
Abstract
Chlorogenic acid, an ester of caffeic acid and quinic acid, is found in foods such as eggplant and peaches. Its role in heart disease remains poorly understood. This study investigated whether chlorogenic acid affects cardiac hypertrophy and fibrosis in animal and cellular models of isoproterenol-induced cardiac hypertrophy. Treatment of isoproterenol-stimulated cardiomyocytes with chlorogenic acid reduced cell size and the expression levels of cardiac hypertrophy-related genes. In the animal model, isoproterenol was delivered via an osmotic minipump for 2 weeks to induce cardiac hypertrophy, and chlorogenic acid was intraperitoneally administered for the same duration. Echocardiographic analysis showed that chlorogenic acid significantly reduced wall thickness in mice. Picrosirius red staining, quantitative reverse transcription polymerase chain reaction, and Western blot analysis revealed that cardiac fibrosis was attenuated by chlorogenic acid. Chlorogenic acid treatment downregulated galectin 3 (Lgals3), a fibrosis-associated gene that had been upregulated by isoproterenol stimulation. Galectin 3 knockdown ameliorated isoproterenol-induced cardiac hypertrophy and reduced the expression levels of COL1A1 and ADAMTS8; galectin 3 overexpression increased cardiomyocyte size and upregulated COL1A1 and ADAMTS8 expression levels. These findings suggest that chlorogenic acid could serve as a novel treatment for cardiac hypertrophy and fibrosis via downregulation of galectin 3.
Similar content being viewed by others


Introduction
Chlorogenic acid, a phenolic compound composed of caffeic acid and quinic acid, is found in vegetables and fruits. It has been reported to exert anti-liver fibrosis1, anti-cancer2, anti-inflammatory3, antioxidant4, and anti-cardiac hypertrophy5,6 effects. A previous study demonstrated that chlorogenic acid inhibited cardiac hypertrophy through suppressing Wnt/beta catenin signaling pathway or endoplasmic reticulum stress; the present study explored its effects on cardiac hypertrophy and fibrosis through a different mechanism.
Cardiac hypertrophy and fibrosis are often caused by various stimuli, including hypertrophic cardiomyopathy, hypertension, myocardial infarction, and aortic stenosis7. When these stimuli persist, the resulting changes can become irreversible, leading to heart failure. In experimental models, isoproterenol administration or transverse aortic constriction (TAC) surgery is commonly used to induce cardiac hypertrophy. TAC is a model of pressure overload-induced cardiac hypertrophy and heart failure that requires skilled investigators. Here, we selected a more consistent animal model involving isoproterenol infusion through an osmotic minipump. Cardiac fibrosis is characterized by increased collagen type I deposition in cardiac muscle, which results in cardiac function abnormalities8. Pathological cardiac hypertrophy and fibrosis impair cardiac function and warrant intervention to mitigate their effects.
Galectin 3 (Lgals3), a member of the β-galactoside-binding protein family, serves diverse biological functions. In cardiovascular diseases, it is regarded as a biomarker of heart failure9,10 and is strongly associated with cardiac remodeling and fibrosis11. Indeed, galectin 3 protein expression is increased in an animal model of isoproterenol-induced heart failure12. Serum galectin 3 levels also have prognostic value for cardiovascular mortality in patients with chronic heart failure13. A disintegrin and metalloproteinase with thrombospondin motifs 8 and 12 (ADAMTS8 and ADAMTS12) are directly involved in fibrosis promotion14,15,16. ADAMTS8 expression was substantially increased in patients with dilated cardiomyopathy, along with the levels of collagen type I and α-smooth muscle actin (SMA). ADAMTS12 expression was elevated in fibroblasts after unilateral ureter obstruction surgery in mouse and human kidneys. Additionally, the genetic loss of ADAMTS12 ameliorated fibrosis in the kidney and heart16.
This study investigated the effects and mechanisms of chlorogenic acid in an isoproterenol-induced cardiac hypertrophy and fibrosis model. To our knowledge, this is the first report to demonstrate that galectin 3 plays a vital role in the development of cardiac hypertrophy with fibrosis and may serve as a novel therapeutic target for these conditions.
Results
Chlorogenic acid prevented cardiac hypertrophy in H9c2 cells
Chlorogenic acid is a phytochemical found in eggplants, peaches, prunes, and coffee beans (Fig. 1A). To determine whether chlorogenic acid plays a role in the treatment of cardiac hypertrophy, its toxicity was assessed in H9c2 cardiomyoblast cells. Chlorogenic acid showed no adverse effect on cell viability at concentrations up to 10 μM for 24 h (Fig. 1B). Texas Red™-X Phalloidin staining revealed that isoproterenol (10 μM) increased cell size, an effect significantly reduced by chlorogenic acid treatment (0.1 μM for 24 h) (Fig. 1C,D). Under these conditions, chlorogenic acid did not induce cytotoxicity (Fig. 1E). NPPA and natriuretic peptide B (NPPB) are well-established hypertrophy markers. No significant differences were observed in Nppa or Nppb expression between the control and chlorogenic acid-treated groups in the absence of isoproterenol stimulation (Fig. 1F,G). However, chlorogenic acid significantly prevented the isoproterenol-induced upregulation of these genes (Fig. 1F,G). Reactivation of fetal gene expression, including Myh7, is a hallmark of cardiac hypertrophy17. Chlorogenic acid treatment (9 h) suppressed the isoproterenol-induced increase in Myh7 expression (Fig. 1H), without causing cytotoxicity under these conditions (Fig. 1I).

