within the alpha-mannosidase middle functional domain, a region essential for mannose metabolism and alpha-mannosidase enzymatic activity. The second variant, c.3292C > T (p.Gln1098Ter), was inherited from the mother and it generated a premature stop codon. These variants resulted in a compound heterozygous condition in the patient. Prediction using the DOMINO tool suggested an autosomal recessive inheritance pattern. Notably, the MAN2A2 gene is highly expressed in several brain regions. The encoded enzyme, an alpha-mannosidase, is localized to the Golgi apparatus, the cellular organelle where the processing and maturation of N-glycans occurs. In silico analyses consistently classified both variants as likely pathogenic, supported by structural prediction analyses that indicated significant disruptions in protein architecture. Glycosylation analyses demonstrated impaired N-glycosylation, evidenced by the accumulation of immature serum glycoprotein N-glycans including disease-specific hybrid-type species. Further investigations are essential to elucidate the role of this gene in ASD and cognitive delay.
Similar content being viewed by others
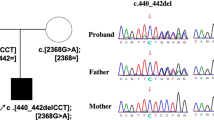

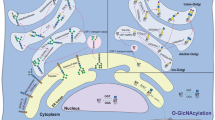
Introduction
Protein glycosylation is a post-translational modification where single monosaccharides or oligosaccharides chains (glycans) are added to proteins, impacting their structure and function. This process plays crucial roles in protein folding and is indispensable for proteins to attain their native structure and acquire traits like solubility, antigenicity, and longevity. Nearly half of the total body proteins, particularly those on cell surfaces and secreted, undergo glycosylation facilitating recognition between cells and with the extracellular matrix1. Glycosylation executes a pivotal role in neurodevelopment, influencing processes like cell signalling, adhesion, and synaptic plasticity. Dysregulation of glycosylation pathways has been implicated in neurodevelopmental disorders, underscoring its significance in brain development and function2,3. Genetic variants in glycosylation pathways that alter glycosylation may result in congenital disorders of glycosylation (CDG) a group of inborn errors of metabolism with prominent central nervous system involvement4. Moreover, aberrant glycosylation contributes to complex disorders such as autism spectrum disorder (ASD), intellectual disability, and schizophrenia5,6,7.
Alpha-Mannosidase II, MAN2A1 and MAN2A2 isozymes, convert precursor oligomannose N-glycans to mature complex-type glycans in the medial Golgi by specifically removing two mannosyl residues from GlcNAc1Man5GlcNAc2 to produce GlcNAc1Man3GlcNAc2 (Fig. 1).

Schematic showing the N-glycan processing pathway including the biosynthetic step catalyzed from MAN2A1 and MAN2A2. Glycan symbols: N-acetylglucosamine, blue square; mannose, green circle.
This is vital for N-glycosylation in the secretory pathway as illustrated by double null Man2a1/ Man2a2 mice which die prematurely and completely lack complex N-glycans8. Tissue specificity and functions of alpha-mannosidase II enzymes have been investigated by respective mice models. Man2a1 null mice synthesize complex N-glycans in most tissues and show a phenotype similar to human congenital dyserythropoietic anemia type II (alpha-mannosidase II deficiency, OMIM 154582) characterized by the lack of complex N-glycans in the erythrocyte surface. Tissue specificity of MAN2A1 deficiency relies on the complementary activity of MAN2A2 enzyme showing identical substrate specificity towards N-glycan structures8. Man2a2 null mice are infertile and are otherwise apparently normal. Aberrant N-glycan structures hamper spermatogenic cells to adhere to Sertoli cells, thus implicating the pivotal role of specific carbohydrate moiety, N-acetylglucosamine-terminated tri-antennary and fucosylated N-glycan structure, for spermatogenesis9. However, the role of MAN2A2 in cells other than spermatogenic germ cells remains unclear. The crystal structure of Golgi α-mannosidase II from Drosophila melanogaster revealed the presence of several structural domains. This enzyme has been extensively characterized in both its unbound state and in complex with the anti-cancer agent swainsonine and the inhibitor deoxymannojirimycin10. The identified functional domains include: the Glycosyl hydrolase family 38 N-terminal domain (IPR000602), the Alpha-mannosidase middle domain (IPR015341), the Lysosomal alpha-mannosidase-like central domain (IPR048534), the Glycosyl hydrolase family 38 C-terminal domain (IPR000602), and the Glycosyl hydrolase family 38 C-terminal sub-domain (IPR041566).
Recently, a novel congenital disorder of glycosylation (CDG) has been identified in two siblings with a homozygous truncating variant p.Val1101Ter in MAN2A2 (MAN2A2-CDG) presenting with prominent neurological involvement, encompassing neurodevelopmental disturbance, psychiatric issues and dysmorphisms. In-depth glycosylation analyses of patients’ lymphoblasts showed distinct changes consistent with MAN2A2 deficiency11.
