
Abstract
Hepatocellular carcinoma (HCC) remains a leading cause of cancer-related mortality worldwide, characterized by frequent recurrence and poor clinical outcomes. This study aimed to develop a mitochondria-related genes (MRGs) signature for prognostic stratification and immunotherapy response prediction in HCC patients. Derived from the TCGA-LIHC cohort and validated across independent ICGC-JP and GEO datasets, the MRGs signature comprised four genes (DTYMK, ABCB6, GOT2, and TOMM40L) that were markedly overexpressed in HCC tissues and strongly associated with adverse prognosis. MRGs-based nomogram exhibited superior predictive accuracy, highlighting their clinical potential for personalized risk assessment. Within the tumor microenvironment, high-MRGs tumors demonstrated significant enrichment of immunosuppressive components, including regulatory T cells, tumor-associated macrophages, and checkpoint molecules PD-1 and CTLA-4. The MRGs-high subgroup showed heightened sensitivity to cisplatin but resistance to erlotinib, and impaired immunotherapy responses, which has potential clinical transformation value in the design of individualized combination therapy. Functional validation revealed ABCB6 as a key oncogenic driver, with genetic depletion significantly attenuating HCC cell proliferation, migration, and invasion in vitro. Collectively, the MRGs signature serves as a better predictor of HCC prognosis and therapeutic resistance, while its core component ABCB6 emerges as a critical regulator of HCC malignancy.
Similar content being viewed by others

Introduction
Hepatocellular carcinoma (HCC) ranks as the third most lethal malignancy worldwide1, with current multimodal therapeutic approaches—including surgical resection, adjuvant chemotherapy and radiotherapy, and molecularly targeted agents—yielding suboptimal outcomes, evidenced by survival rates of 36% at 1 year, 17% at 3 years2, and < 10% at 5 years3, alongside a 5-year recurrence rate of 40–60%4. These poor outcomes stem from HCC’s intrinsic tumor aggressiveness, characterized by rapid proliferation, invasive potential, and immune dysregulation5,6, underscoring the critical need for novel therapeutic targets and strategies to improve clinical management.
Emerging evidence demonstrates mitochondria’s central role in tumorigenesis through dysregulated cellular energy metabolism, reactive oxygen species (ROS) generation, and apoptotic control7,8. As metabolic hubs, mitochondrial reprogramming is governed by multifaceted regulatory mechanisms—microenvironmental stress and genetic alterations drive overexpression of glycolysis-related enzymes and glucose transporters, enabling rapid ATP replenishment under hypoxia9. Mitochondria-derived ROS, critical byproducts of oxidative metabolism10, are elevated in tumors, triggering compensatory upregulation of antioxidant systems11, while oncogenes like K-Ras, B-raf, and c-Myc further exploit these mechanisms to promote malignancy12. Beyond bioenergetics, mitochondria orchestrate oxidative phosphorylation, metabolite synthesis, oxidative stress responses, and cell death pathways13. Despite these multifaceted contributions, the prognostic relevance of mitochondria-related genes in HCC remains poorly defined.
This study developed a mitochondria-related genes (MRGs) prognostic signature using The Cancer Genome Atlas-Liver cancer (TCGA-LIHC) cohort, with rigorous validation of its clinical relevance through multi-cohort analyses in the International Cancer Genome Consortium (ICGC) and Gene Expression Omnibus (GEO) datasets, confirming MRGs overexpression and survival correlations. A MRGs-integrated nomogram demonstrated superior prognostic precision for HCC patient survival stratification. Functional profiling revealed MRGs roles in tumor biology, chemosensitivity, immunosuppressive tumor microenvironment (TME) modulation, genomic instability, and immunotherapy. Mechanistic validation identified ATP-Binding Cassette Subfamily B Member 6 (ABCB6) as a core oncogenic driver, with in vitro knockdown experiments confirming its critical role in sustaining HCC proliferation, migration, and invasiveness.
Materials and methods
Data source
The HCC cohorts utilized in this study were sourced from the TCGA (https://portal.gdc.cancer.gov/), ICGC (https://dcc.icgc.org/), GEO (http://www.ncbi.nlm.nih.gov/geo/), and KM Plotter (https://kmplot.com/) databases, encompassing expression matrices, clinical profiles, and immunotherapy datasets. Detailed information on the cohorts is summarized in Table 1.
Data processing
The TCGA-LIHC dataset in HTSeq-Counts format was utilized for differential gene expression analysis between HCC and normal liver tissues, HTSeq-Counts data were normalized using DESeq2’s median-of-ratios method. Data originally in HTSeq FPKM format were converted to transcripts per million (TPM) for downstream analyses. For the ICGC and GEO microarray datasets, probe-level expression data were processed using the R package “affy” to generate probe set expression summaries via the Robust Multichip Analysis (RMA) pipeline, which integrates background correction, quantile normalization, and summarization of probe intensities into gene-level values. Platform-specific normalization was performed for KM Plotter clinical data. Prior to analysis, RNA-seq data were subjected to log2 conversion. Missing values were defined as missing or unknown clinical characteristics. This study analyzed data from publicly available databases and did not necessitate a re-evaluation of medical ethics.
Constructing a MRGs in HCC
In the TCGA-LIHC cohort, RNA-seq count data from 373 HCC tissues and 50 adjacent non-tumor tissues were analyzed to identify differentially expressed genes (DEGs) between tumor and normal samples. Differential expression analysis was performed using the R package “DESeq2”, with volcano plot generated via “ggplot2” to visualize DEGs meeting thresholds of |log2 FC|≥ 1 and adjusted p value < 0.05. Tumor-specific DEGs were intersected with 1136 mitochondrial genes from the MitoCarta3.0 database (https://www.broadinstitute.org/mitocarta/), yielding mitochondria-related DEGs. Lasso regression analysis was then applied to these candidate genes using the “glmnet” and “survival” packages, implementing a robust ten-fold cross-validation framework (seed set to 2024) to systematically identify the optimal lambda (λ) value that minimized the cross-validation error. Subsequently, univariate and multivariate Cox proportional hazards regression analyses were performed to establish a MRGs prognostic signature, weighted by regression coefficients. Forest plots depicting hazard ratios with 95% confidence intervals (95% CI) were generated using the R package “ggplot2”. To enhance the clinical relevance of survival analysis, the final multivariate Cox model integrating significant clinical covariates underwent rigorous false discovery rate (FDR) adjustment (Benjamini–Hochberg procedure) with a significance threshold set at q-value < 0.1, effectively controlling for multiple comparisons.