Chlorogenic acid attenuated cardiac hypertrophy in H9c2 cells. (A) Chemical structure of chlorogenic acid. (B) Cell viability following treatment with chlorogenic acid (0–10 μM for 24 h). (C) Representative images of H9c2 cells stained with Texas Red™-X Phalloidin after isoproterenol treatment (10 μM, 24 h) with or without chlorogenic acid (0.1 μM, 24 h). Scale bar = 100 μm. (D) Quantification of cell size. (E) Cytotoxicity under the same conditions as in (C). (F–H) mRNA expression levels of Nppa, Nppb, and Myh7 in H9c2 cells treated with isoproterenol (10 μM) with or without chlorogenic acid (0.1 μM) for 9 h. Gapdh was used for normalization. (I) Cytotoxicity under the same conditions as in (F–H). ***P < 0.001; ###P < 0.001. ISO; isoproterenol, CGA; chlorogenic acid, DAPI; 4’,6-diamidino-2-phenylindole.
Chlorogenic acid prevented cardiac hypertrophy in a mouse model of isoproterenol-induced hypertrophy
To evaluate whether chlorogenic acid influences cardiac hypertrophy in mice, we used an isoproterenol-treated mouse model. As shown in Fig. 2A–C, isoproterenol administration increased gross heart weight, as well as the heart weight-to-body weight (HW/BW) and heart weight-to-tibia length (HW/TL) ratios. These increases were attenuated by chlorogenic acid treatment. Hematoxylin and eosin (H&E) staining, along with wheat germ agglutinin staining, demonstrated that chlorogenic acid reduced the isoproterenol-induced enlargement of cardiomyocyte cross-sectional area (Fig. 2D–F). To further assess the effects of chlorogenic acid on cardiac remodeling, we performed echocardiography. Figure 3A presents representative M-mode echocardiograms. As expected, isoproterenol treatment significantly elevated heart rate compared with the sham group, and this increase was reduced by chlorogenic acid (Fig. 3B). The thicknesses of the left ventricular interventricular septum and posterior wall were significantly increased in the isoproterenol-treated group relative to the sham group. These increases were attenuated by chlorogenic acid treatment (Fig. 3C,D). In cardiac hypertrophy, the lumen typically narrows. The left ventricular internal diameters at systole and diastole were decreased by isoproterenol treatment and restored by chlorogenic acid (Fig. 3E,F). Left ventricular ejection fraction and fractional shortening did not significantly differ among the three groups (Fig. 3G,H).

Chlorogenic acid prevented cardiac hypertrophy in a mouse model of isoproterenol-induced cardiac hypertrophy. (A) Representative gross heart image. Scale on the ruler represents 1 mm. (B–C) Heart weight-to-body weight ratio and heart weight-to-tibia length ratio in three animal groups (n = 9–10 per group). ***P < 0.001; ###P < 0.001. (D) Representative H&E-stained heart tissues (n = 5–6 per group). Top panel: Low magnification (Scale bar = 1000 μm). Bottom panel: High magnification (Scale bar = 50 μm). (E) Quantification of cardiomyocyte area in heart tissues (n = 5–6 per group). (F) Representative wheat germ agglutinin-stained heart tissues. Top panel: Low magnification (Scale bar = 1000 μm). Bottom panel: High magnification (Scale bar = 50 μm). ***P < 0.001; ###P < 0.001. ISO; isoproterenol, CGA; chlorogenic acid.

Chlorogenic acid improved cardiac remodeling in isoproterenol-induced cardiac hypertrophy. (A) Representative M-mode echocardiographic images of the sham, isoproterenol, and isoproterenol + chlorogenic acid groups (n = 8 per group). (B–H) Quantification of echocardiographic parameters. IVSd (mm): interventricular septum thickness; LVPWd (mm): left ventricular posterior wall thickness; LVIDs (mm): left ventricular internal diameter at systole; LVIDd (mm): left ventricular internal diameter at diastole; LVFS (%): fractional shortening; LVEF (%): ejection fraction. ***P < 0.001; ##P < 0.01 and ###P < 0.001. ISO; isoproterenol, CGA; chlorogenic acid.
Chlorogenic acid mitigated the expression of cardiac hypertrophic genes and galectin 3 in an isoproterenol-induced hypertrophy model
Next, we investigated the effects of chlorogenic acid on cardiac hypertrophy marker genes. Nppa, Nppb, and Lgals3 are well-established cardiac hypertrophy and heart failure markers18,19. Expression levels of these genes were significantly increased in the isoproterenol group compared with the sham group; these increases were attenuated by chlorogenic acid (Fig. 4A–C). Chlorogenic acid also reduced the isoproterenol-induced elevation in Myh7 mRNA levels (Fig. 4D). Osteopontin (i.e., SPP1) is implicated in cardiac remodeling and fibrosis20,21. Chlorogenic acid treatment reversed the isoproterenol-induced increase in Spp1 mRNA expression (Fig. 4E). Western blot analysis further confirmed that the protein expression levels of NPPA, LGALS3, and SPP1 were significantly decreased by chlorogenic acid (Fig. 4F–I), consistent with the qRT-PCR results.

Chlorogenic acid attenuated cardiac hypertrophy-related gene expression in isoproterenol-induced cardiac hypertrophy mice. (A–E) mRNA expression levels of Nppa, Nppb, Lgals3, Myh7, and Spp1 in heart tissues, measured by qRT-PCR (n = 6–10 per group). Actb was used for normalization. (F) Western blot images of the three experimental groups. ACTB was used as a loading control. The blots above and below the green line are different gels. (G − I) Protein levels were quantified using ImageJ software (n = 4–6 per group). **P < 0.01 and ***P < 0.001; #P < 0.05, ##P < 0.01, and ###P < 0.001. ISO; isoproterenol, CGA; chlorogenic acid.