The present study provides a comprehensive description of the clinical, molecular and glycosylation findings in a novel patient with novel MAN2A2 variants. Furthermore, it revises the patient with the previously reported cases11 in the context of MAN2A2 function within neurodevelopment.
Results
Clinical report
The now 16-year-old patient was born full term by spontaneous vaginal delivery, after uneventful pregnancy to a mother affected with borderline personality disorder. His neonatal history was unremarkable, except for patent foramen ovale. At age 12 months, developmental delay, lack of contact, and apathetic behaviour were evident. The patient was able to walk independently at 1 year and 6 months, he started speaking his first words at the age of 3 years. Upon entering school, he developed learning and attention disorders, behavioral complaints and difficulties in social interaction. He was formally diagnosed with autism spectrum disorder (ASD) and moderate Intellectual Disability (ID). Due to the presence of aggressive behavior, he underwent antipsychotic therapy (risperidone), discontinued due to excessive drowsiness. At study time, pertinent physical findings included below average height (3° pc), and dysmorphic facial features with thick eyebrows, high nasal bridge, prominent columella, beaked nose, full lips. He had right-convex scoliosis of the dorsal-lumbar spine. Abdomen ultrasound showed biliary lithiasis and was otherwise normal. The neuropsychological evaluation highlighted severe intellectual disability (full scale IQ:35 on the Wechsler Intelligent Scale for Children, fourth edition) moderate adaptive functioning with respect to communication, but profoundly low as to socialization skills. The patient had moderate-severe autism clinical symptoms (Autism Diagnostic Observation Schedule-Second Edition-ADOS-2 total score:8). Neurological examination showed impaired gross and fine motor skills, attentional and inhibitory dysfunctions and recurrent repetitive behaviours with varying intensity depending on his social interactional context. His expressive verbal language was also, repetitive, and non-contextualized and his facial expressions were often inadequate. Mood was described by caregivers as very unstable, characterized by sudden irritability and aggressive acting out.
Next generation sequencing
WES analysis identified two heterozygous variants within MAN2A2 gene (NM_006122) localized in chromosome 15. The first variation (c.1679G > A), was inherited by the father and it was a missense mutation involving the change of a Arginine (Arg) with Glutamine (Gln) at position 560 (p.Arg560Gln).
Contrarily, the second variant (c.3292C > T) was inherited from the mother and it generated a premature stop codon at position 1098 (p.Gln1098Ter). Both the identified variants were confirmed by conventional Sanger sequencing. The first variant was located within the alpha mannosidase middle domain (from aa 502 to aa 605). In contrast, the second variant was localized near the site of glycosylation Asn1093. Both the variants have been submitted to ClinVar database with the accession entries VCV003766448.1 and VCV003766447.1, for and the variants p.Arg560Gln and p.Gln1098Ter respectively.
Multiple in silico algorithms described both the variants as likely pathogenic (Table S1). Notably, diverse tools classified both the variants as pathogenic. According to GnomAD database, the allele frequency exhibited a very low value (0.00000137) for the variant c.1679G > A, while it was not found for the truncating variant c.3292C > T (Supplementary File 1, Table S1).
According to DOMINO inheritance prediction tool, MAN2A2 shows a very like recessive inheritance pattern with a predicted score of 0.058. Both variants identified exhibited high conservation parameters in PhastCons100way and PhyloP100way analyses, confirming the high conservation of the mutated sites (Table 1).
The structure prediction analysis using the AlphaFold3 algorithm revealed differences between the mutated and wild-type protein models. AlphaFold3 generated five models for each predicted protein. Figure S1 in Supplementary File 1 illustrates the alignment of all predicted protein structures and highlights the best-selected model.
The comparison between wild-type Arg560 and mutated Gln560 revealed important differences in hydrogen bond patterns. The complete set of hydrogen bonds identified across all five AlphaFold3-predicted models for the wild-type both the mutated MAN2A2 proteins is shown in Supplementary File 2. Figure 2 illustrates the predicted models of both wild-type and mutated Arg560Gln MAN2A2 proteins, highlighting functional domains, N-linked glycosylation sites and the nucleophile site Asn95 in different colors.

Structural prediction analysis of wild-type and Arg560Gln-mutated MAN2A2 proteins. (a) Graphical representation of the wild-type MAN2A2 protein. (b) Graphical representation of the Arg560Gln-mutated MAN2A2 protein. In both the Figures the panel highlights different functional domains and regions using distinct colors: blue for extra domain regions, orange for the N-terminal catalytic domain of Golgi alpha-mannosidase IIx (residues 165–508), green for the Alpha-mannosidase middle domain (502–605), and red for the Glycosyl hydrolases family 38 C-terminal domain (765–970). N-linked glycosylation sites are marked with pink squares, while the nucleophile site Asn95 is indicated by a dark gray circle. (c) Close-up view of the wild-type Arg560 residue (cyan), showing seven hydrogen bonds: three with Glu529, one with Gly564, one with Leu556, and one with Thr1059. (d) Close-up view of the mutated Gln560 residue (cyan), forming three hydrogen bonds with Glu529, Gly564, and Leu556. (e) Graphical representation of hydrogen bonds within the Alpha-mannosidase middle domain (502–605) in the wild-type protein. (f) Graphical representation of hydrogen bonds within the Alpha-mannosidase middle domain (502–605) in the Arg560Gln-mutated protein.