Investigation of the MRGs using the TCGA-LIHC cohort
Pairwise correlations among Deoxythymidylate Kinase (DTYMK), ABCB6, Glutamic-Oxaloacetic Transaminase 2 (GOT2), and Translocase of Outer Mitochondrial Membrane 40 Homolog Like (TOMM40L) were analyzed in the TCGA-LIHC cohort, with correlation matrices visualized using the R package “ggplot2”. Receiver operating characteristic (ROC) curve generated via the “pROC” package were employed to evaluate the diagnostic potential of the MRGs for HCC, and results were graphically represented with “ggplot2”. HCC patients in TCGA-LIHC cohort were stratified into MRGs-high and -low subgroups based on median MRGs expression levels. To comprehensively assess clinical implications, three analytical approaches were implemented. Scatter plots visualized with “ggplot2” depicting MRGs expression distributions; Kaplan–Meier curves analyzed with the “survival” package and plotted via “survminer”; Time-dependent ROC curves computed using the “timeROC” package to evaluate dynamic prognostic performance, with visualization again performed in “ggplot2”.
Validation of the MRGs using the independent cohorts
The ICGC-JP, GSE14520, and GSE112790 cohorts served as expression validation cohorts to assess differential expression of the four MRGs-incorporated genes (DTYMK, ABCB6, GOT2, and TOMM40L) between HCC and normal tissues, while the ICGC-JP and GSE14520 cohorts were further utilized as prognostic validation cohorts to evaluate the clinical relevance of the MRGs signature. Analytical methods for generating ROC curves, scatter plots, Kaplan–Meier survival curves, and time-dependent ROC curves—employed to quantify diagnostic accuracy, expression distribution, survival stratification, and dynamic prognostic performance, respectively—were implemented as described in the preceding methodology section.
Construction and evaluation of the nomogram model
The TCGA-LIHC cohort was primarily employed to evaluate associations between MRGs and clinicopathological characteristics in HCC patients and to establish a prognostic nomogram. The ICGC-JP and GSE14520 cohorts were subsequently used for external validation of the nomogram’s predictive accuracy. The nomogram model integrated MRGs and pathological stage variables through multivariate Cox regression, with calibration curves plotted via the “rms” and “survival” packages to compare predicted versus observed survival probabilities. To ensure model robustness, we conducted comprehensive internal validation via bootstrap resampling (n = 1000 replicates), deriving an optimism-adjusted Harrell’s concordance index (C-index) to mitigate overfitting effects. Time-dependent ROC analysis was further applied to assess dynamic predictive performance as described above.
Enrichment analysis of MRGs-related genes
Initially, HCC patients in the TCGA-LIHC cohort were stratified into MRGs-high and -low subgroups based on median MRG expression levels, followed by differential gene expression analysis (MRG-high vs. -low) as previously described. MRGs-related genes (ranked by absolute log2 FC values) were subjected to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses14,15 using the R package “clusterProfiler”, with results visualized via “GOplot”. GO functional analysis encompassed three domains—cellular components (CC), molecular functions (MF), and biological processes (BP). For gene set enrichment analysis, pathway significance was assessed using the Benjamini–Hochberg method to control the false discovery rate (FDR < 0.05) across all tested pathways.
Correlation of MRGs with HRD and prediction of drug sensitivity.
We acquired homologous recombination deficiency (HRD) scores for HCC patients in the TCGA-LIHC cohort from a prior publication16, followed by integrating HRD scores with MRGs expression levels to explore potential therapeutic implications through correlation analysis. Drug sensitivity predictions for cisplatin (a platinum-based chemotherapeutic) and erlotinib (an EGFR inhibitor) were generated for TCGA-LIHC samples using the Genomics of Cancer Drug Sensitivity (GDSC, https://www.cancerrxgene.org) database. These predictions were computed via the R package “pRRophetic”, which estimates the half-maximal inhibitory concentration (IC50) through ridge regression-based algorithms, enabling quantification of tumor-specific drug response profiles.
Role of MRGs in the TME
Transcriptomic data from HCC patients in the TCGA-LIHC cohort were analyzed using the CIBERSORTx platform (https://cibersortx.stanford.edu) to calculate immune cell infiltration scores through the CIBERSORT algorithm. Due to the cohort’s limited sample size, deconvolution p value-based sample filtering was not implemented. RNA-seq profiles of four immunosuppressive checkpoints (PD-1, CTLA-4, PD-L1, and PD-L2) were simultaneously extracted from the same dataset, while immune subtype classifications were adopted from prior research16. Single-cell RNA sequencing data from the GSE98638 cohort in GEO database were processed via the TISCH2 portal (http://tisch.comp-genomics.org). Additionally, we identified specific DEGs that were down-regulated in proliferating T cells (Tprolif) and up-regulated in exhausted T cells (Tex). Functional enrichment analysis of these DEGs was performed using Metascape (https://metascape.org/gp/), followed by correlation studies between these DEGs and MRGs members.
Relationship of MRGs to genetic mutations and tumor stemness
The association between MRGs and genetic mutations as well as tumor stemness was explored in the TCGA-LIHC cohort. Genetic mutation profiles were processed and visualized using the R package “MAfTools”, identifying the top 20 most frequently mutated genes in MRGs-high versus -low subgroups. Expression differences of DTYMK, ABCB6, GOT2, and TOMM40L were further compared between TP53-mutated (TP53-mut) and TP53-wild-type (TP53-wt) subgroups. Tumor stemness indices (DNAss and RNAss) were obtained from published data17, and their correlations with MRGs were systematically evaluated through integrated analysis of both DNAss and RNAss metrics.
MRGs predicted response to immunotherapy
The relationship between MRGs and tumor mutation burden (TMB), microsatellite instability (MSI), and immunotherapy response was comprehensively assessed in the TCGA-LIHC cohort. TMB values were calculated using the R package “MAfTools”, while MSI scores were sourced from a prior study18. Tumor immune dysfunction and exclusion (TIDE) scores, predictive of immunotherapy efficacy, were generated through the TIDE algorithm (http://tide.dfci.harvard.edu/). Integrated analysis of TMB, MSI, and TIDE metrics was conducted to compare their distributions between MRGs-high and -low subgroups. Additionally, clinical data and expression profiles of 173 nivolumab-treated and 112 ipilimumab-treated patients were retrieved from the KM Plotter database, along with 101 anti-PD-1-treated and 25 adoptive cell therapy (ACT)-treated melanoma patients from the GEO database. These cohorts were stratified into MRGs-high and -low subgroups based on median expression levels of MRGs, respectively, and Kaplan–Meier curves were employed to assess survival differences between subgroups using the methodology described above.
Cell culture
The human hepatocellular carcinoma cell lines Huh7 and HepG2 were obtained from Zhejiang Meisen Cell Technology Co., Ltd. (Hangzhou, Zhejiang, China) and cultured in DMEM medium (Gibco, Cat# 11965092) supplemented with 10% fetal bovine serum (FBS; PAN Biotech, Cat# P30-2602) and 1% penicillin–streptomycin (Gibco, Cat# 15140148), under standardized conditions.