Chlorogenic acid attenuated cardiac fibrosis and inflammation in the isoproterenol-induced hypertrophy model
The effects of chlorogenic acid on cardiac fibrosis were assessed in the isoproterenol-induced hypertrophy model. Picrosirius red staining revealed that collagen deposition (pink color) was reduced in the chlorogenic acid-treated isoproterenol group relative to the vehicle-treated isoproterenol group (Fig. 5A,B). Fibrosis-related genes were analyzed by qRT-PCR. Col1a1 and Ccn2 (CTGF) mRNA expression levels were significantly increased in the isoproterenol group relative to the sham group (Fig. 5C,D). These increases were attenuated by chlorogenic acid treatment. ADAMTS proteins are known to function as extracellular matrix remodeling enzymes22. Adamts8 mRNA levels were upregulated in the isoproterenol group, and this increase was reduced by chlorogenic acid (Fig. 5E). Adamts12 mRNA levels were elevated in the isoproterenol group compared with the sham group; however, chlorogenic acid treatment did not alter this increase (Fig. 5F). Additionally, chlorogenic acid reduced COL1A1 protein expression levels in the isoproterenol-treated group (Fig. 5G,H). To determine whether inflammation contributed to the chlorogenic acid-mediated reduction in galectin 3, we analyzed the expression of inflammation-related genes. Chlorogenic acid significantly reduced the isoproterenol-induced upregulation of Nos2, Tnf, and Il1b (Fig. 5I–K).

Chlorogenic acid suppressed isoproterenol-induced cardiac fibrosis and inflammation in mice. (A) Representative picrosirius red-stained images of heart tissues. Top: Low magnification (Scale bar = 1000 μm). Bottom: High magnification (Scale bar = 50 μm). Pink staining indicates collagen deposition. (B) Quantification of cardiac fibrosis in low-magnification images (n = 5 per group). (C–F) mRNA levels of Col1a1, Ccn2, Adamts8, and Adamts12 were measured in heart tissues by qRT-PCR (n = 4–8 per group). (G and H) Representative Western blot images of COL1A1 in heart tissues (n = 6–8 per group). (I–J) mRNA expression levels of Nos2, Tnf, and Ilb in heart tissues, measured by qRT-PCR (n = 7–9 per group). **P < 0.01 and ***P < 0.001; #P < 0.05, ##P < 0.01, and ###P < 0.001. ISO; isoproterenol, CGA; chlorogenic acid.
Galectin 3 knockdown prevented isoproterenol-induced cardiac hypertrophy
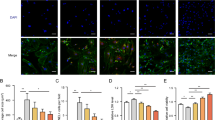
To determine whether galectin 3 plays a role in cardiac hypertrophy, we conducted galectin 3 knockdown experiments in H9c2 cells. Transfection of galectin 3 siRNA at concentrations of 10–100 nM significantly reduced Lgals3 mRNA levels; the greatest effect was observed at 100 nM (Fig. 6A). This result was consistent with the Western blot findings (Fig. 6B). To determine whether galectin 3 influences cardiomyocyte hypertrophy, we measured cell size after isoproterenol stimulation (10 μM, 24 h) following galectin 3 siRNA transfection. Texas Red™-X Phalloidin staining demonstrated that galectin 3 knockdown attenuated the isoproterenol-induced increase in cell size (Fig. 6C,D). Furthermore, galectin 3 knockdown completely suppressed the isoproterenol-induced increases in Lgals3 and Nppa mRNA levels (Fig. 6E,F). Western blot results were consistent with qRT-PCR findings (Fig. 6G,H).

Galectin 3 knockdown reduced isoproterenol-induced cardiomyocyte hypertrophy. (A–B) Galectin 3 mRNA and protein expression after galectin 3 knockdown at 10–100 nM concentrations. (C) Representative fluorescence microscopy images of H9c2 cells stained with Texas Red™-X Phalloidin. Cardiomyocytes were treated as follows: control siRNA (siCont), Galectin 3 siRNA (siGal3), control siRNA with isoproterenol (siCont + ISO), and Galectin 3 siRNA with isoproterenol (siGal3 + ISO). Red color indicates F-actin in the cytoskeleton; blue color indicates cell nuclei. Scale bar = 100 μm. (D) Quantification of cell surface area for each group shown in C. (E–F) Galectin 3 was silenced and stimulated with isoproterenol in H9c2 cells. mRNA levels of Lgals3 and Nppa were measured by RT-PCR. Gapdh was used for normalization. (G–H) Representative Western blot analysis of LGALS3 expression in H9c2 cells transfected with siCont or siGal3, with or without isoproterenol treatment (10 μM for 24 h). **P < 0.01 and ***P < 0.001; ###P < 0.001; @@@P < 0.001. ISO; isoproterenol, CGA; chlorogenic acid.
Galectin 3 regulated fibrosis-related gene expression
Next, we examined the involvement of galectin 3 in cardiac fibrosis. Galectin 3 knockdown did not directly affect COL1A1 protein expression; however, it reduced COL1A1 expression in the presence of isoproterenol stimulation (Fig. 7A,B). ADAMTS8 protein levels were significantly increased in isoproterenol-treated cells, and these increases were attenuated by galectin 3 siRNA (Fig. 7C,D). Additionally, galectin 3 knockdown directly suppressed Spp1 mRNA expression in H9c2 cells and mitigated the isoproterenol-induced increase in Spp1 expression (Fig. 7E).