In the N-linked glycosylation sites (Asn95, Asn305, Asn1093, and Asn1131), the structure prediction analysis did not reveal any differences in the hydrogen bonding patterns between the wild-type and mutated Arg560Gln proteins. Conversely, the nucleophile site Asp289 formed an additional hydrogen bond with Arg313 in the mutated Arg560Gln protein compared to the wild-type. The predicted hydrogen bonding patterns of the N-linked glycosylation sites are illustrated in Supplementary File 1, Figure S2.
Focusing on the missense mutation site, the wild-type Arg560 formed seven hydrogen bonds: three with Glu529, one with Gly564, one with Leu556, and one with Thr1059 (Fig. 1). In contrast, the mutated Gln560 formed only three hydrogen bonds with Glu569, Gly564, and Leu556. Table 2 presents a comprehensive comparison of hydrogen bonding networks between wild-type and Arg560Gln mutant MAN2A2 within the alpha-mannosidase middle domain (residues 502–605).
This functionally critical domain, annotated in InterPro (IPR015341), mediates both mannose metabolic process (GO:0006013) and in alpha-mannosidase activity (GO:0004559). The analysis includes all five predicted models for each variant to ensure statistical rigor in identifying mutation-induced bonding changes. A comprehensive comparison of hydrogen bonding patterns across the entire alpha-mannosidase middle domain (residues 502–605) is available in Supplementary File 3.
Comparison of MAN2A2 and MAN2A1 expression database analysis
The human brain expression profiles of MAN2A2 and MAN2A1 were obtained from the BrainSPAN and BrainRNAseq databases. According to BrainSPAN, both genes are expressed across various cerebral structures, sharing similar localization but differing in expression levels. Notably, MAN2A2 exhibited higher expression in multiple brain regions, with peak levels observed across the ages of 13 and 40 years, compared to MAN2A1 (Fig. 3).

Heatmaps of MAN2A2 and MAN2A1 expressions in the human brain. (a) Heatmap depicting the MAN2A2 expression profile (measured in reads per kilobase per million, RPKM) across various brain regions at different developmental stages, indicated by post-conceptional weeks (pcw). (b) Heatmap illustrating the MAN2A1 expression profile across brain structures, also represented in RPKM and categorized by donor age in post-conceptional weeks (pcw). Data were obtained from the BrainSPAN database, by various donors with different ages.
The BrainRNAseq database also confirmed a higher expression of MAN2A2 compared to MAN2A1 in the human brain. Notably, MAN2A2 exhibited the highest expression in human neurons (Fig. 4).

Comparison of MAN2A2 (red) and MAN2A1 (blue) expression profiles in human brain cells. Data were obtained from the BrainRNAseq database and are expressed in fragments per kilobase million (FPKM).
Glycosylation analyses
Serum transferrin glycoform analyses
Transferrin analyses by CE showed a main component corresponding to fully sialilated 4-sialo transferrin (84%; n.v.84.0 ± 1.1%) and normal amounts of 3-sialo (3%; n.v. 4,5 ± 1,9%) and 2-sialo (0,7%; n. v. 0.6 ± 0,5%) indicating normal serum transferrin glycosylation.
Serum N-glycosylation analysis by MALDI-TOF MS
MALDI-TOF MS profile of patient serum N-linked glycans showed a mildly abnormal pattern with respect to three age-matched healthy controls (Supplementary File 1, Figure S3). Both control and MAN2A2 spectra are dominated by the biantennary disialo-glycoform with the complex-type structures accounting for 95–96% of the total N-glycans in the controls. No substantial change in the overall amount of complex-type N-glycans was observed for our MAN2A2 patient. Furthermore, differences were found in the relative amount of truncated species, which increased in the patient to 44% (reference range 32–34%), mostly due to a slight total increase of biantennary mono-sialylated, mono-galactosilated and a-galactosylated structures. Conversely, the fully sialylated complex N-glycans were reduced from 61–64% to 50%. In addition, the oligomannose glycoforms Man5GlcNAc2 (Man 5) and Man6GlcNAc2 (Man 6), belonging to a minor population of the total serum N-glycans (< 4%), were found slightly increased. Besides these species, a plethora of minor peaks, including a number of disease-specific hybrid structures were also detectable in serum glycosylation profiles, though the relative changes were difficult to investigate due to their very low intensity (< 1% of the total N-glycan content).