Lentiviral transfection
shRNA lentiviral vectors targeting ABCB6 (shABCB6) and non-targeting controls (shCtrl), designed and packaged by Genecoopia Biotechnology Co., Ltd. (Guangzhou, China), were utilized for gene silencing (specific sequences detailed in Table 2). Following thawing of viral aliquots on ice, the infection protocol involved aspirating existing culture medium and introducing fresh medium containing lentiviral stock at 50% working volume. After 4-h incubation at 37 °C with 5% CO2, replacement of fresh complete medium. Transduction efficiency was verified through GFP fluorescence intensity assessment via inverted fluorescence microscopy at 48 h post-infection. Stable cell pools were subsequently selected through sequential puromycin treatment (2 μg/mL) with media replacement every 48 h until non-transduced control cells demonstrated complete mortality.
Real-time fluorescence quantitative PCR
Total RNA was isolated from cells using the Oriscience Prime RNA Extraction Kit (Oriscience Biotechnology Co., Ltd., Chengdu, China) following the manufacturer’s protocol, with RNA concentration and purity quantified via UV spectrophotometry (A260/A280 ratio ≥ 1.8). Reverse transcription of 1 μg RNA into cDNA was performed using a PrimeScript RT reagent kit (Takara Bio, Japan). Quantitative PCR amplification was carried out on an ABI QuantStudio 6 Flex system (Thermo Fisher Scientific, USA) with Taq Pro SYBR qPCR Master Mix (Vazyme Biotech, Nanjing, China), employing the following cycling parameters: 95 °C for 30 s, 40 cycles of 95 °C for 5 s, and 60 °C for 30 s. GAPDH served as the endogenous control, and all primer pairs are listed in Table 2, with melt curve analysis confirming amplification specificity.
Western blot
Protein extraction and immunoblotting were performed as follows: Cells in logarithmic growth phase were harvested, washed twice with ice-cold PBS, and lysed in RIPA buffer (Beyond Biotechnology, Shanghai, China) supplemented with 1 mM PMSF protease inhibitor (Beyotime Biotechnology, China). Lysates were boiled at 100 °C for 15 min in 4× SDS-PAGE loading buffer. Protein quantification was conducted using a BCA assay kit (Beyond Biotechnology) with microplate reader absorbance measurement at 562 nm. Equal protein aliquots (30 μg) and pre-stained molecular weight markers (4 μL, Thermo Fisher) were resolved individually through 8–15% gradient SDS-PAGE gels under 80 V constant voltage for 2 h, followed by semi-dry electrophoretic transfer (25 V, 30 min) to PVDF membranes (Millipore, USA). Membranes were blocked with 5% BSA in TBST for 2 h at room temperature, then incubated overnight at 4 °C with primary antibodies against ABCB6 (1:1000, ab47837, Abcam) and β-actin (1:2000, AF2811, Beyotime) diluted in blocking buffer. After three TBST washes (10 min each), membranes were probed with HRP-conjugated secondary antibodies (1:5000, Cell Signaling Technology) for 1 h at room temperature. Protein signals were developed using Oriscience ECL Substrate (Chengdu, China) and visualized through a ChemiDoc Imaging System (Bio-Rad), with β-actin serving as loading control.
CCK-8
Cell viability was assessed using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) according to standardized protocols. Briefly, 5 × 103 cells/well were seeded in 96-well plates containing 100 μL complete DMEM medium per well and allowed to adhere overnight. Following 24, 48, and 72-h incubation periods under 37 °C/5% CO2 conditions, culture medium was replaced with 100 μL fresh serum-free DMEM containing 10% CCK-8 reagent (v/v). Plates were further incubated for 2 h under standard culture conditions. Optical density (OD) values were quantified at 450 nm using a microplate reader (BioTek Synergy H1) with reference wavelength set at 650 nm. Three technical replicates were performed per condition, with blank wells (medium + CCK-8 without cells) serving as background controls.
Clone formation
Stable transfected cells were seeded at 1 × 103 cells/well in 12-well plates and cultured for 7–10 days under 37 °C/5% CO2 conditions until macroscopic colonies (> 50 cells/colony) formed. Colonies were fixed with 4% paraformaldehyde (PFA; Solarbio, China) for 20 min at room temperature, followed by three 5-min PBS washes. Staining was performed with 0.1% crystal violet (Beyotime, China) in 20% ethanol for 15 min. After thorough deionized water rinsing to remove residual dye, plates were air-dried and scanned using a GelCount colony counter (Oxford Optronix, UK). Quantitative analysis was performed by threshold-based image processing (ImageJ v1.53), counting colonies with diameters ≥ 50 μm. Experiments included triplicate wells per condition, with untransfected parental cells serving as baseline controls.
Cell cycle
Cell cycle distribution was assessed via flow cytometry (BD FACSCanto II, USA) using the following protocol. Cells were harvested via trypsin–EDTA digestion, washed twice with ice-cold PBS, and fixed in 70% ethanol (− 20 °C) for ≥ 18 h. Fixed cells were centrifuged (300×g, 5 min), permeabilized with 0.25% Triton X-100/PBS for 15 min on ice, and stained with BD Pharmingen™ PI/RNase Staining Buffer (Cat# 550825) containing 50 μg/mL propidium iodide and 100 μg/mL RNase A for 30 min at 37 °C in darkness. Cell suspensions were filtered through 40 μm nylon mesh (Falcon) to eliminate aggregates prior to acquisition. Flow cytometry analysis was performed with 10,000 events recorded per sample using 488 nm excitation and 585/42 nm emission filters. Cell cycle phase quantification (G0/G1, S, G2/M) was conducted through ModFit LT 5.0 software (Verity Software House) employing the Dean-Jett-Fox algorithm, with ≥ 95% confidence in model fitting.
Wound healing
Cells were seeded in 6-well plates at 5 × 105 cells/well and cultured in complete medium until reaching 90–95% confluency under 37 °C/5% CO2 conditions. Three parallel linear wounds per well were created using sterile 200 μl pipette tips (Axygen), followed by three gentle PBS washes to remove detached cells. To minimize proliferation interference, cells were maintained in serum-starved DMEM (2% FBS; Gibco) for 24 h. Time-lapse imaging was initiated immediately post-scratching using an Olympus IX83 live-cell imaging system equipped with a 10× objective, capturing images at 0 and 24-h intervals. Wound closure quantification was performed through ImageJ v1.53 software (NIH) by measuring the residual wound area relative to initial dimensions (T0), with three technical replicates per condition.
