
Abstract
West Africa has been experiencing a resurgence of Marburg virus disease (MVD), a zoonotic pathogen that causes severe hemorrhagic fever in both humans and primates. Regretfully, there are not any effective medications on the market right now. The binding interactions between MARV VP40, a protein essential to viral replication, and a commercial medication, estradiol benzoate, and a natural substance, procyanidin, were examined in this work. Ten hydrogen bonds and hydrophobic interactions with important residues (Ala316, Val317, Lys230, Glu89, Asn87) stabilized procyanidin’s superior binding affinity (− 11.3 kcal/mol) over estradiol benzoate (− 8.9 kcal/mol), according to molecular docking. With a lower radius of gyration (1.94 nm) and RMSD (0.28 nm) than estradiol benzoate (RMSD: 0.37 nm, Rg: 1.97 nm), procyanidin formed a more stable complex, according to molecular dynamics simulations conducted over 200 ns. Additionally, procyanidin showed decreased solvent accessibility and increased intermolecular hydrogen bonding (average 3.50 bonds), suggesting stronger binding. Procyanidin’s superior drug-likeness, decreased cardiotoxicity, and decreased carcinogenicity potential were all shown by ADMET analysis. Its higher binding energy (− 57.82 KJ/mol) was further validated by free energy calculations (MM-PBSA). According to these results, procyanidin, a natural substance shows promise as an antiviral medication against MARV targeting its VP40 protein.
Similar content being viewed by others

Introduction
Filoviruses were first implicated in 1967 as causative agents for an outbreak of hemorrhagic fever among laboratory personnel in Europe who had been in contact with imported African green monkey blood and tissues from Uganda containing the Marburg virus (MARV). This was followed by more sporadic cases of humans suffering from Marburg hemorrhagic fever in Kenya and Zimbabwe1. Reports from Germany and Yugoslavia 1967 by CDC, mentioned 31 cases and 7 deaths with 23% fatality rate was the first ever outbreak of Marburg virus2. Later, on March 21, 2023, Tanzania government officials declared the country’s first Marburg outbreak. The outbreak was declared over on May 31, 2023, with a total of nine cases reported, one probable and eight confirmed, of which six died with 67% case fatality rate. All the cases were reported from the country’s northwest Kagera region2.
MARV is a pleomorphic virus observed in filamentous, circular, U-shaped, rod-like, and other forms. It typically measures 80 nm in diameter and averages 790 nm in length. The surface of MARV virions is covered with 5–10 nm-long glycoprotein spikes spaced approximately 10 nm apart, essential for host cell attachment3. MARV is a non-segmented, negative-sense RNA virus with a 19.1 kb genome encoding seven genes in a linear order: 3’-NP-VP35-VP40-GP-VP30-VP24-L-5’. These genes are flanked by conserved transcription start and stop signals, with long noncoding regions containing cis-acting elements critical for replication, transcription, and genome packaging. Except for VP24 and VP30, which share a five-nucleotide overlapping sequence (UAAUU), the genes are separated by intergenic regions of 4–97 nucleotides4. The genome is encapsulated in a nucleocapsid complex composed of NP, VP35, VP30, and L proteins, which are essential for viral replication and transcription. The L protein functions as an RNA-dependent RNA polymerase, while VP35 serves as a polymerase cofactor. The virion is enveloped in a host-derived membrane with a matrix formed by VP40, facilitating virion budding. GP spikes enable host cell attachment, and VP24 assists in virion release. MARV structural proteins are crucial for its replication cycle and pathogenicity5.
The major matrix protein VP40 of Marburgvirus plays the most essential roles in the virus budding, by intercepting the Jak1-dependent signaling pathway in infected cells6. MARV VP40 deactivates the tyrosine phosphorylation activity of Jak1 and Tyk2, STAT1, STAT27. Filovirus VP40 protein appears to be centrally implicated in assembling and budding for the virus itself, and such expression results in cell surface release of VLP8. This action is mediated by late domains, which is essential for the late stages of viral budding and short amino acid sequence segments (16PPPY19). As a for example, in the case of Ebola virus VP40, two late motifs; PTAP and PPXY motifs, interact with certain host proteins associated with the budding process through Tsg101, Nedd4, and Rsp59. In case of MARV VP40, only one PPPY motif is present which interacts with Tsg101. Moreover, conserved motifs, such as EBOV VP40’s 96LPLGVA101 and MARV VP40’s 84LPLGIM89, are required for efficient VLP release10.