Serum N-glycosylation analysis by UPLC-ESI MS
To gain more insights into relative variations of diagnostic low abundance N-glycan structures, we performed serum N-glycosylation analysis by HILIC-UPLC-ESI MS after PNGase release, RFMS derivatization and purification of the whole N-glycans pool12.
Figure 5a shows the superimposed Extracted Ion Chromatograms (EICs) of the oligomannose structures from two age-matched controls and from MAN2A2 defective patient, this last showing increased intensity of all the Man5-9GlcNAc2 structures, especially as regards Man 5 (m/z 773.811) and Man 6 (m/z 854.838), as previously observed by MALDI MS analysis.

UHPLC-ESI MS analysis of MAN2A2 diagnostic species in patient serum sample compared to two age-matched controls. (a) Overlaid Extracted ion chromatograms (EICs) of RFMS labelled oligomannose serum N-glycans showing in the patient (red line) the increase of all the species from Man 5 to Man 9 with respect to controls (blue and green line). (b) Overlaid EICs of RFMS-labelled hybrid-type N-glycans with their major assignments, showing in the patient the accumulation of some specific disease-associated structures (as highlighted by red circles), especially the penta-mannosylated ones with shorter elongations at α1-3 mannose branch. Each chromatographic profile is normalized with respect to the dominant disialo-biantennary structure. Glycan symbols: N-acetylglucosamine, blue square; mannose, green circle; galactose: yellow circle; sialic acid: purple diamond (+ 45°: α2-6 linked; -45°: α2-3 linked); fucose: red triangle.
The superimposed EICs associated to the hybrid-type glycans in Fig. 5b show in the patient a remarkable accumulation of Man5GlcNAc3 (m/z 875.351), the disease-specific pentamannosylated structure with the α1-3 antenna bearing only the GlcNAc residue, together with Man5GlcNAc3Fuc1 (m/z 948.380) corresponding to its fucosylated analog. Minor increase in the same structures with an additional galactose unit (Gal1Man5GlcNAc3; m/z 956.377) and its fucosylated analog (Gal1Man5GlcNAc3Fuc1; m/z 1029.406) were also detected. Conversely, major hybrid-type sialylated structures did not accumulate in patient serum with respect to the controls, with the exception of two low abundant fucosylated species at m/z 1039.928 (NeuAc1Gal1Man4GlcNAc3Fuc1) and at m/z 1174.954 (NeuAc1Gal1Man5GlcNAc3Fuc1) owning four and five mannose residues respectively.
Discussion
As extensively documented, disruption of glycosylation process may significantly impact neurodevelopmental and cognitive functions, potentially contributing to ASD and intellectual disability5,6. In this study, we describe clinical, molecular and glycosylation insights of an adolescent patient diagnosed with autism spectrum disorder (ASD) and low cognitive functioning. WES Trio analysis identified two heterozygous variants in the MAN2A2 gene, inherited from each parent, resulting in a compound heterozygous condition. This gene follows an autosomal recessive inheritance pattern. NGS analysis didn’t reveal any genetic variants in additional genes associated with patient’s phenotype. However, it is important to note that the contribution of other genetic variants undetectable by WES cannot be entirely excluded as potential contributors to the patient phenotype. Furthermore, clinical and serum glycosylation analyses are consistent with MAN2A2 deficiency in the present patient.
Only two affected brothers have been reported so far with a homozygous truncating variant p.Val1101Ter in MAN2A211 (Supplementary File 1, Table S2). Compared to the reported sibs, our patient showed similar minor facial dysmorphisms (thick eyebrows, broad nose, full lips) and below average height. Likewise, the elder brother previously described11, the present patient developed speech delay, social difficulties in childhood and unstable mood with aggressive behaviour managed with pharmacological antipsychotic treatment. Altogether, previously reported11 and the present patient show that MAN2A2-CDG is characterized by neurodevelopmental disturbance with mild/moderate intellectual disability, autism spectrum disorder, and additional psychiatric issues such as bipolar disorder, social withdrawal and emotive dysregulation thus supporting a role for MAN2A2 in the CNS. Evidence from previous studies11, in conjunction with the present results, indicates that in patients with MAN2A2-CDG, multisystem involvement, which are prevalent in the majority of CDGs, are typically absent or minimal. MAN2A2 is primarily expressed in the brain, with almost fourfold higher expression than MAN2A1 in humans11,13 According to the BrainSPAN database, MAN2A2 exhibits higher expression levels across several brain regions than MAN2A1, particularly between the ages of 13 and 40 years (Fig. 3). Furthermore, data from the BrainRNAseq database confirm this trend, showing a higher expression of MAN2A2 in neurons compared to MAN2A1 (Fig. 4).