Transwell assay
Cell invasion capacity was evaluated using Corning Transwell chambers (8 μm pore, Cat# 3422) pre-coated with 300 μg/mL Matrigel matrix (BD Biosciences, Cat# 356234) diluted in serum-free DMEM. Briefly, 1 × 104 cells suspended in 200 μL serum-free medium were seeded into the upper chamber, while 600 μL complete medium containing 10% FBS (Gibco) was added to the lower compartment as chemoattractant. Following 48-h incubation at 37 °C with 5% CO2, non-invading cells were removed from the upper membrane surface using cotton swabs. Invaded cells on the lower membrane were fixed with 4% paraformaldehyde (15 min), stained with 0.1% crystal violet (Beyotime, Cat# C0121) for 20 min, and washed thrice with PBS. Five randomly selected fields per membrane were imaged using an Olympus IX73 microscope (20× objective), with cell counts quantified through ImageJ v1.53 software.
Statistical analysis
All statistical analyses in this study were performed using R Studio, with a statistical significance threshold set at P < 0.05. Comparative analyses between groups were implemented through the Wilcoxon rank-sum test for continuous variables and the chi-square test for categorical variables. Correlation patterns were quantified via the Spearman rank correlation method. Prognostic evaluations leveraged Cox proportional hazards regression models for continuous parameters and log-rank testing for dichotomized variables, ensuring methodological alignment with data types.
Results
Development of a MRGs prognostic signature in HCC
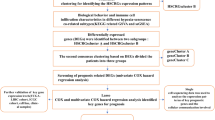
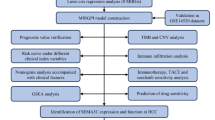
Figure 1 illustrated the overall workflow of this study. Differential gene expression analysis in the TCGA-LIHC cohort identified 3299 up-regulated and 1232 down-regulated DEGs in tumor tissues compared to normal controls (Fig. 2A). By intersecting these DEGs with 1136 mitochondrial genes from the MitoCarta3.0 database, we identified 171 overlapping mitochondrial-associated DEGs (Fig. 2B). Subsequent Lasso regression analysis with ten-fold cross-validation narrowed this set to 16 candidate genes (Fig. 2C,D). Univariate and multivariate Cox regression analyses further refined the model, revealing DTYMK, ABCB6, and TOMM40L as independent prognostic risk factors, while GOT2 emerged as a protective factor in HCC (Fig. 2E,F). Using regression coefficients derived from the TCGA-LIHC training subset, we constructed a mitochondrial-related genes (MRGs) signature with the formula: MRGs = (0.288 × DTYMK) + (0.733 × ABCB6) + (0.256 × TOMM40L) − (0.324 × GOT2).

Flow chart of this study.

Construction of a HCC-associated MRGs signature utilizing the TCGA-LIHC cohort. (A) The volcano plot demonstrating differentially expressed genes between HCC and normal tissues using the TCGA-LIHC cohort. (B) The Venn diagram showing the intersection of HCC-associated DEGs with human mitochondria-associated genes. Lasso (C,D), univariate (E), and multivariate Cox regression analyses (F) were performed to screen and construct a MRGs signature in HCC. (G) Heat map showing correlation between members of MRGs. (H,I) Unpaired and paired analyses were performed to assess the differential expression of members of MRGs in HCC and normal tissues. (J) ROC curve demonstrating the accuracy of MRGs in diagnosing HCC tissue. (K) The scatterplot demonstrating the difference in survival time and number of deaths in HCC patients in the MRGs-high and -low groups. (L) Kaplan–Meier survival curve showing the difference in overall survival of HCC patients in the MRGs-high and -low groups. (M) Time-dependent ROC curves demonstrating the accuracy of MRGs in predicting 1-, 3-, and 5-year survival in HCC patients. **P < 0.01, ***P < 0.001.
Correlation analysis demonstrated strong positive associations among DTYMK, ABCB6, and TOMM40L, whereas all three genes showed inverse correlations with GOT2 (Fig. 2G). Expression profiling revealed significant up-regulation of DTYMK, ABCB6, and TOMM40L in HCC tissues, contrasting with GOT2 down-regulation (Fig. 2H,I). The MRGs signature exhibited exceptional diagnostic potential for HCC, achieving an AUC of 0.986 in tumor discrimination (Fig. 2J).
Risk stratification and Kaplan–Meier survival analyses confirmed that high MRGs correlated with reduced overall survival and elevated mortality (Fig. 2K,L). Time-dependent ROC curves demonstrated better prognostic accuracy for the MRGs signature, with AUC values of 0.749, 0.710, and 0.689 for 1-, 3-, and 5-year survival predictions, respectively (Fig. 2M).
Validation of MRGs expression and prognostic significance
To confirm the expression patterns of the MRGs signature, we analyzed three independent datasets, including ICGC-JP, GSE14520, and GSE112790. Consistent with prior findings, DTYMK, ABCB6, TOMM40L, and the MRGs were significantly up-regulated in HCC tumor tissues compared to normal controls, whereas GOT2 expression was markedly reduced (Fig. 3A–C). The MRGs signature demonstrated robust diagnostic performance, with AUC values of 0.923 (ICGC-JP), 0.864 (GSE14520), and 0.926 (GSE112790) in distinguishing HCC from normal tissues (Fig. 3D–F).

Validation of MRGs expression levels and prognostic value using multiple independent cohorts. (A–C) ICGC-JP, GSE14520, and GSE112790 cohorts were used to validate the expression levels of DTYMK, ABCB6, GOT2, TOMM40L, and MRGs in HCC and normal tissues, respectively. (D–F) ROC curves demonstrating the accuracy of MRGs in diagnosing HCC tissue. The scatterplot of MRGs, ICGC-JP (G) and GSE14520 (J) cohorts were the validation subset 1 and validation subset 2, respectively. (H,K) Kaplan–Meier survival analyses were performed to demonstrate the difference in overall survival of HCC patients between the MRGs-high and -low groups. (I,L) Time-dependent ROC curves demonstrating the accuracy of MRGs in predicting 1- and 3-year survival in HCC patients. *P < 0.05, **P < 0.01, ***P < 0.001.
We further evaluated the prognostic utility of the MRGs signature using the ICGC-JP and GSE14520 datasets. Risk stratification analysis revealed that patients in the MRGs-high group exhibited higher mortality rates and shorter median survival times compared to the -low group (Fig. 3G,J). Kaplan–Meier curves underscored a striking divergence in outcomes, with the MRGs-high group showing significantly poorer overall survival (Fig. 3H,K). Time-dependent ROC curves confirmed the predictive reliability of the MRGs signature, achieving AUCs > 0.6 for both 1-year and 3-year survival probabilities across validation cohorts (Fig. 3I,L).