Galectin 3 regulated COL1A1 and ADAMTS8 in cardiomyocytes. (A–D) Western blot analysis of COL1A1 and ADAMTS8 in galectin 3-transfected cells in the presence or absence of isoproterenol (10 μM). ACTB was used as a loading control. B and D show quantification of COL1A1 and ADAMTS8 expression. (E) Spp1 mRNA levels in Galectin 3 siRNA-transfected H9c2 cells in the presence or absence of isoproterenol (10 μM for 24 h). (F–I) mRNA levels of Lgals3, Col1a1, Adamts8, and Spp1 in pCMV3-N-GFPSpark-galectin 3-transfected H9c2 cells. (J–M) Representative immunoblots and quantification in H9c2 cells with galectin 3 overexpression. *P < 0.05, **P < 0.01, and ***P < 0.001; #P < 0.05 and ###P < 0.001; @@P < 0.01 and @@@P < 0.001. ISO; isoproterenol.
We investigated whether galectin 3 overexpression plays a role in fibrosis promotion. Transfection of the pCMV3-N-GFPSpark-galectin 3 plasmid visibly increased Lgals3 mRNA levels in H9c2 cells (Fig. 7F). Galectin 3 overexpression significantly increased the mRNA expression levels of Col1a1 and Adamts8 but not Spp1 (Fig. 7G–I). Protein expression from the pCMV3-N-GFPSpark-galectin 3 plasmid was detected at 56 kDa using an anti-GFP antibody (Fig. 7J). LGALS3 expression increased by more than twofold when detected using an anti-galectin 3 antibody (Fig. 7K). Furthermore, galectin 3 overexpression significantly upregulated the protein expression levels of ADAMTS8 and COL1A1 (Fig. 7L,M).
Galectin 3 overexpression increased cardiomyocyte size
Next, we examined the direct effect of Galectin 3 on cell size. After transfection of the GFP vector and pCMV3-N-GFPSpark-galectin 3 plasmid in H9c2 cells, we measured cell size using Texas Red™-X Phalloidin staining. Galectin 3 overexpression significantly increased cell size (Fig. 8A–B). Additionally, galectin 3 overexpression upregulated mRNA and protein expression levels of NPPA (Fig. 8C–E).

Galectin 3 overexpression increased cardiomyocyte size. (A–B) Representative images of cells overexpressing pCMV3-N-GFPSpark-galectin 3 and quantification of cell size. Green fluorescence indicates GFP expression. Each merged image represents a combination of the three individual channels. Scale bar = 100 μm. (C) Nppa mRNA levels in galectin 3-overexpressing cells. (D–E) Representative immunoblot images and quantification of NPPA expression in galectin 3-overexpressing cells. **P < 0.01 and ***P < 0.001. GFP; green fluorescent protein. (F) Schematic diagram of the proposed mechanism showing how chlorogenic acid regulates cardiac hypertrophy and fibrosis.
Discussion
This study demonstrated that chlorogenic acid mitigated cardiac hypertrophy and fibrosis in isoproterenol-treated mice and cardiomyocytes. The isoproterenol-induced upregulation of galectin 3, SPP1, and ADAMTS8 was associated with cardiac fibrosis. These effects were attenuated by chlorogenic acid treatment. Notably, galectin 3 induced cardiac hypertrophy and increased the expression levels of fibrosis-related genes, including COL1A1 and ADAMTS8 (Fig. 8F). These findings align with a previous study demonstrating that chlorogenic acid pretreatment prevents isoproterenol-induced hypertrophy in neonatal rat myocytes by reducing inflammation and reactive oxygen species production23. Moreover, Ping et al. reported that chlorogenic acid attenuates cardiac hypertrophy by activating the AMP-activated protein kinase/sirtuin 1 pathway in a rat model of isoproterenol-induced hypertrophy6.
Our study differs from previous research in that it demonstrated chlorogenic acid-mediated inhibition of fibrosis and cardiac hypertrophy; it also identified a novel mechanism for fibrosis inhibition. Indeed, chlorogenic acid attenuated COL1A1 mRNA and protein expression levels, along with collagen deposition. We also examined the role of galectin 3 in animal models of cardiac hypertrophy and fibrosis. Galectin 3 expression was increased in heart tissues under isoproterenol stimulation. Galectin 3 is recognized as a biomarker in various diseases, including heart failure24, and is implicated in fibrogenesis. Although no prior studies have revealed an effect of galectin 3 on cardiac hypertrophy, it has been associated with fibrosis. For instance, fibrosis was reduced in galectin 3 knockout mice receiving angiotensin II or TAC. Additionally, fibrosis was attenuated by galectin 3 inhibition using N-acetyllactosamine in TGR(mREN2)27 heart failure rats25. These findings indicate that galectin 3 may promote fibrosis. In the present study, galectin 3 knockdown did not affect fibrosis-related genes, including COL1A1 and ADAMTS8, in the absence of isoproterenol stimulation. However, these genes were significantly downregulated by galectin 3 knockdown in the presence of hypertrophic stimulation. Our findings suggest that galectin 3 indirectly induces fibrosis under cardiac hypertrophic stress stimuli. In addition to its contributions to cardiac fibrosis, galectin 3 is overexpressed in patients with chronic kidney disease and idiopathic pulmonary fibrosis, where it plays a critical role in the fibrotic process26,27,28. Galectin 3 has also been implicated in both inflammatory and fibrotic responses24. It is primarily secreted by macrophages and has been shown to influence infarct wall thickness after myocardial infarction29. In our study, expression levels of inflammatory markers, including Lgals3, Nos2, Il1b, and Tnf, were elevated after isoproterenol stimulation and reduced by chlorogenic acid treatment. These results indicate that chlorogenic acid suppresses isoproterenol-induced upregulation of galectin 3 via modulation of the inflammatory response.