Despite over five decades of research, no vaccine or medication has been proven effective in preventing Marburg virus disease. However, in the absence of well-established therapeutic options, antiviral drugs like favipiravir and remdesivir, commonly used for Ebola treatment, show potential in managing Marburg virus infections11. Drug discovery, however, is both resource-intensive and time-consuming. To address these challenges, two strategies have gained prominence: computer-aided drug discovery and drug repurposing. The exploration of potential inhibitors of the VP40 protein has expanded to include both plant-derived and commercially available compounds. Plant-derived compounds are particularly valuable in drug development due to their diverse chemical structures and well-documented biological activities. Traditional medicine plays a significant role in healthcare, particularly in remote regions, by harnessing natural resources for treatment. Himalayan medicinal plants, in particular, are highly regarded for their bioactive compounds, offering promising opportunities for drug discovery. Our computational studies have identified procyanidin, a compound found in Fagopyrum acutatum (Tartary buckwheat), as a promising drug candidate for Marburg virus treatment. Procyanidin’s strong potential highlights the importance of leveraging plant-based compounds in the development of effective antiviral therapies.
Results
Active site residues of MARV VP40
The active site residue in pocket 1 and pocket 2 for binding of natural and commercial compound are present in c-terminal end of the target protein as represented in Supplementary File: Supplementary Fig. 1. Pocket 1 consists of 36 amino acids, with a pocket score of 17.83, a probability score of 0.807, and an average conservation score of 2.519. Among, 36 amino acids in active site of protein Leu215, Pro216, Ile102, Arg150, Arg153, and Ser315, which were majorly involved in binding to proposed drug procyanidin. Pocket 2 comprise of 37 amino acids with a pocket score of 15.61, a probability score of 0.761 and an average conservation score of 2.308. Among, 37 residues, Trp193, Pro216, Val219, Gln157, and Phe289 were linked to the commercial drug estradiol benzoate.
Molecular docking analysis
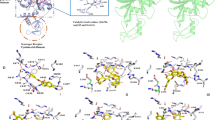
In an effort to identify a lead candidate, 142 (138 natural and 4 commercials) compounds were submitted to virtual screening via a docking approach. Multiple conformations of each ligand were generated during the docking process, and their binding energy profiles ranged from − 11.3 to − 4.0 kcal/mol. Based on the docking analysis, good interactions were observed with estradiol benzoate and procyanidin. The binding energy value of the commercial drug estradiol benzoate was found to be − 8.9 kcal/mol. This binding is attributed to the formation of one hydrogen bond in estradiol benzoate complexes (Fig. 1a). The binding energy of the MARV VP40-procyanidin complex was calculated to be − 11.3 kcal/mol. These interactions were stabilized by the formation of ten hydrogen bonds and other non-covalent interactions between procyanidin and the protein’s active site residues during docking (Fig. 1b). The predicted binding energy values for all 145 docked compounds are presented in Supplementary File: Supplementary Table 1.

(a) protein-ligand interaction, 2D diagram and binding affinity of natural compound Procyanidin. (b) protein-ligand interaction, 2D diagram and binding affinity of commercial compound estradiol benzoate.
Protein–ligand interactivity analysis
For the control drug estradiol benzoate, the analysis revealed the formation of eight hydrophobic interactions, one hydrogen bond, and one salt bridge. The hydrophobic interactions were observed with the following residues: Pro99, Phe159, Trp193, Pro195, Pro216, Val219, Val287, and Gln290, involving ligand atoms 2, 9, 24, 24, 3, 24, 23, and 10, respectively. The hydrogen bond was formed through residue Gln157, interacting with the ligand atom at a distance of 3.12 Å. Additionally, a salt bridge was identified with residue Arg194 at a distance of 4.94 Å (Fig. 1a; Table 1). In contrast, the natural compound procyanidin formed five hydrophobic interactions with residues Ile102, Phe159, Leu215, Pro216, and Phe289, involving ligand atoms 20, 17, 14, 24, and 23, respectively. Additionally, ten hydrogen bonds were identified with residues Ile102, Pro148, Arg150, Arg153, Arg194, and Ser315, with interaction distances ranging from 1.74 to 3.54 Å (Fig. 1b; Table 1).