Clinically integrated MRGs-based nomogram for HCC prognosis
A general overview of the relationship between the MRGs and clinicopathologic parameters using the TCGA-LIHC cohort (Fig. 4A). Multivariate analysis revealed significant associations between high MRGs and advanced clinicopathological parameters, including pathologic stage (TCGA: stage II vs. I, P < 0.01; stage III vs. I, P < 0.01; ICGC: stage III vs. I, P < 0.05; stage IV vs. I, P < 0.05), tumor stage (TCGA: T2 vs. T1, P < 0.001; T3 vs. T1, P < 0.01), histologic grade (TCGA: G3 vs. G1, P < 0.001; G4 vs. G1, P < 0.05; G3 vs. G2, P < 0.001), elevated AFP levels (TCGA: > 400 vs. ≤ 400 ng/mL, P < 0.001), and residual tumor status (TCGA: R1 vs. R0, P < 0.05) (Fig. 4B–G).

Construction and validation of a HCC-related nomogram model based on MRGs. (A) A general overview of the association of MRGs with clinicopathologic characteristics of patients with HCC using TCGA-LIHC cohort. MRGs were related to pathological stage in TCGA-LIHC (B) and ICGC cohorts (C), T stage (D), histologic grade (E), AFP concentration (F), and residual tumor (G). (H) A nomogram model was constructed combining MRGs and pathological stage by utilizing the TCGA-LIHC cohort. (I) The calibration curves of the model were shown for 1-, 3-, and 5-year, respectively. (J–L) Time-dependent ROC curves demonstrated the ability of the model to predict 1-, 3-, and 5-year overall survival in HCC patients using the TCGA-LIHC, ICGC-JP, and GSE14520 cohorts, respectively. *P < 0.05, **P < 0.01, ***P < 0.001.
To enhance prognostic precision, we integrated the MRGs signature with pathologic stage to develop a clinically actionable nomogram using the TCGA-LIHC cohort (Fig. 4H). The model demonstrated a good predictive performance, achieving a concordance index (C-index) of 0.711 (95% CI: 0.684–0.738) and well-calibrated survival estimates across time points (Fig. 4I). Time-dependent ROC analysis further validated its accuracy, with AUCs of 0.781 (1-year), 0.764 (3-year), and 0.725 (5-year) for survival prediction (Fig. 4J). The nomogram retained strong prognostic utility in independent cohorts (ICGC and GSE14520), consistently outperforming individual clinical variables (Fig. 4K,L), underscoring its generalizable clinical applicability.
Functional annotation and therapeutic implications of the MRGs
HCC patients in the TCGA-LIHC cohort were stratified into MRGs-high and -low groups using the median MRGs, revealing 471 up-regulated and 415 down-regulated DEGs (Fig. 5A). Subsequently, MRGs-related DEGs were subjected to GO and KEGG enrichment analysis. At the cellular level, these DEGs were predominantly localized in the collagen-containing extracellular matrix, apical plasma membrane, ion channel complex, transmembrane transporter complex, and so on. In terms of molecular function, these DEGs were primarily responsible for signaling receptor activator activity, receptor ligand activity, gated channel activity, passive transmembrane transporter activity, and other functions (Fig. 5B). In biological processes, MRGs-related genes positively modulated cell fate and angiogenesis, while negatively regulating detoxification of inorganic compounds and copper ions and homeostasis of zinc ions (Fig. 5C). KEGG pathway analysis further linked these DEGs to critical oncogenic pathways, including Neuroactive ligand-receptor interaction, Cytokine-cytokine receptor interaction, Xenobiotic/drug metabolism via cytochrome P450, and Chemical carcinogenesis (Fig. 5D).

Biological functions of MRGs. (A) The volcano map showing differentially expressed genes between MRGs using the TCGA-LIHC cohort. MRGs related DEGs were subjected to GO enrichment analysis, including MF, CC (B), and BP (C). (D) KEGG analysis of MRGs related DEGs. (E) The scatterplot demonstrating the correlation between MRGs and HRD using the TCGA-LIHC cohort. (F) Comparison of the difference in IC50 between MRGs-high and -low groups for cisplatin and erlotinib. ***P < 0.001.
Given the strong enrichment in drug metabolism pathways, we investigated the MRGs signature’s association with HRD, a pan-cancer biomarker predictive of platinum chemotherapy response. The MRGs exhibited a significant positive correlation with HRD (r = 0.385, P < 0.001; Fig. 5E). Consistently, drug sensitivity analysis revealed that high-MRGs tumors showed increased sensitivity to cisplatin (lower IC50, P < 0.001) but resistance to erlotinib (higher IC50, P < 0.001; Fig. 5F), suggesting high-MRGs patients may preferentially benefit from platinum-based regimens.
MRGs signature shapes an immunosuppressive tumor microenvironment
Given the MRGs signature’s association with cytokine-cytokine receptor interactions and chemokine signaling, we profiled immune infiltration using the CIBERSORTx algorithm. High-MRGs tumors exhibited elevated infiltration of M0 macrophages, regulatory T cells (Tregs), dendritic cells resting, T cells follicle helper, and activated CD4+ memory T cells, whereas low-MRG tumors were enriched in naïve B cells, resting CD4+ memory T cells, and activated NK cells (Fig. 6A,B). The MRGs positively correlated with key immunosuppressive checkpoints, including PD-1 (r = 0.263, P < 0.001) and CTLA-4 (r = 0.318, P < 0.001) (Fig. 6C). Immune subtyping revealed prognostic stratification, with high-MRGs tumors predominantly clustered into C1/C2 subtypes (poor prognosis), whereas low-MRGs tumors exhibited preferential segregation into the C3 cluster (favorable prognosis) (Fig. 6D).

Role of MRGs in the tumor microenvironment. (A) The heatmap showing the relationship between MRGs and immune cells on the basis of the Cibersort algorithm. (B) Differences in infiltration of immune cells between MRGs-high and -low groups. (C) Chord plot demonstrating the correlation of MRGs with four immunosuppressive checkpoints. (D) Differences in immune subtypes of HCC patients between MRGs-high and -low groups. C1: wound healing; C2: IFN-gamma dominant; C3: inflammatory; C4: lymphocyte depleted; C6: TGF-β dominant. (E,F) Single-cell analysis demonstrated the expression abundance of DTYMK, ABCB6, GOT2, and TOMM40L on immune cells using the GSE98638 cohort. (G) Volcano diagram showing differentially expressed genes on Tprolif and Tex cells. (H) Functional enrichment of the differentially expressed genes. (I,J) Heatmap showing the correlation of MRGs members with Tprolif-upregulated and Tex-downregulated genes. *P < 0.05, **P < 0.01, ***P < 0.001.