MALDI MS analysis showed no substantial decrease in the overall amount of complex species in serum, as observed for lymphoblasts from the described brothers with a homozygous variant p.Val1101Ter11. This may suggest a minor impact of this novel MAN2A2 variants (p.Arg560Gln and p.Gln1098Ter) on serum protein glycosylation. On the other hand, we observed an increase of major biantennary truncated N-glycans lacking terminal sialic acid(s) and/or galactose and the reduction of the fully sialylated species. Such structural changes, although reliable, do not appear to be directly connected to MAN2A2 deficiency.
Additional investigations by the more sensitive MS strategy based on RFSM labelling of serum N-glycans and subsequent analysis by HILIC UHPLC-ESI MS, allowed us to improve our diagnostic capability, revealing also accumulation of glycoforms present in very low abundance as Man 5, Man 6 and additional oligomannose species ranging from Man 7 to Man 9, together with an evident enhancement of disease-specific N-glycans with special regard to pentamannosylated hybrid-type asialo-structures. The slight accumulation of unprocessed oligomannose glycans may be explained with a decreased minor dysregulation of MGAT1 and the other mannosidases upstream of MAN2A2 (Fig. 1). A significant accumulation of pentamannosylated hybrid N-glycans was found also in lymphoblasts from the younger brother with homozygous variant p.Val1101Ter, whereas no significant increase was observed for the oligomannose species in both these patients11. Previous research has shown that inhibition of mannosidases may result in accumulation of oligomannose structure and disarrangement of complex type N-glycan synthesis14,15. Glycosylation of liver PTPRJ, a member of protein tyrosine phosphatases (PTPs), in mice under high fat intake showed an abundance of oligomannose structures, decrease of complex fucosylated as well as sialylated bi- and triantennary N-glycans that coincided with a reduction in MAN2A1 protein16.
The presence of subtle serum glycosylation changes in the present patient is consistent with glycome profiles in Man2a2-null mice and points to the roles and tissue specificities of MAN2A1/MAN2A2 enzymes. As previously demonstrated by MALDI-TOF MS analyses8, in both wild-type and Man2a2-null mice N-glycans are mainly complex-type. They also contained oligomannose type but not significant levels of hybrid-type N-glycans. In contrast, Man2a1-null embryos showed high levels of hybrid-type N-glycans and reduced levels of complex-type N-glycans. Double-null embryos had a significant increase of hybrid-type N-glycans and lack complex-type N-glycans.
Evidence has been provided that the MAN2A1 enzyme is likely the major contributor to N-glycan processing in many cell types, with MAN2A2 playing a subsidiary role in this process in mouse tissues8. Despite the role of MAN2A1 in most tissues, it is plausible that even subtle N-glycan changes caused by MAN2A2 deficiency may be detrimental to the SNC in patients with MAN2A2-CDG.
Exploration by cerebrospinal fluid (CSF) glycome profiling and/or autoptic brain tissues from MAN2A2-CDG patients or investigations of human iPSC derived neurons or brain organoids may address this issue.
The structure prediction analysis carried out using AlphaFold 3 algorithm revealed significant structural differences between the wild-type MAN2A2 protein, and the mutated Arg560Gln (Fig. 1). The prediction revealed significant alterations in the hydrogen bond pattern at position 560. Specifically, the mutated Gln560 lost four hydrogen bonds compared to the wild-type Arg560, including its interaction with Thr1059. Notably, this mutation disrupted the hydrogen bond network within the annotated Alpha-mannosidase middle domain (502–605) (IPR015341), a region essential for mannose metabolism (GO:0006013) and alpha-mannosidase activity (GO:0004559). Within this context, Table 2 illustrates the differences in hydrogen bonding observed in the mutated protein within this specific domain that may represent structural adaptations to the mutation, potentially influencing enzyme stability or function.
According to the UniProt database, the Arg560 residue is highly conserved across various vertebrates, including Homo sapiens, Mus musculus, Rattus norvegicus, Macaca mulatta, Macaca fascicularis, Sus scrofa, Bos taurus, and Castor canadensis. Notably, the overall MAN2A2 protein sequence exhibits a high degree of conservation, with identity percentages ranging from 89.73% in M. musculus to 98.09% in both M. mulatta and M. fascicularis. The identity matrix of the above-mentioned protein as well as the close up on the residue Arg560 is illustrated in Supplementary File 1, Figure S4.
By performing a structural alignment between our best prediction model of the wild-type MAN2A2 enzyme and the crystal structure of α-mannosidase II from Drosophila melanogaster (PDB ID: 1HTY)10, we observed a low degree of structural divergence. As expected, not all residues aligned perfectly. A total of 799 residues were aligned (out of 1015 in 1HTY and 1150 in our MAN2A2 model), resulting in a root-mean-square deviation (RMSD) of 0.773 Å, indicating a high degree of structural similarity. A visual representation of the D. melanogaster 1HTY model, along with the structural overlay of the two compared models, is provided in Figure S5.