Pharmacokinetics analysis
A critical physicochemical and pharmacokinetic profile assessment is vital for rational drug design and development of new drugs to guarantee their potential efficacy12. The analysis of drug candidates gives important insights into the biological activity, efficacy, and safety through the assessment of their pharmacokinetic properties. Procyanidin, a natural product, possesses a molecular weight of 594.14 g/mol, and the commercial product estradiol benzoate possesses a molecular weight of 372.6 g/mol. According to Lipinski’s Rule of Five, a molecular weight below 500 g/mol is favourable for oral bioavailability13.
Lipophilicity, as represented by LogP values, is different among the compounds. Procyanidin has a LogP value of 0.748, while estradiol benzoate has a LogP value of 5.077. Increased lipophilicity of estradiol benzoate may enhance membrane permeability but may compromise solubility and enhance plasma protein binding14. Alternatively, procyanidin is more hydrophilic, with implications for increased solubility and possibly enhanced absorption.
Hydrogen bond donors (HBD) and acceptors (HBA) are crucial for drug-receptor interactions. Lipinski’s Rule of Five sets the recommended limits at HBD ≤ 5 and HBA ≤ 10. Procyanidin has 1 HBD and 3 HBA, while estradiol benzoate has 10 HBD and 13 HBA. Since Lipinski’s rule allows one violation, procyanidin meets the drug-likeness criteria13.
tPSA values are also critical for oral bioavailability. Procyanidin is reported to possess a tPSA value of 46.53 Å2which falls below the prescribed value of 140 Å2, whereas estradiol benzoate possesses a greater tPSA value of 229.99 Å2. Based on Veber’s Rule, decreased tPSA values are associated with enhanced oral bioavailability15. Fsp³, an indicator of molecular complexity, is 0.48 for procyanidin and 0.22 for estradiol benzoate, implying that procyanidin is likely to possess greater biological interaction potential16.
QED score for the drug-likeness of procyanidin is 0.584 and for estradiol benzoate is 0.153, with better drug-likeness represented by higher values. ADMET studies of absorption suggest that procyanidin is more permeable in the Caco-2 assay (6.559) than estradiol benzoate (4.854), indicating better absorption in the intestines. P-glycoprotein (P-gp) interactions are significant for drug absorption and disposition17. Estradiol benzoate is a P-gp substrate, which can result in decreased absorption by efflux mechanisms and in multidrug resistance. Procyanidin, on the other hand, is a P-gp inhibitor, which increases absorption but should be used with caution in multidrug therapy conditions18.
Distributionally, plasma protein binding (PPB) determines the drug’s availability at the site of action. Procyanidin has a PPB of 89.5%, while that of estradiol benzoate is 91.6%. Increased PPB decreases the proportion of unbound drugs available for therapeutic effect, which could reduce efficacy19. Procyanidin has a volume of distribution (VDss) of 0.023 L/kg, while estradiol benzoate has 0.264 L/kg.
Excretion profiles, such as clearance and half-life, are important for establishing dosing frequency and risk of accumulation. Procyanidin has a plasma clearance (CLplasma) of 6.65 ml/min/kg, whereas estradiol benzoate is 9.67 ml/min/kg, meaning that estradiol benzoate is cleared faster. The half-life (T₁/₂) of procyanidin is 2.92 h, whereas that of estradiol benzoate is 0.83 h, meaning that procyanidin could have a more convenient dosing schedule with a lower risk of accumulation.
Toxicity assessment is important in establishing a compound’s safety profile. One of the most significant concerns is cardiotoxicity, especially via blockade of the human Ether-à-go-go-Related Gene (hERG) potassium channel. Procyanidin has a lesser chance of hERG inhibition (0.12) than estradiol benzoate (0.3), hence a lower risk of cardiotoxicity20. The determination of genotoxicity and carcinogenicity is also important. Procyanidin has a higher genotoxic probability (0.978) than estradiol benzoate (0.347), yet its carcinogenic probability (0.068) is much lower than estradiol benzoate (0.926). Though it has the possibility of mutation, procyanidin seems to have a lower overall risk of being carcinogenic, but estradiol benzoate’s high carcinogenic probability is cause for concern21.
Other toxicity indexes were examined. Procyanidin has a lesser drug-induced liver injury (DILI) likelihood of 0.27, while estradiol benzoate possesses a higher likelihood of 0.401, signifying a higher risk of hepatotoxicity with increased exposure22. Procyanidin also demonstrates less ototoxicity (0.349) than estradiol benzoate (0.966). These results point toward the need for more studies to ascertain safety profiles and maximize therapeutic uses.