In the GSE98638 single-cell cohort, MRGs components (DTYMK, ABCB6, GOT2, and TOMM40L) were broadly expressed across immune cells, with DTYMK, ABCB6, and GOT2 showing pronounced enrichment in proliferating T cells (Tprolif) (Figs. 6E,F). Furthermore, given the critical role of exhausted CD8+ T cells (Tex), we analyzed differentially expressed genes between Tprolif and Tex populations, identifying 11 significantly down-regulated genes in Tprolif and 50 up-regulated genes in Tex (Fig. 6G). Functional enrichment linked these genes to antigen presentation, immune response regulation, and CD8+ T cell exhaustion pathways (Fig. 6H). Correlation analysis further revealed DTYMK positively associated with Tex-upregulated and Tprolif-downregulated genes, ABCB6 and TOMM40L correlated with Tex-upregulated genes, while GOT2 inversely correlated with exhaustion markers (Fig. 6I,J).
MRGs signature predicts genomic instability and immunotherapy resistance
To investigate the relationship between MRGs and tumor biology, we analyzed the top 20 mutated genes in HCC, revealing significantly higher TP53 mutation rates in high-MRGs tumors compared to low-MRGs counterparts (Fig. 7A). MRGs components demonstrated mutation-dependent expression patterns, with DTYMK, ABCB6, and TOMM40L showing up-regulation in TP53-mutant tumors while GOT2 exhibited suppression (Fig. 7B). The MRGs signature correlated strongly with tumor stemness, displaying significant positive association with RNAss (r = 0.383, P < 0.001) but no correlation with DNAss (Fig. 7C). Furthermore, high-MRGs tumors demonstrated elevated genomic instability markers, including increased TMB and MSI scores relative to low-MRGs tumors, suggesting heightened genomic instability. (Fig. 7D,E).

MRGs with genetic mutations and immunotherapy. (A) The oncoplot depicted gene mutation differences in HCC patients between MRGs-high and -low groups. (B) Differential expression of DTYMK, ABCB6, GOT2 and TOMM40L between TP53-mut and -wt groups. (C) The scatterplot demonstrated the correlation of MRGs with tumor stemness indicators, DNAss and RNAss. (D–F) Differences in TMB, MSI, and TIDE scores of HCC patients between MRGs-high and -low groups. (G,H) Kaplan–Meier curves demonstrated the difference in progression-free survival (PFS) of patients treated with nivolumab or ipilimumab between the MRGs-high and -low groups. (I,J) Kaplan–Meier curves demonstrated the difference in PFS of melanoma patients treated with anti-PD-1 or ACT between the MRGs-high and -low groups using the GSE91061 and GSE100797 cohort, respectively. **P < 0.01, ***P < 0.001.
To assess MRG’s predictive value for immunotherapy response, TIDE algorithm analysis showed higher immune evasion potential in high-MRGs HCC patients, indicating poorer immunotherapy responsiveness (Fig. 7F). Given limited HCC immunotherapy cohorts, we extended our validation to pan-cancer datasets. In nivolumab-treated (n = 173) and ipilimumab-treated (n = 112) cohorts, high-MRGs patients showed significantly shorter progression-free survival (Fig. 7G,H). Consistent results emerged from survival analysis of 101 anti-PD-1-treated melanoma patients (GSE91061 cohort), where high-MRGs patients had markedly reduced survival (Fig. 7I). Similarly, among 25 adoptive cell therapy (ACT)-treated melanoma patients, high-MRGs expression correlated with diminished therapeutic response (Fig. 7J).
Core gene ABCB6 knockdown suppresses HCC malignancy
To identify pivotal MRGs components, time-dependent ROC analysis highlighted ABCB6 as the most critical gene in the model (Fig. 8A). Clinical-pathological correlation analysis revealed that elevated ABCB6 expression significantly associated with advanced T-stage, tumor burden status, and vascular invasion in HCC patients (Fig. 8B-D), indicating that ABCB6 may drive tumor progression.

Knockdown of ABCB6 significantly inhibited HCC cell proliferation, migration, and invasion in vitro. (A) Time-dependent ROC curves were utilized to screen core genes for MRGs. (B–D) Differential expression of ABCB6 in different T-stages, tumor status and vascular invasion. (E) WB and qPCR assays were performed to validate the knockdown efficiency of ABCB6 in HepG-2 and Huh-7 cells. (F,G) The CCK-8 and colony formation assays demonstrated the effect of knockdown of ABCB6 expression on the proliferation of HCC cells. (H) Flow cytometry showing the effect of ABCB6 abrogation on the HCC cell cycle. (I,J) Wound healing and transwell assays exhibited the effects of ABCB6 knockdown on HCC cell migration and invasion. *P < 0.05, **P < 0.01, ***P < 0.001.
Subsequent functional validation confirmed efficient ABCB6 knockdown in HepG2 and Huh7 cells via western blotting and qRT-PCR (Fig. 8E). ABCB6 depletion profoundly inhibited HCC cell proliferation, as evidenced by CCK-8 assays and colony formation suppression (Fig. 8F,G). Cell cycle analysis demonstrated G2/M phase arrest in ABCB6-deficient cells (Fig. 8H). Furthermore, wound healing and transwell assays revealed marked impairment of migration and invasion capabilities following ABCB6 silencing (Fig. 8I,J). These findings collectively establish ABCB6 as a key driver of HCC progression.
Discussion
HCC remains a major contributor to global cancer-related mortality, with its poor prognosis largely attributed to metastatic progression and chemoresistance despite advancements in diagnostic and therapeutic strategies in recent years19,20. Elucidating the pathophysiological processes and molecular mechanisms driving HCC pathogenesis is therefore essential for developing effective clinical interventions21.
Dysregulated cellular energy metabolism, a hallmark of cancer progression, is closely linked to mitochondrial dysfunction arising from mtDNA mutations, enzyme deficiencies, or dysregulated oncogenes/tumor suppressors22. Mitochondria-associated genes have emerged as promising prognostic biomarkers across multiple tumor types23,24. In addition, they play pivotal roles in modulating tumor immune evasion mechanisms. For instance, PCK1 suppressed GTP-dependent cGAS activation, impairing STING-mediated immune cell infiltration and activation in the tumor microenvironment, thereby facilitating immune escape and tumor progression25. Similarly, MFN1 downregulation in tumor-associated macrophages disrupted mitochondrial dynamics to induce anti-tumor immune responses26, while DTYMK deficiency drives mitochondrial dysfunction and immunosuppression27. Despite these advances, current research predominantly focuses on isolated MRGs rather than comprehensive analyses of mitochondrial gene networks. A systematic evaluation of MRGs in HCC is therefore imperative to elucidate their collective impact on prognosis and therapeutic resistance.