ADAMTS8 expression was increased in myocardial infarction, a TAC-induced cardiac fibrosis rat model, angiotensin II-treated cardiac fibroblasts, and human dilated cardiomyopathy with severe cardiac fibrosis. ADAMTS8 overexpression promoted the expression of α-SMA, COL1A1, and CTGF, whereas ADAMTS8 knockdown inhibited their expression14. Another study demonstrated that ADAMTS8 knockdown attenuated angiotensin II-induced cardiac fibrosis and dysfunction in mice30. ADAMTS8 has been implicated in fibrosis-related diseases, including pulmonary arterial hypertension and myocardial infarction14,31. Further studies are needed to determine whether ADAMTS8 represents a potential therapeutic target for cardiac fibrosis and dysfunction.
Hoeft et al. reported that ADAMTS12 knockout mice were protected from kidney and cardiac fibrosis16. In the present study, ADAMTS8 and ADAMTS12 expression levels increased in response to isoproterenol-induced cardiac fibrosis. These two ADAMTS proteins were selected for investigation because preliminary RNA-seq results revealed a more than twofold increase in expression in the isoproterenol group compared with the control group. As described above, the reduction in ADAMTS8 expression after galectin 3 knockdown suggests that the galectin 3–ADAMTS8 axis is involved in the fibrosis process.
Conversely, galectin 3 knockdown suppressed SPP1 in both the presence and absence of isoproterenol stimulation. SPP1 was identified as a key gene in fibrotic endothelial-mesenchymal transition among patients with degenerative ascending aortic aneurysm20. SPP1 knockdown inhibited TGF-β1-induced COL1A1 and α-SMA expression, whereas SPP1 overexpression promoted fibroblast proliferation32. These findings suggest that SPP1 also plays a role in the fibrosis process.
Taken together, our results imply that galectin 3 acts directly on SPP1 and indirectly on ADAMTS8 and COL1A1. To further investigate the role of galectin 3, we performed overexpression experiments, revealing that galectin 3 overexpression increased the expression levels of ADAMTS8 and COL1A1 but not SPP1. Notably, galectin 3 overexpression also increased cardiomyocyte size, which was accompanied by the upregulation of NPPA expression.
In summary, this study demonstrated that chlorogenic acid attenuates cardiac hypertrophy and fibrosis via downregulation of the galectin 3–ADAMTS8/SPP1 axis. Targeting galectin 3, ADAMTS8, or SPP1 could serve as a therapeutic strategy for cardiac fibrosis with hypertrophy.
Materials and methods
Reagents
Isoproterenol (cat. no. I5627) was obtained from Sigma-Aldrich Co. (St. Louis, MO, USA). Chlorogenic acid (cat. no. J60457.03) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Antibodies against β-actin (ACTB, cat. no. sc-47778), type I collagen alpha 1 (COL1A1, cat. no. sc-293182), galectin-3 (LGALS3, cat. no. sc-32790), ADAMTS8 (cat. no. sc-514717), green fluorescent protein (GFP, cat. no. sc-9996), and secreted phosphoprotein 1 (SPP1, cat. no. sc-21742) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). The anti-atrial natriuretic peptide antibody (NPPA, cat. no. GTX639071) was obtained from GeneTex Inc. (Irvine, CA, USA). Texas Red™-X Phalloidin (cat. no. T7471) was acquired from Invitrogen (Carlsbad, CA, USA).
Ethics statement
All experiments were approved by the Ethics of Animal Experimental Committee at Chonnam National University Medical School (CNUH IACUC-24033). All animal experiments were conducted in strict adherence to relevant ethical and regulatory guidelines, including the ARRIVE guidelines (https://arriveguidelines.org).
Animal model of cardiac hypertrophy
We purchased icrTac:ICR outbred mice (8 weeks old, weighting 33–35 g) from Damool Science Laboratory Animal Technology (Daejeon, Korea) because this age group exhibits a well-established cardiac hypertrophy response. All mice were housed in an environment with free access to food and water under the proper temperature (22–25 °C) and relative humidity control (55–65%) with a 12:12 h light–dark cycle. The mice were given one week to acclimatize. Male mice were anesthetized via intraperitoneal injection of ketamine (36–45 mg/kg) and xylazine (17–21 mg/kg). The animals were subcutaneously implanted with osmotic minipumps and monitored for 14 days. The minipumps were filled with either vehicle (0.9% NaCl) or isoproterenol (25 mg/kg body weight/day). The study consisted of three groups: sham + vehicle (n = 10), isoproterenol + vehicle (n = 9), and isoproterenol + chlorogenic acid (n = 9). Chlorogenic acid (10 mg/kg/day) was administered intraperitoneally once daily for 14 days, beginning 24 h after minipump implantation.