Molecular dynamic simulation
RMS deviation
RMS deviations were determined by aligning the MD trajectories to the initial structures and analyzing them over time. Figure 2 illustrates variations in the backbone dynamics across the trajectories of protein-ligand complexes during 200 ns simulations. In Fig. 2a, a deviation between 25 and 50 ns and 15–200 ns of the simulation was observed in the VP40_estradiol benzoate complex. However, in case of VP40_procyanidin, lesser deviation was detected compared to estradiol benzoate complex. The average RMS deviations of the backbone atoms of VP40_procyanidin 0.28 ± 0.08 nm in contrast to 0.37 ± 0.04 nm noted for VP40_estradiol benzoate.

(a) Calculations of backbone RMS deviations for each trajectory of VP40 with natural compound Procyanidin run1 (Violet), run2 (Magenta), and run3 (Green) and commercial compound estradiol benzoate run1 (Red), run2 (Yellow), and run3 (Orange) from the initial position till 200 ns MD simulations. (b) Calculations of backbone RMS fluctuation for each trajectory of VP40 with natural compound Procyanidin run1(Violet), run2 (Magenta), and run3 (Green) and commercial compound estradiol benzoate run1 (Red), run2 (Yellow), and run3 (Orange) from the initial position till 200 ns MD simulations. (c) Fluctuating residue of protein when attached to procyanidin. (d) Fluctuating residue of protein when attached to estradiol benzoate.
RMS fluctuation
Root-mean-square fluctuation (RMSF) quantifies the fluctuations of residues from their initial positions during the simulation period and thus measures protein flexibility. The calculation of RMSF from the 200 ns MD trajectory revealed that the RMSF value was lower in the VP40_procyanidin (0.13 ± 0.05) and VP40_estradiol benzoate (0.15 ± 0.08) (Fig. 2b). Common fluctuating residues in between 3 runs from procyanidin with VP40 were Ala316, Val317, and Lys230 (Fig. 2c), whereas for estradiol benzoate between 3 runs Glu89, Asn87, Ala316, Val317, and Lys230 as shown in Fig. 2d.
Radius of gyration
The radius of gyration (ROG) serves as a measure to assess the compactness of proteins during simulations. We calculated the ROG for both VP40_procyanidin as well as VP40_estradiol benzoate trajectories to know the impact of flexibility on compactness for residues, as shown in Fig. 3a. The procyanidin bound state showed significantly (p < 0.05) lower radius of gyration trajectory (1.94 ± 0.02 nm) when compared with that of estradiol benzoate bound state (1.97 ± 0.01 nm).

(a) The radius of gyration measuring compactness of the envelope protein during 200 ns MD simulations. lower gyrations were observed from the initial conformations till the end of the simulations for natural compound run1 (Violet), run2 (Magenta), and run3 (Green) and higher gyration in commercial compound estradiol benzoate run1 (Red), run2 (Yellow), and run3 (Orange) from the initial position till 200 ns MD simulations. (b) Intermolecular H-bonding interactions of the protein–ligand complexes corresponding to 200 ns MD simulations. Lesser number of H-bonds were found in commercial compound estradiol benzoate (black) and higher in natural compound Procyanidin (red).
Intermolecular hydrogen bonding analysis
The intermolecular H-bonds formed during 200 ns MD simulation were measured to analyze the binding affinity and stability of the ligands with the VP40 protein. Figure 3b represents the average of triplicate run of intermolecular H-bonding analysis for procyanidin visualized in red and estradiol benzoate visualized in black with a cutoff value of 0.35 nm. VP40_procyanidin formed 0–9 bonds with an average of 3.50 bonds respectively. Whereas VP40_estradiol benzoate formed 0–4 with an average of 1.06 bonds. The complex VP40_procyanidin was further stabilized by the continuous formation of H-bonds, which supported the predictions from docking. Hydrogen bonds enhance rigidity and provide specificity to protein interactions, playing a crucial role in stabilizing secondary structures such as α-helices and β-sheets. Alterations in hydrogen bonding can significantly impact conformational dynamics, stability, and, most importantly, the binding specificity between proteins and their ligands23. Evidence from docking, RMSD, RMSF, and Rg analyses proved that procyanidin have tight binding at the active sites than estradiol benzoate.