Conversely, the sequence similarity analysis revealed that MAN2A2 shares 54.08% identity with MAN2A1. Notably, the amino acid Arg560 is conserved in MAN2A1.
In alignment with the structure prediction analyses the hydrogen bonds associated with the mutated Gln560 residue exhibited a notable variation compared to the wild-type Arg560. Furthermore, MuPRO tool indicated a decreased protein stability for both the observed variants. The variant p.Gln1098Ter, being a truncating variant, suggests a potential scenario of incomplete protein translation. Particularly noteworthy is its proximity to the glycosylation site at position Asn 1093. The premature stop codon results in the non-translation of the latter portion of the MAN2A2 protein, thereby affecting another glycosylation site at position 1131, corresponding to N-linked asparagine. As documented, mRNAs with premature termination codons (PTCs) are typically degraded through nonsense-mediated mRNA decay (NMD)17,18. Western blot analysis would be necessary to confirm the presence of the truncated protein in the patient’s fibroblasts; however, this was not feasible in the present study.
Further analyses are needed to validate the protein function, in particular we underscore that functional studies in in vivo models are necessary for confirming the effect of both variant in the protein structure and in patient’s phenotype.
Materials and methods
Next generation sequencing
This study was conducted in accordance with the Declaration of Helsinki 1964 and approved by the local Ethics Committee “Comitato Etico IRCCS Sicilia-Oasi Maria SS”, Prot. CE/193, as of 5 April 2022, approval code: 2022/04/05/CE-IRCCS-OASI/52. Written informed consent has been obtained from the patient and both the parents. Genomic DNA was extracted from peripheral blood leukocytes retrieved from the patient and both the parents, as previously described19. Library preparation (TRIOS) and exome enrichment were conducted adopting the Agilent SureSelect V7 kit in accordance to manufacturer instructions. The sequencing run was performed on Illumina HiSeq 3000—Agilent SureSelect V7 instrument. This approach achieved 97% of regions covered at least 20x. The reference genome used for the alignment was HG38. The identified variants were filtered in alignment to the following criteria: (i) recessive/de novo/X-linked pattern of inheritance, (ii) allele frequencies (minor allele frequency, MAF) < 1% using as reference the following genomic datasets: 1000 Genomes, ESP6500, ExAC, GnomAD. For confirming both the observed variants, Sanger sequencing was performed using the BigDye™ Terminator v1.1 Cycle Sequencing Kit (Life Technologies, CA, USA) with the ABI 3130 instrument (Life Technologies, CA, USA). For the variant c.1679G > A the sequences of the primers adopted for the Sanger experiment were forward: 5′-ACCGCACTCATCTTACACCG-3; reverse: 5′-CCTGCCATTCCTAGGTGCTC -3′. On the other hand, the primers employed for the confirmation of the variant c.3292C > T were forward: 5′-GCTGGCAGCTAACTTCTCTCT-3; reverse: 5′-AAAAGAGAAGGCTCCCACCT-3′.
Data analysis
Pathogenic variants were investigated on the Human Gene Mutation Database (HGMD Professional 2023). Diverse VarAFT20 filters were adopted on the vcf files. DOMINO web server (https://domino.iob.ch/) (accessed on 10 January 2025) was used for predicting the probability of the MAN2A2 autosomal recessive inheritance pattern. In the DOMINO analysis, the gene name was used as the query, and the output was a score ranging from 0 (indicative of an autosomal recessive inheritance) to 1 (indicative of an autosomal dominant inheritance). The observed variant was described according to the “American College of Medical Genetics” (ACMG) guidelines21 and was performed with VarSome according to a previous study22 and other evidence from the literature. PhastCons100way and PhyloP100way scores (from VarSome analysis), were used for analyzing the conservation tendency of the specific mutation region. Multiple algorithms were employed for the variants classification, including PolyPhen 2 (http://genetics.bwh.harvard.edu/, accessed on 10 January 2025) and Mutation Taster (https://www.mutationtaster.org/, accessed on 10 January 2025). ProtVar web server (https://www.ebi.ac.uk/ProtVar/) (accessed on 10 January 2025) was used for the prediction with AlphaMissense algorithm. Uniprot database (https://www.uniprot.org/, accessed on 10 January 2025) was used for retrieving the MAN2A2 protein details related to the wild-type protein sequence. Furthermore, the details related to the functional domains were provided by InterPro (https://www.ebi.ac.uk/interpro, accessed on 10 January 2025) database. Gene ontology terms (GO) were retrieved from QuickGO database (https://www.ebi.ac.uk/QuickGO/, accessed on 10 January 2025).