Echocardiography
Mouse echocardiography is a research technique that enables assessments of heart muscle and valve function, as well as evaluations regarding the effects of genetic, pharmacological, and mechanical interventions on the left ventricle (e.g., coronary artery ligation and transverse aortic stenosis). This method can detect various heart diseases, including coronary artery disease, heart failure, and myocardial disorders. Echocardiograms were obtained using the Vivid S5 system (GE Healthcare, Chicago, IL, USA). Mice (n = 8 per group) were anesthetized with an intraperitoneal injection of 2,2,2-tribromoethanol (Avertin; 146 mg/kg) before imaging. Data acquisition included M-mode images at the level of the papillary muscles and short-axis views. Measurements included left ventricular posterior wall thickness, interventricular septal thickness, left ventricular end-diastolic diameter, and left ventricular end-systolic diameter. Ejection fraction and fractional shortening were calculated and evaluated. Echocardiographic assessments were performed by an expert following established guidelines33.
Histological analysis, picrosirius red staining, and wheat germ agglutinin staining
Mice were euthanized via CO2 inhalation. Heart tissues were fixed with 4% paraformaldehyde, embedded in paraffin, and sectioned at a thickness of 3 μm. For H&E staining, sections were deparaffinized twice with xylene and rehydrated through a graded ethanol series (100–70% ethanol). The sections were incubated with hematoxylin for 4 min, rinsed with tap water for 5 min, incubated with eosin and phloxine for 1 min, and dehydrated with 90–100% ethanol. Finally, the sections were cleared with xylene and mounted. NIS Elements Software Version AR 3.0 (Nikon, Japan) was used to measure cross-sectional area. Wheat germ agglutinin staining was performed to visualize cell membranes and delineate individual cardiomyocytes. After deparaffinization and blocking with 3% normal goat serum in PBS for 30 min, sections were incubated with wheat germ agglutinin conjugated to Alexa Fluor 488 (1:100 dilution) for 30 min and examined under a fluorescence microscope (Nikon, Japan).
To determine cardiac fibrosis, picrosirius red staining was performed using a kit (ab246832, Abcam, Cambridge, UK). Heart sections were deparaffinized and hydrated with distilled water. The sections were incubated with picrosirius red solution for 1 h, then briefly rinsed with 0.5% acetic acid solution. The sections were subsequently rinsed with 100% ethanol, dehydrated with ethanol, and mounted. Images were captured using a fluorescence microscope (Eclipse 80i, Nikon, Japan).
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
RNA was extracted from neonatal cardiac fibroblasts of rats, H9c2 cells, and heart tissue of mice that had undergone osmotic minipump implantation. Extraction was performed using TRIzol reagent (Invitrogen), a standard method for RNA preparation. The RNA concentration in each sample was measured using an ASP-2680 spectrophotometer (ACTGene, Piscataway, NJ, USA) to ensure accurate quantification for comparative analysis in qRT-PCR. For cDNA synthesis, 1 μg of RNA from each sample was reverse transcribed using TOPscript RT DryMIX (Enzynomics, Daejeon, South Korea). Relative mRNA levels were determined using the 2−ΔΔCt method, with Gapdh or Actb as housekeeping genes for normalization. PCR primer sequences are listed in Table 1.
Western blotting
Total protein was extracted from mouse heart tissues using radioimmunoprecipitation assay lysis buffer, which contained 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 50 mM Tris–HCl (pH 7.5), 2 mM ethylenediaminetetraacetic acid, 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, 1 mM Na3VO4, and 5 mM NaF, along with a protease inhibitor cocktail (Calbiochem/EMD Millipore, Billerica, MA, USA). Protein concentrations were measured using a bicinchoninic acid protein assay kit from Thermo Fisher Scientific. In total, 40 µg of protein were loaded onto a 10% sodium dodecyl sulfate–polyacrylamide gel for electrophoresis. The proteins were subsequently transferred to polyvinylidene difluoride membranes, blocked with 5% skim milk, and incubated overnight at 4 °C with primary antibodies diluted 1:1000.
After three washes with Tris-buffered saline plus Tween (20 mM Tris, 200 mM NaCl, and 0.04% Tween 20) for 5 min at 25 °C, the blots were treated with anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibodies at a dilution of 1:2000 for 1 h at 25 °C. Protein bands were detected using Immobilon Western blotting reagents (EMD Millipore), and their intensities were measured using ImageJ 1.49 software (https://imagej.net/).
Transfection
To knock down specific genes, H9c2 cells were transfected with control short-interfering RNA (siRNA), galectin3 siRNA, or Adamts8 siRNA (Bioneer, Daejeon, South Korea) at a concentration of 100 nM for 2 days, using RNAiMAX reagent in accordance with the manufacturer’s instructions. To overexpress galectin 3, the pCMV3-N-GFPSpark-mouse galectin 3 plasmid (Sino Biological Inc.) was transfected into H9c2 cells as specified by the manufacturer.
Cell culture and cell size measurement
H9c2 cells were used to validate in vivo findings because they exhibit hypertrophic stimuli responses similar to those of primary cardiomyocytes34,35. H9c2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Cell size was measured under three different experimental conditions. In the first condition, H9c2 cells were serum-starved for 24 h, then stimulated with isoproterenol (10 μM) and simultaneously treated with chlorogenic acid (0.1 μM) for 24 h. In the second, isoproterenol treatment followed transfection of si-control or si-galectin 3. In the third, cells were transfected with the GFP vector or pCMV3-N-GFPSpark-galectin 3. All cells were washed twice with warm phosphate-buffered saline (PBS), fixed in warm 1.85% paraformaldehyde solution for 20 min, and permeabilized with 0.1% Triton X-100 in PBS for 10 min at 25 °C. After two washes with PBS, cells were blocked with 3% normal goat serum for 20 min, incubated with Texas Red™-X Phalloidin (1:300) for 1 h in the dark, and then stained using ProLong™ Gold Antifade Mountant with DAPI. Images were captured using a microscope (Nikon, Tokyo, Japan); cell size was quantified using NIS Elements Software Version AR 3.0 (Nikon, Japan).