Solvent accessible surface area of VP40
This study utilized SASA to assess the impact of ligand binding on VP40’s conformational behaviour. Figure 4a represents the solvent accessibility of VP40 upon binding of natural compound (procyanidin) and commercial compound (estradiol benzoate) over 200 ns MD simulations. By comparing the SASA plots, we found that both natural and commercial compound exhibited almost similar trajectory patterns on binding to target. However, the accessibility of VP40 increased upon binding to commercial compound compared to that of natural compounds. SASA value for VP40_estradiol benzoate complex was found to be in the range between 145.33 and 153.76 nm2 with an average of 147.43 nm2. In case of natural compound, the SASA value of VP40_procyanidin complex was found to be in the range of 140.47 to 151.89 nm2 with an average of 142.64 nm2. Procyanidin transitions into the binding pocket, enhancing interactions and reducing solvent accessibility. In the estradiol benzoate complex, residues become more exposed to the surrounding water molecules and are weakly involved in binding, while in the procyanidin; residues show decreased accessibility due to increased binding interactions with the target protein, leading to more compactness and stability. In Fig. 4b the average of all the 3 runs for procyanidin and estradiol benzoate it can be seen that the protein has reached at an equilibrium state at the end of 200ns simulation.

(a) SASA plot analysis corresponding to 200 ns MD simulations of natural compound Procyanidin run1 (Violet), run2 (Magenta), and run3 (Green) and higher gyration in commercial compound estradiol benzoate run1 (Red), run2 (Yellow), and run3 (Orange) from the initial position till 200 ns MD simulations. (b) Average SASA plot analysis corresponding to 200 ns MD simulations of natural compound Procyanidin run1 (Violet), run2 (Magenta), and run3 (Green) and higher gyration in commercial compound estradiol benzoate run1 (Red), run2 (Yellow), and run3 (Orange) from the initial position till 200 ns MD simulations.
Free energy calculations using MM-PBSA
Decompositions of binding energies were performed to gain insight into protein–ligand binding interactions by analyzing their individual energy components—including van der Waals, electrostatic, polar solvation, and SASA (non-polar solvation) energies. The binding free energy of estradiol benzoate was calculated to be approximately − 11.75 kcal/mol, comprising − 1.80 kcal/mol (van der Waals), − 3.19 kcal/mol (electrostatic), − 38.11 kcal/mol (polar solvation), and − 3.42 kcal/mol (SASA/non-polar solvation). In contrast, procyanidin showed a much stronger binding affinity, with a total binding free energy of approximately − 45.47 kcal/mol, consisting of 7.56 kcal/mol (van der Waals), − 9.74 kcal/mol (electrostatic), − 39.69 kcal/mol (polar solvation), and − 3.61 kcal/mol (SASA/non-polar solvation). It is important to note that these binding free energies were computed without including the entropy contribution (–TΔS). When an estimated entropy correction of 12.35 kcal/mol is considered, the corrected binding free energies become approximately − 24.10 kcal/mol for estradiol benzoate and − 57.82 kcal/mol for procyanidin (Table 2).
Principal component analysis (PCA) and free energy landscape (FEL) analysis of VP40 protein complexes
PCA was utilized to analyze large-scale motions of VP40 protein in binding with procyanidin (natural product) and estradiol benzoate (commercial drug) on varying simulation time scales. The procedure utilized covariance matrices based on Cα atomic fluctuations followed by diagonalization of MD trajectory data. Collective motions were presented by displacements along each eigenvector. Projections of Cα trajectories onto PC1 and PC2 were studied in both complexes. Larger motion amplitudes were indicated by higher covariance values for estradiol benzoate: 9.59 nm² (trajectory 1) and 9.89 nm² (trajectory 2); 10.14 nm² (trajectory 3) and for procyanidin 4.05 nm² (trajectory 1); and 4.75 nm² (trajectory 2) and 7.31 nm² (trajectory 3) in run 1 (Figs. 5a and 6a). In this study, FEL models (Figs. 5b, and 6b) were constructed based on projections along the first two principal components (PC1 and PC2), revealing scattered energy basins for estradiol benzoate indicative of flexible conformations while centralized and deeper basins for procyanidin suggest tighter and more stable conformational states. With energy values ranging from 0-15.4KJ/mol (Run 1), 0-14.7 KJ/mol (Run 2) and 0-14.5KJ/mol in case of estradiol benzoate, whereas, in case of procyanidin the energy value ranged from 0-13.3KJ/mol (Run 1), 0-13.8KJ/mol and 0-14.5KJ/mol.