This study identified a MRGs signature—comprising DTYMK, ABCB6, GOT2, and TOMM40L—as a robust prognostic biomarker for HCC. MRGs expression was markedly upregulated in HCC tissues and correlated with advanced TNM stages, suggesting their tumor-promoting function in driving disease progression. Specifically, elevated DTYMK and TOMM40L levels were linked to poorer survival outcomes and associated with cell cycle dysregulation and metabolic reprogramming28. The MRGs-high subgroup exhibited significantly reduced overall survival, while the MRGs-derived nomogram demonstrated high predictive accuracy for clinical outcomes. These findings advance the understanding of mitochondrial biomarkers in HCC prognosis and therapeutic stratification.
Emerging evidence indicates that mitochondrial dysregulation drives oncogenic progression through mechanisms including anti-apoptotic signaling and chemoresistance29. Notably, upregulated DTYMK and TOMM40L in HCC cells have been shown to induce resistance to sorafenib and 5-fluorouracil, respectively30,31. Intriguingly, our study revealed a paradoxical association of MRGs with chemotherapeutic responses: while negatively correlated with inorganic detoxification pathways, MRGs exhibited strong positive correlations with HRD—a genomic instability marker guiding platinum-based therapy. Computational drug response profiling further predicted enhanced cisplatin sensitivity but erlotinib resistance in MRGs-high HCC patients. These findings provide compelling mechanistic evidence linking mitochondrial gene networks to the efficacy of specific chemotherapeutic agents.
The ATP-binding cassette (ABC) transporter superfamily, utilizing ATP hydrolysis to drive substrate transport across cellular membranes, plays fundamental roles in cellular homeostasis32. Among these, ABCB6—a core MRG—belongs to the subfamily B of ABC transporters and has recently been implicated in diverse biological processes including DNA repair, translational regulation, and epigenetic modulation33,34. Despite these pleiotropic functions, the tumorigenic role of ABCB6 in HCC remains poorly characterized. Our functional investigations revealed that ABCB6 knockdown in HepG-2 and Huh-7 cells substantially suppressed proliferation by inducing G2/M phase arrest, while concurrently impairing migration and invasion capacities compared to controls. These in vitro findings collectively demonstrate the oncogenic dependency of HCC cells on ABCB6-mediated regulatory networks, highlighting its potential as a therapeutic vulnerability in HCC.
Tumorigenesis initiates through cancer cells’ evasion of immune surveillance—a process marked by depletion of cytotoxic immune populations, expansion of immunosuppressive cells, and overexpression of checkpoint molecules35,36,37. Our analysis demonstrated that MRGs-high tumors exhibit enhanced immunosuppressive landscapes, characterized by infiltrated Tregs and macrophages, upregulated checkpoint genes (PD-1 and CTLA-4), and diminished activated NK cells, collectively fostering protumoral niche formation. Mechanistically, tumor stem-like cells, which drive therapeutic resistance and recurrence38,39, showed significant correlation with elevated MRGs expression and RNAss, while MRGs-high subgroups exhibited intrinsic immunotherapy refractoriness. However, MRGs have certain limitations in predicting HCC immunotherapy, as in this study we used the TIDE algorithm with the TCGA-LIHC cohort to infer the predictive capability of MRGs for immunotherapy, while validation data primarily comes from melanoma cohorts. Additionally, we confirmed through a pan-cancer cohort treated with nivolumab and ipilimumab that high MRGs are indeed associated with poor immunotherapy response, which to some extent indicates the potential of MRGs for future clinical translation in HCC.
Collectively, our integrative analysis delineates MRGs as a systemic regulatory axis in HCC, with prognostic relevance spanning therapeutic response prediction, tumor microenvironment modulation, and genomic instability interplay. Through rigorous model evaluation via time-dependent ROC analysis, ABCB6 emerged as the central hub gene within the MRGs network. Functional validation demonstrated ABCB6’s essential role as an oncogenic driver, with genetic ablation in HCC cell lines markedly suppressing proliferative capacity through G2/M arrest and impairing metastatic competence via migration/invasion pathway disruption. These mechanistic insights position ABCB6 not merely as a biomarker but as a clinically actionable target, offering novel therapeutic entry points for precision oncology frameworks in liver cancer management.
However, our study still has several limitations. First, the analysis is based on retrospective cohorts, and prospective clinical validation is required to confirm the utility of the mitochondrial-related gene signature in real-world settings. Additionally, the insufficient predictive power of the current model represents one of the core limitations of this study. In future work, we will focus on improving model performance—for example, by expanding the sample size, incorporating more comprehensive multi-omics data, and exploring more advanced modeling techniques. Functional validation of the mitochondrial-related gene ABCB6, particularly in vivo, is another critical step to further strengthen the biological relevance of these markers.
Conclusion
This study demonstrates the translational potential of the MRGs comprising four key genes for prognostic stratification and immunotherapy response prediction in HCC. Crucially, functional characterization revealed ABCB6—the central MRGs component—as an oncogenic linchpin, with its genetic ablation in HCC models markedly suppressing malignant phenotypes including proliferation, migration, and invasive capacity through cell cycle disruption. Our findings bridge mechanistic insights with clinical applications, simultaneously advancing the MRGs framework as a predictive biomarker platform and establishing ABCB6-driven pathways as actionable targets for therapeutic development in hepatocarcinogenesis.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 71, 209–249. https://doi.org/10.3322/caac.21660 (2021).
El-Serag, H. B. Hepatocellular carcinoma: Recent trends in the United States. Gastroenterology 127, S27–S34. https://doi.org/10.1053/j.gastro.2004.09.013 (2004).
Xu, I. M. et al. Transketolase counteracts oxidative stress to drive cancer development. Proc. Natl. Acad. Sci. USA 113, E725–E734. https://doi.org/10.1073/pnas.1508779113 (2016).
Venook, A. P., Papandreou, C., Furuse, J. & de Guevara, L. L. The incidence and epidemiology of hepatocellular carcinoma: A global and regional perspective. Oncologist 15(Suppl 4), 5–13. https://doi.org/10.1634/theoncologist.2010-S4-05 (2010).
Lu, J. et al. Management of patients with hepatocellular carcinoma and portal vein tumour thrombosis: comparing east and west. Lancet Gastroenterol. Hepatol. 4, 721–730. https://doi.org/10.1016/S2468-1253(19)30178-5 (2019).
Cheng, A. L. et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 10, 25–34. https://doi.org/10.1016/S1470-2045(08)70285-7 (2009).
Wallace, D. C. Mitochondria and cancer. Nat. Rev. Cancer 12, 685–698. https://doi.org/10.1038/nrc3365 (2012).
Yang, Y. et al. Mitochondria and mitochondrial ROS in cancer: Novel targets for anticancer therapy. J. Cell Physiol. 231, 2570–2581. https://doi.org/10.1002/jcp.25349 (2016).