BrainSPAN (https://www.brainspan.org/) (accessed on 18 March 2025) and BrainRNAseq (https://brainrnaseq.org/) (accessed on 18 March 2025) databases were used for comparing the expression profile of MAN2A2 and MAN2A1.
Protein structure predictions were performed using the AlphaFold3 algorithm via the webserver (https://alphafoldserver.com/) (accessed on 18 March 2025). The algorithm generated five models per prediction for the wild-type MAN2A2 and the mutated Arg560Gln and Gln1098Ter variants. For analysis, the “best model” of each prediction was selected based on the predicted local distance difference test (pLDDT), prioritizing the highest confidence score (ranging from 0 to 100). The pLDDT values were processed using the Rstudio software version 3.4.3 with the jsonlite, bio3d, and ggplot2 packages. Line plots depicting the pLDDT values for each model are presented in Figure S6, while Figure S7 displays the heatmap of the predicted alignment error for each selected “best model.” Protein structures modelling was performed employing UCSF ChimeraX software version 1.7 (https://www.cgl.ucsf.edu/chimerax/). The complete set of hydrogen bonds identified across all five AlphaFold3-predicted models for the wild-type and both the mutated MAN2A2 proteins is shown in Supplementary File 2. Additionally, MuPRO tool was used for estimating the impact of the mutations on protein stability http://mupro.proteomics.ics.uci.edu/, accessed on 10 January 2025) by score predicting the value and sign of energy change (delta delta G), spanning from − 1 to 123. STRING database (https://string-db.org/, accessed on 10 January 2025) was used for investigating about the specific MAN2A2 protein pathways.
Glycosylation analyses
Serum transferrin glycoform analysis
Separation and detection of serum Trf glycoforms were carried out using the CE Capillarys CDT system (Sebia, France), as previously described24.
Serum N-glycosylation analyses
N-glycan profiling of total serum glycoproteins was performed either by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI TOF MS) and by ultra-high-performance liquid chromatography-electrospray mass spectrometry (UHPLC-ESI MS).
Sample preparation for MALDI-TOF analysis, accomplished on 10 µl of serum, consisted of protein denaturation by RapiGest™ SF surfactant (Waters corporation, Milford, MA USA) followed by reduction, alkylation and deglycosylation by PNGase F (Roche Merk KGaA) as already described in detail25. The released N-glycans were purified, permethylated and analyzed in positive polarity on a 4800 Proteomic Analyzer (AB Sciex).
UHPLC-ESI MS analysis of total serum glycoproteins was performed as thoroughly described in Messina et al.12. Briefly, the N-linked glycans were enzymatically released from 7.5 µl of serum and labelled by the GlycoWorks RapiFluor MS™ (RFMS) N-Glycan kit (Waters Corporation, Milford, MA, USA). Samples were therefore separated by an UHPLC THERMO system Vanquish equipped with an ACQUITY UPLC Glycan BEH Amide 130 Å, 2.1 mm × 150 mm, 1.7 μm hydrophilic interaction liquid chromatography (HILIC) column (Waters Corporation Milford, MA, USA) and analyzed by an Orbitrap Exploris™ 120 mass spectrometer (Thermo Fisher Scientific Inc., Bremen, Germany). We performed three runs for each analysis, and we found not substantial differences between chromatograms and spectra recorded.
Conclusion
The present study documents a novel patient with MAN2A2-CDG presenting with both autism spectrum disorder (ASD), intellectual disability and subtle dysmorphic facial features. WES Trio analysis revealed in the patient two heterozygous variants within MAN2A2 gene inherited from each parent, leading to a condition of compound heterozygosity. To date, no OMIM record links this gene to disease. In silico predictions deemed both the variants as likely pathogenic while the conservation parameters demonstrated high conservation for both mutated sites across diverse vertebrate species. Protein structure prediction analyses indicated notable changes from the wild-type protein to both the mutated forms. Glycosylation analyses demonstrated impaired N-glycosylation, with accumulation of immature serum glycoprotein N-glycans including disease-specific hybrid-type species. Further analyses are imperative to better understand how MAN2A2 dysfunction contributes to human disease phenotype.
Data availability
Both the variants that support the findings of this study have been deposited in the ClinVar Database with the primary accession codes VCV003766448.1 and VCV003766447.1, for and the variants p.Arg560Gln and p.Gln1098Ter respectively.
References
Apweiler, R. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta (BBA) 1473, 4–8 (1999).
Haltiwanger, R. S. & Lowe, J. B. Role of glycosylation in development. Annu. Rev. Biochem. 73, 491–537 (2004).
Schjoldager, K. T., Narimatsu, Y., Joshi, H. J. & Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell. Biol. 21, 729–749 (2020).
Ng, B. G., Freeze, H. H., Himmelreich, N., Blau, N. & Ferreira, C. R. Clinical and biochemical footprints of congenital disorders of glycosylation: Proposed nosology. Mol. Genet. Metab. 142, 108476 (2024).