(a) 2D projections of the principal eigenvectors (PC1 and PC2) of commercial compound estradiol benzoate run1 (Red), run2 (Yellow), and run3 (Orange) from the initial position till 200 ns MD simulations. (b) Principal component analysis (PCA) and free energy landscape (FEL) models of the commercial compound estradiol benzoate run1 (Red), run2 (Yellow), and run3 (Orange) from the initial position till 200 ns MD simulations.

(a) 2D projections of the principal eigenvectors (PC1 and PC2) of natural compound Procyanidin run1 (Violet), run2 (Magenta), and run3 (Green) from the initial position till 200 ns MD simulations. (b) Principal component analysis (PCA) and free energy landscape (FEL) models of the natural compound Procyanidin run1 (Violet), run2 (Magenta), and run3 (Green) from the initial position till 200 ns MD simulations.
Discussion
There has been a need for discovering efficient drugs to combat the Marburg virus (MARV) disease and eradicate future outbreaks. This aspect is crucial in bringing a halt to the epidemic disaster that has ravaged healthcare systems and populations in respective regions24. The drug discovery to control and prevent the MARV disease is challenging due to the need to execute a preclinical research in the BSL-4 facility25. Computational drug design is among the promising solutions to all these challenges, offering windows for accelerating the development of anti-MARV medicines. In-silico drug discovery has great scope and can be useful in any stage of a candidate drug’s preclinical development. It is more affordable and less time-consuming in comparison with traditional drug development methods26.
We screened 138 natural compounds and found that procyanidin has the highest binding affinity to VP40 MARV (PDB ID: 5B0V), with a strong association supported by a high pairwise cross-correlation coefficient. This compound showed effective intermolecular interactions and strong binding affinity with the VP40 protein of MARV. The study used various computational drug design techniques, including molecular docking, post-docking MM-PBSA calculations, and molecular dynamics (MD) simulations. A compound’s potential and drug-likeness properties are determined by its binding efficiency, stability, and toxicity27. Advances in computational methods have enabled the in-silico evaluation of toxicity, which has been used to assess the safety profiles of target compounds27. Without such approaches, such substances might pose risks to humans and animals. Therefore, it is important to evaluate the toxicity of a compound before its oral administration to target cells27. In this study, the oral toxicity of the compounds was assessed using the ProTox-II server, which identified procyanidin as safe. It was predicted to be tolerable at higher doses (LD50) compared to the commercial drug estradiol benzoate (LD50). Active binding pockets were identified using PrankWeb, including key residues such as Leu215, Pro216, Ile102, Arg150, Arg153, and Ser315, which were mostly involved in binding to proposed drug procyanidin and Trp193, Pro216, Val219, Gln157, and Phe289 were found to be bound to the commercial drug estradiol benzoate. Natural compounds were found to be more potent in binding and selective toward MARV VP40 as observed through in-silico docking analysis. Among the selected candidates, procyanidin showed stable interactions with the lowest binding energy of − 11.3 kcal/mol, whereas estradiol benzoate showed a binding energy of − 8.9 kcal/mol. Recent research on 2,569 natural compounds targeting MARV VP40 identified NPL130 (CHEMBL2087156) as a strong candidate with optimal binding affinity and stable complex formation, along with Nilotinib, which had a binding score of − 8 kcal/mol28.
The analysis of the physicochemical and ADMET properties of the antiviral compounds provided important insights into their potential as Marburg VP40 inhibitor, and are a crucial step in analyzing the pharmacokinetic profile of new drugs. Procyanidin and estradiol benzoate showed marked differences in their physicochemical, pharmacokinetic, and toxicity profiles. The better oral bioavailability, solubility, and drug-likeness of procyanidin are revealed by its low molecular weight, high hydrophilicity (LogP 0.748), and QED value (0.584). Its good pharmacokinetic profile, including high permeability (Caco-2: 6.559), P-gp inhibition, low PPB (89.5%), and long half-life (2.92 h), indicates better absorption, distribution, and dosing convenience. However, it has a slightly higher genotoxicity risk (0.978). Estradiol benzoate, on the other hand, has a greater lipophilicity, LogP (5.077), that would increase the membrane permeability but decrease the solubility and increase protein binding. Higher tPSA, 229.99 Å2 with a lower QED, 0.153, decreases the oral bioavailability. It is a P-gp substrate and therefore may show reduced absorption. Despite having less genotoxicity, estradiol benzoate had greater carcinogenicity, 0.926, and liver injury risks, 0.401. Thus, procyanidin promises more in terms of ADMET properties compared to its commercial counterpart.