Han, T. H. et al. Stilbenoid derivatives as potent inhibitors of HIF-1alpha-centric cancer metabolism under hypoxia. Biomed. Pharmacother. 176, 116838. https://doi.org/10.1016/j.biopha.2024.116838 (2024).
Poljsak, B. & Milisav, I. Decreasing intracellular entropy by increasing mitochondrial efficiency and reducing ROS formation-the effect on the ageing process and age-related damage. Int. J. Mol. Sci. 25, 6321. https://doi.org/10.3390/ijms25126321 (2024).
Yi, L. et al. Inflammation-mediated SOD-2 upregulation contributes to epithelial-mesenchymal transition and migration of tumor cells in aflatoxin G(1)-induced lung adenocarcinoma. Sci. Rep. 7, 7953. https://doi.org/10.1038/s41598-017-08537-2 (2017).
DeNicola, G. M. et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109. https://doi.org/10.1038/nature10189 (2011).
Fan, S. et al. Shikonin blocks CAF-induced TNBC metastasis by suppressing mitochondrial biogenesis through GSK-3beta/NEDD4-1 mediated phosphorylation-dependent degradation of PGC-1alpha. J. Exp. Clin. Cancer Res. 43, 180. https://doi.org/10.1186/s13046-024-03101-z (2024).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
Ogata, H. et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34. https://doi.org/10.1093/nar/27.1.29 (1999).
Thorsson, V. et al. The immune landscape of cancer. Immunity 48, 812–830. https://doi.org/10.1016/j.immuni.2018.03.023 (2018).
Malta, T. M. et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 173, 338–354. https://doi.org/10.1016/j.cell.2018.03.034 (2018).
Bonneville, R. et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis. Oncol. https://doi.org/10.1200/PO.17.00073 (2017).
Ozer, E. D., Suna, N. & Boyacioglu, A. S. Management of hepatocellular carcinoma: Prevention, surveillance, diagnosis, and staging. Exp. Clin. Transplant. 15, 31–35. https://doi.org/10.6002/ect.TOND16.L9 (2017).
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 73, 17–48. https://doi.org/10.3322/caac.21763 (2023).
He, Y. et al. Biomarkers and future perspectives for hepatocellular carcinoma immunotherapy. Front Oncol. 11, 716844. https://doi.org/10.3389/fonc.2021.716844 (2021).
Hsu, C. C., Tseng, L. M. & Lee, H. C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. (Maywood) 241, 1281–1295. https://doi.org/10.1177/1535370216641787 (2016).
Herkenne, S. et al. Developmental and tumor angiogenesis requires the mitochondria-shaping protein Opa1. Cell Metab. 31, 987–1003. https://doi.org/10.1016/j.cmet.2020.04.007 (2020).
Park, S. H. et al. TOMM20 as a potential therapeutic target of colorectal cancer. Bmb Rep. 52, 712–717. https://doi.org/10.5483/BMBRep.2019.52.12.249 (2019).
Qin, W. et al. PCK1 inhibits cGAS-STING activation by consumption of GTP to promote tumor immune evasion. J. Exp. Med. https://doi.org/10.1084/jem.20240902 (2025).
Zhao, M. et al. Engineering nanoparticles boost TNBC therapy by CD24 blockade and mitochondrial dynamics regulation. J. Control Release 355, 211–227. https://doi.org/10.1016/j.jconrel.2023.01.075 (2023).
Felizardo, T. C. et al. Harnessing autophagy for cell fate control gene therapy. Autophagy 9, 1069–1079. https://doi.org/10.4161/auto.24639 (2013).
Guo, Y. et al. DTYMK expression predicts prognosis and chemotherapeutic response and correlates with immune infiltration in hepatocellular carcinoma. J. Hepatocell Carcinoma 8, 871–885. https://doi.org/10.2147/JHC.S312604 (2021).
Lee, J. Y., Ham, J., Lim, W. & Song, G. Apomorphine induces mitochondrial-dysfunction-dependent apoptosis in choriocarcinoma. Reproduction 160, 367–377. https://doi.org/10.1530/REP-20-0230 (2020).
Gu, X., Li, S., Ma, X., Huang, D. & Li, P. Heterogeneity characterization of hepatocellular carcinoma based on the sensitivity to 5-fluorouracil and development of a prognostic regression model. Front Pharmacol. 14, 1252805. https://doi.org/10.3389/fphar.2023.1252805 (2023).
Sun, F. et al. Inhibition of DTYMK significantly restrains the growth of HCC and increases sensitivity to oxaliplatin. Cell Death Dis. 12, 1093. https://doi.org/10.1038/s41419-021-04375-3 (2021).
Xiang, L. et al. HIF-1-dependent heme synthesis promotes gemcitabine resistance in human non-small cell lung cancers via enhanced ABCB6 expression. Cell Mol. Life Sci. 79, 343. https://doi.org/10.1007/s00018-022-04360-9 (2022).
Emadi-Konjin, H. P. et al. Isolation of a genomic clone containing the promoter region of the human ATP binding cassette (ABC) transporter, ABCB6. Biochim. Biophys. Acta 1574, 117–130. https://doi.org/10.1016/s0167-4781(01)00340-2 (2002).
Xiao, H. N., Zhao, Z. Y., Li, J. P. & Li, A. Y. Comprehensive pan-cancer analysis: Essential role of ABCB family genes in cancer. Transl. Cancer Res. 13, 1642–1664. https://doi.org/10.21037/tcr-23-2050 (2024).
Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 12, 31–46. https://doi.org/10.1158/2159-8290.CD-21-1059 (2022).
Senga, S. S. & Grose, R. P. Hallmarks of cancer-the new testament. Open Biol. 11, 200358. https://doi.org/10.1098/rsob.200358 (2021).
Rui, R., Zhou, L. & He, S. Cancer immunotherapies: Advances and bottlenecks. Front Immunol. 14, 1212476. https://doi.org/10.3389/fimmu.2023.1212476 (2023).
Prasetyanti, P. R. & Medema, J. P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 16, 41. https://doi.org/10.1186/s12943-017-0600-4 (2017).
Sancho, P., Barneda, D. & Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 114, 1305–1312. https://doi.org/10.1038/bjc.2016.152 (2016).
Author information
Authors and Affiliations
Contributions
Hongli Shu conducted experimental investigations, performed data analysis and interpretation, and drafted the manuscript; Rui Li and Xiaolong Tang provided critical analytical tools and contributed essential datasets; Qiongfang Zhang conceptualized the research framework, supervised the study design, and ensured methodological rigor. All authors reviewed the final manuscript, approved its intellectual content, and endorsed its submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shu, H., Li, R., Tang, X. et al. A mitochondrial-related gene signature predicts prognosis and immunotherapy response in hepatocellular carcinoma. Sci Rep 15, 25435 (2025). https://doi.org/10.1038/s41598-025-09464-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-09464-3