Dwyer, C. A. & Esko, J. D. Glycan susceptibility factors in autism spectrum disorders. Mol. Asp. Med. 51, 104–114 (2016).
Liu, Y. et al. Altered expression of glycan patterns and glycan-related genes in the medial prefrontal cortex of the valproic acid rat model of autism. Front. Cell. Neurosci. 16, 1057857 (2022).
Pradeep, P., Kang, H. & Lee, B. Glycosylation and behavioral symptoms in neurological disorders. Transl Psychiatry 13, 154 (2023).
Akama, T. O. et al. Essential and mutually compensatory roles of α-mannosidase II and α-mannosidase IIx in N-glycan processing in vivo in mice. Proc. Natl. Acad. Sci. 103, 8983–8988 (2006).
Akama, T. O. et al. Germ cell survival through carbohydrate-mediated interaction with sertoli cells. Science 1979(295), 124–127 (2002).
Van den Elsen, J. M., Kuntz, D. A. & Rose, D. R. Structure of Golgi alpha-mannosidase II: A target for inhibition of growth and metastasis of cancer cells. EMBO J. 20, 3008–3017 (2001).
Mahajan, S. et al. Homozygous truncating variant in MAN2A2 causes a novel congenital disorder of glycosylation with neurological involvement. J. Med. Genet. 60, 627–635 (2023).
Messina, A. et al. HILIC-UPLC-MS for high throughput and isomeric N-glycan separation and characterization in congenital disorders glycosylation and human diseases. Glycoconj J. 38, 201–211 (2021).
Fagerberg, L. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406 (2014).
Choi, H.-Y. et al. N-glycan remodeling using mannosidase inhibitors to increase high-mannose glycans on acid α-glucosidase in transgenic rice cell cultures. Sci. Rep. 8, 16130 (2018).
Huang, A. et al. Disrupting N-glycosylation using type I mannosidase inhibitors alters B-cell receptor signaling. ACS Pharmacol. Transl. Sci. 5, 1062–1069 (2022).
Ulke, J. et al. High-fat diet alters N-glycosylation of PTPRJ in murine liver. J. Nutr. Biochem. 123, 109500 (2024).
Patro, I. et al. Nonsense-mediated mRNA decay: Mechanistic insights and physiological significance. Mol. Biotechnol. 66, 3077–3091 (2024).
Lykke-Andersen, S. & Jensen, T. H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell. Biol. 16, 665–677 (2015).
Vinci, M. et al. Bioinformatic evaluation of KLF13 genetic variant: Implications for neurodevelopmental and psychiatric symptoms. Genes (Basel) 15, 1056 (2024).
Desvignes, J.-P. et al. VarAFT: A variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. 46, W545–W553 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Kopanos, C. et al. VarSome: The human genomic variant search engine. Bioinformatics 35, 1978–1980 (2019).
Cheng, J., Randall, A. & Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins: Struct. Funct. Bioinform. 62, 1125–1132 (2006).
Sturiale, L. et al. ALG12-CDG: Novel glycophenotype insights endorse the molecular defect. Glycoconj J. 36, 461–472 (2019).
Sturiale, L. et al. Aberrant sialylation in a patient with a HNF1α variant and liver adenomatosis. iScience 24, 102323 (2021).
Acknowledgements
We acknowledge Domenico Garozzo, CNR—Catania (Italy) for helpful insights in glycosylation analyses. Furthermore, special acknowledgements for this manuscript to Eleonora Di Fatta for her valuable assistance in the translation, preparation and formatting of the text. Additionally, we would like to sincerely thank Angelo Gloria, Rosanna Galati Rando and Alda Ragalmuto for their technical contribution
Funding
This research was partially funded by the European Union – Next Generation EU, Mission 4 Component 1, CUP E53D23009730006. Ministry of University and Research—PRIN (Research Projects of National Relevant Interest) 2022—(ID: 202255RLB4). This work was partially supported by the Italian Ministry of Health “Ricerca Corrente 2017–2023” and 5 × mille.
Author information
Authors and Affiliations
Contributions
Conceptualization: S.T., M.V., R.B. and F.C.; Methodology: S.T., M.V., L.C., A.Mu., A.P., F.P., A.Me., V.C., L.S. F.C.; Clinical assessment: L.C. and F.P.; Data interpretation: S.T., M.V., L.S., A.P., A.Mu., R.B. and F.C. Visualization: S.T., M.V., L.S., S.S., F.C. and R.B.; writing: S.T., A.P., A.Me., L.S., F.C. and R.B. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Treccarichi, S., Vinci, M., Cirnigliaro, L. et al. MAN2A2-related glycosylation defects in autism and cognitive delay. Sci Rep 15, 24471 (2025). https://doi.org/10.1038/s41598-025-09400-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-09400-5