These studies suggest that the ligand was able to stabilize the structure of the target protein. Also, the results were further corroborated by carrying out MD simulations in triplicate for 200 ns using the GROMACS 2023.2 simulation package29. Trajectory from a single independent system was analyzed, and MARV VP40 complexes with commercial as well as natural compounds presented quite different RMSD trajectories. Estradiol benzoate had more deviation of 0.37 ± 0.04 nm, whereas procyanidin presented smaller RMSD values of 0.28 ± 0.08 nm, indicating a greater structural stability for procyanidin. The RMSF values for VP40_procyanidin (0.13 ± 0.05) was smaller than those for VP40_estradiol benzoate (0.15 ± 0.08), meaning the complexes were more rigid and stable with natural compounds. Zhu et al.30 reported RMSF values of procyanidin-SARS-Cov-2 complexes at around 0.12 to 0.14 nm, while the RMSF of natural compounds was lower than that of synthetic compounds. Their results for synthetic drug complexes (like ribavirin) had RMSF values within the range of 0.16 to 0.18 nm, thereby suggesting higher flexibility and reduced stability. The Rg values for the VP40 complexes were measured to understand the compactness of the protein-ligand complex. For the natural compounds, procyanidin (1.94 ± 0.02) showed a smaller radius compared to estradiol benzoate (1.97 ± 0.01) had higher Rg values30. The investigation of protein-ligand complexes for viral targets reported that compounds that possessed tight binding to the protein typically had Rg values between 1.80 and 1.90 nm31. Hydrogen bonding analysis showed that VP40_procyanidin proceeded on average to form 3.75 bonds whereas VP40_estradiol benzoate boding at a less amount than this, at 1.56 bonds. Procyanidin was calculated to be binding with a free energy of -190.28 kJ/mol, while estradiol benzoate binding energies were much weaker, at 49.16 kJ/mol. PCA and free energy landscape (FEL) analyses showed that VP40 was more compact and stable in the presence of procyanidin, with restricted covariance matrix values and well-defined, centralized energy basins, whereas estradiol benzoate treated proteins were broader with less stable states. The distinction between various ligands underlines the importance of ligand structure in VP40 dynamics, which may guide the focused design of effective drugs. The molecular dynamics simulations played a pivotal role in validating the stability, flexibility, and binding strength of the VP40-ligand complexes. By analyzing RMSD, RMSF, radius of gyration, hydrogen bonding, PCA, and FEL profiles over extended simulation time, the study confirmed that procyanidin maintained a more compact and stable interaction with VP40 compared to estradiol benzoate. These simulations provided critical insight into the dynamic behavior of the complexes, reinforcing the reliability of docking predictions and MM-PBSA results. Thus, MD simulations served as a powerful approach to assess the therapeutic potential and mechanistic interactions of candidate compounds against MARV VP40.
Methodology
Target selection for the study
From the RCSB Protein Data Bank (PDB) (https://www.rcsb.org/), the full-length VP40 dimer structure of MPXV was taken using PDB entry 5B0V, with chain A chosen for analysis. The protein has 317 amino acid residues. Hydrogen atoms were added to the PDB coordinates, and missing partial atomic charges were added; then the structure was minimized energetically via SwissPDB Viewer32. PDB ID 5B0V was selected due to its high-resolution crystal structure and its functional relevance in the viral assembly and egress processes of Marburg virus.
Ligand 3D structures curation
We have selected ligands from 12 plant species native to the Himalayan regions of Sikkim, India, located at high altitude up to 10,000 feet above sea level. Fagopyrum acutatum, Diploknema butyracea, Saussurea costus, Nardostachys jatamansi, Astilbe rivularis, Bergenia ciliata, Aconitum heterophyllum, Tinospora sinensis, Allium wallichii, Panax pseudoginseng, Aconitum ferox, and Heracleum wallichii33. The selection of phytochemical constituents was based on their prevalence in different parts of the plant, such as the bark, leaves, roots, and other physiological components. In total, 142 compounds in which 138 were natural compounds and 4 were commercial drug were selected for virtual screening against the VP40 MARV protein. The compounds were sourced from IMPPAT 2.0 software (https://cb.imsc.res.in/imppat/home) (Table 1). This in-silico study included a control group of four widely repurposed antiviral drugs targeting VP35 of MARV, with the compounds sourced from PubChem (Table 1)34. The ligand structure files were converted into PDBQT using the Open Babel tool35. Missing H-atoms and geometry of the ligand structures were added using Avogadro v.1.2.1936.