Cell viability
H9c2 cells were serum-starved for 24 h in a 96-well plate, then treated with varying concentrations of chlorogenic acid (0, 0.1, 1, 3, and 10 μM) for either 9 or 24 h. After treatment, cells were incubated with 25 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution for 2 h to allow formation of insoluble formazan crystals. Cell viability was quantified by dissolving the formazan in dimethyl sulfoxide and measuring absorbance at 570 nm.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA). Data are presented as mean ± standard error of the mean. Comparisons between two groups were conducted using either Student’s t-test or the Mann–Whitney U test, depending on distribution normality assessed by the Shapiro–Wilk test. For comparisons among three or more groups, one-way analysis of variance followed by Tukey’s post hoc test or the Kruskal–Wallis test was applied, as appropriate based on the Shapiro–Wilk test results.
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
References
Yang, F. et al. Chlorogenic acid inhibits liver fibrosis by blocking the miR-21-regulated TGF-beta1/Smad7 signaling pathway in vitro and in vivo. Front. Pharmacol. 8, 929. https://doi.org/10.3389/fphar.2017.00929 (2017).
Gupta, A., Atanasov, A. G., Li, Y., Kumar, N. & Bishayee, A. Chlorogenic acid for cancer prevention and therapy: Current status on efficacy and mechanisms of action. Pharmacol. Res. 186, 106505. https://doi.org/10.1016/j.phrs.2022.106505 (2022).
Hwang, S. J., Kim, Y. W., Park, Y., Lee, H. J. & Kim, K. W. Anti-inflammatory effects of chlorogenic acid in lipopolysaccharide-stimulated RAW 2647 cells. Inflamm. Res. 63(1), 81–90. https://doi.org/10.1007/s00011-013-0674-4 (2014).
Liang, N. & Kitts, D. D. Role of chlorogenic acids in controlling oxidative and inflammatory stress conditions. Nutrients https://doi.org/10.3390/nu8010016 (2015).
He, K., Wang, X., Li, T., Li, Y. & Ma, L. Chlorogenic acid attenuates isoproterenol hydrochloride-induced cardiac hypertrophy in AC16 cells by inhibiting the Wnt/beta-catenin signaling pathway. Molecules https://doi.org/10.3390/molecules29040760 (2024).
Ping, P. et al. Chlorogenic acid attenuates cardiac hypertrophy via up-regulating Sphingosine-1-phosphate receptor1 to inhibit endoplasmic reticulum stress. ESC Heart Fail. 11(3), 1580–1593. https://doi.org/10.1002/ehf2.14707 (2024).
Caturano, A. et al. Cardiac hypertrophy: From pathophysiological mechanisms to heart failure development. Rev. Cardiovasc. Med. 23(5), 165. https://doi.org/10.31083/j.rcm2305165 (2022).
Hinderer, S. & Schenke-Layland, K. Cardiac fibrosis: A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 146, 77–82. https://doi.org/10.1016/j.addr.2019.05.011 (2019).
Felker, G. M. et al. Galectin-3 in ambulatory patients with heart failure: Results from the HF-ACTION study. Circ. Heart Fail. 5(1), 72–78. https://doi.org/10.1161/CIRCHEARTFAILURE.111.963637 (2012).
Zaborska, B. et al. The role of galectin-3 in heart failure-the diagnostic, prognostic and therapeutic potential-where do we stand?. Int. J. Mol. Sci. https://doi.org/10.3390/ijms241713111 (2023).
Gehlken, C., Suthahar, N., Meijers, W. C. & de Boer, R. A. Galectin-3 in heart failure: An update of the last 3 years. Heart Fail. Clin. 14(1), 75–92. https://doi.org/10.1016/j.hfc.2017.08.009 (2018).
Zhou, H. et al. YAK577 attenuates cardiac remodeling and fibrosis in isoproterenol-infused heart failure mice by downregulating MMP12. Korean Circ. J. https://doi.org/10.4070/kcj.2024.0093 (2024).
Cheng, Z. et al. Prognostic value of serum galectin-3 in chronic heart failure: A meta-analysis. Front. Cardiovasc. Med. 9, 783707. https://doi.org/10.3389/fcvm.2022.783707 (2022).
Zha, Y. et al. ADAMTS8 promotes cardiac fibrosis partly through activating EGFR dependent pathway. Front. Cardiovasc. Med. 9, 797137. https://doi.org/10.3389/fcvm.2022.797137 (2022).
Allison, S. J. ECM remodelling by ADAMTS12 in fibrosis. Nat. Rev. Nephrol. 20(12), 770. https://doi.org/10.1038/s41581-024-00905-2 (2024).
Hoeft, K. et al. ADAMTS12 promotes fibrosis by restructuring extracellular matrix to enable activation of injury-responsive fibroblasts. J. Clin. Invest. https://doi.org/10.1172/JCI170246 (2024).
Lopez, J. E. et al. beta-myosin heavy chain is induced by pressure overload in a minor subpopulation of smaller mouse cardiac myocytes. Circ. Res. 109(6), 629–638. https://doi.org/10.1161/CIRCRESAHA.111.243410 (2011).
Hara, A. et al. Galectin-3: A potential prognostic and diagnostic marker for heart disease and detection of early stage pathology. Biomolecules https://doi.org/10.3390/biom10091277 (2020).
Wright, G. A. & Struthers, A. D. Natriuretic peptides as a prognostic marker and therapeutic target in heart failure. Heart 92(2), 149–151. https://doi.org/10.1136/hrt.2003.018325 (2006).
Freiholtz, D. et al. SPP1/osteopontin: A driver of fibrosis and inflammation in degenerative ascending aortic aneurysm?. J. Mol. Med. (Berl.) 101(10), 1323–1333. https://doi.org/10.1007/s00109-023-02370-z (2023).
Zhou, M. et al. Fibroblast-secreted phosphoprotein 1 mediates extracellular matrix deposition and inhibits smooth muscle cell contractility in Marfan syndrome aortic aneurysm. J. Cardiovasc. Transl. Res. 15(5), 959–970. https://doi.org/10.1007/s12265-022-10239-8 (2022).
Tang, B. L. ADAMTS: A novel family of extracellular matrix proteases. Int. J. Biochem. Cell Biol. 33(1), 33–44. https://doi.org/10.1016/s1357-2725(00)00061-3 (2001).
Li, Y. et al. Chlorogenic acid prevents isoproterenol-induced hypertrophy in neonatal rat myocytes. Toxicol. Lett. 226(3), 257–263. https://doi.org/10.1016/j.toxlet.2014.02.016 (2014).
Bouffette, S., Botez, I. & De Ceuninck, F. Targeting galectin-3 in inflammatory and fibrotic diseases. Trends Pharmacol. Sci. 44(8), 519–531. https://doi.org/10.1016/j.tips.2023.06.001 (2023).
Yu, L. et al. Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ. Heart Fail. 6(1), 107–117. https://doi.org/10.1161/CIRCHEARTFAILURE.112.971168 (2013).
Ou, S. M. et al. Urinary galectin-3 as a novel biomarker for the prediction of renal fibrosis and kidney disease progression. Biomedicines https://doi.org/10.3390/biomedicines10030585 (2022).
Ou, S. M. et al. Identification of galectin-3 as potential biomarkers for renal fibrosis by RNA-sequencing and clinicopathologic findings of kidney biopsy. Front. Med. (Lausanne) 8, 748225. https://doi.org/10.3389/fmed.2021.748225 (2021).
Nishi, Y. et al. Role of galectin-3 in human pulmonary fibrosis. Allergol. Int. 56(1), 57–65. https://doi.org/10.2332/allergolint.O-06-449 (2007).
Chalise, U. et al. Macrophages secrete murinoglobulin-1 and galectin-3 to regulate neutrophil degranulation after myocardial infarction. Mol. Omics. 18(3), 186–195. https://doi.org/10.1039/d1mo00519g (2022).
Yao, L., Shao, W., Chen, Y., Wang, S. & Huang, D. Suppression of ADAM8 attenuates angiotensin II-induced cardiac fibrosis and endothelial-mesenchymal transition via inhibiting TGF-beta1/Smad2/Smad3 pathways. Exp. Anim. 71(1), 90–99. https://doi.org/10.1538/expanim.21-0064 (2022).
Omura, J. et al. ADAMTS8 promotes the development of pulmonary arterial hypertension and right ventricular failure: A possible novel therapeutic target. Circ. Res. 125(10), 884–906. https://doi.org/10.1161/CIRCRESAHA.119.315398 (2019).
Yue, B. et al. SPP1 induces idiopathic pulmonary fibrosis and NSCLC progression via the PI3K/Akt/mTOR pathway. Respir. Res. 25(1), 362. https://doi.org/10.1186/s12931-024-02989-7 (2024).
Lindsey, M. L., Kassiri, Z., Virag, J. A. I., de Castro Bras, L. E. & Scherrer-Crosbie, M. Guidelines for measuring cardiac physiology in mice. Am. J. Physiol. Heart Circ. Physiol. 314(4), H733–H752. https://doi.org/10.1152/ajpheart.00339.2017 (2018).
Ryu, Y. et al. Gallic acid prevents isoproterenol-induced cardiac hypertrophy and fibrosis through regulation of JNK2 signaling and Smad3 binding activity. Sci. Rep. 6, 34790. https://doi.org/10.1038/srep34790 (2016).
Zhao, T. et al. Selective HDAC8 inhibition attenuates isoproterenol-induced cardiac hypertrophy and fibrosis via p38 MAPK pathway. Front. Pharmacol. 12, 677757. https://doi.org/10.3389/fphar.2021.677757 (2021).
Funding
This study was funded by the National Research Foundation of Korea, supported by the Korean government (RS-2022-NR069133). Additionally, funding was provided by Chong Kun Dang, the Korean Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (RS-2023-00302146 and RS-2023-00302153), and Chonnam National University Hospital Biomedical Research Institute (BCRI24016). The funders had no role in the study design, conduct, data analysis, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
Seong Hoon Kim: Conceptualization, Investigation, Writing-Original Draft. Hae Jin Kee: Conceptualization, Investigation, Visualization, Supervision, Funding Acquisition, Writing-Original Draft Hongyan Zhou: Investigation Hyukjin Park: Investigation Seung Hun Lee: Writing-Review and Editing Doo Sun Sim: Writing-Review and Editing Myung Ho Jeong: Writing-Review and Editing Young Joon Hong: Supervision, Funding Acquisition, Writing-Review and Editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, S.H., Kee, H.J., Zhou, H. et al. Chlorogenic acid attenuates cardiac hypertrophy and fibrosis by downregulating galectin 3. Sci Rep 15, 26925 (2025). https://doi.org/10.1038/s41598-025-12222-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-12222-0