Protein’s active sites prediction
Ligand-binding pockets of the VP40 MARV protein were identified using PrankWeb (https://prankweb.cz/). The 3D structure of the VP40 MARV protein (PDB ID: 5B0V) was uploaded to the server, and active pockets were predicted based on rankings, pocket scores, probability scores, and conservation values.
Docking-based screening
A docking-based investigation identified MARV VP40 inhibitors with 4 commercial compounds and 138 natural phytochemicals. To begin with, the MARV VP40 crystal structure (PDB ID: 5B0V) was prepared, and AutoDock Vina within PyRx was used to perform the molecular docking. Top ligands were selected based on binding energy profiles, and the ligands were studied using BIOVIA Discovery Studio and PLIP37.
Pharmacokinetics analysis
An extensive analysis was performed using computational tools to assess the drug-likeness, pharmacokinetics, and ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties of procyanidin and estradiol benzoate. For this analysis, the canonical SMILES and SDF files for each drug were retrieved from databases such as PubChem (https://pubchem.ncbi.nlm.nih.gov/). Various web-based platforms were utilized to predict drug-likeness and ADMET-related characteristics, including ADMETlab 3.0 (https://admetlab3.scbdd.com/) and cross checked against, FAF-Drugs 4.1 (https://mobyle2.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::FAF-Drugs4), Deep-PK (https://biosig.lab.uq.edu.au/deeppk/), pkSM (https://biosig.lab.uq.edu.au/pkcsm/prediction), vNN-ADMET (https://vnnadmet.bhsai.org/vnnadmet/home.xhtml), Pred-hERG 5.0 (http://predherg.labmol.com.br/), and ADVERPred (https://www.way2drug.com/adverpred/). These tools provided a comprehensive evaluation of the compounds’ properties.
Simulations setup and post MD analysis
MD simulations were performed in triplicate using GROMACS 2023.238 with ligand topologies generated via the GROMOS 54A7 force field38. The 38,872-atom system was solvated in the SPC water model, neutralized, and energy minimized before 200 ns simulations. Trajectories were analyzed using XMGRACE39and binding free energies were computed with the g_mmpbsa tool based on MM-PBSA40. PCA was conducted on estradiol benzoate and procyanidin-bound complexes using the gmx covar and gmx anaeig41 tool to extract dominant motions. Eigenvectors and eigenvalues were obtained, with PC1 and PC2 capturing most of the conformational space. FEL models were built to map stable protein states onto Gibbs free energy, using PC1 and PC2 projections42. These analyses provided insights into the dynamic behavior and stability of the protein-ligand complexes.
Conclusion
In conclusion, procyanidin showed significant potential as a potent inhibitor of the MARV VP40 protein. It exhibited the strongest binding affinity (− 11.3 kcal/mol) and greater structural stability to VP40 compared to synthetic drugs, as evidenced by lower RMSD, RMSF, Rg, SASA, and enhanced hydrogen bonding. PCA and FEL analyses revealed that the protein adopts more stable and energetically favourable conformational states upon binding to procyanidin compared to other proposed compounds. Further, procyanidin demonstrated superior physicochemical and pharmacokinetic properties, suggesting better therapeutic potential than estradiol benzoate. Toxicity analysis confirmed procyanidin’s safety profile supporting its viability as a therapeutic candidate. Given its promising in-silico results, procyanidin represents a valuable candidate for future investigation in the development of novel, effective, and safe antiviral drug against MARV.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Mehedi, M., Groseth, A., Feldmann, H. & Ebihara, H. Clinical aspects of Marburg hemorrhagic fever. Future Virol. 6, 1091–1106. https://doi.org/10.2217/fvl.11.79 (2011).
CDC. History of Marburg Outbreaks_Marburg_CDC (2025).
Welsch, S. et al. Electron tomography reveals the steps in filovirus budding. PLoS Pathog. 6, 1–9 (2010).
Feldmann, H. et al. Marburg virus, a filovirus: messenger rnas, gene order, and regulatory elements of the replication cycle. Virus Res. 24, 1 (1992).
Yu, D. S. et al. The lifecycle of the Ebola virus in host cells. Oncotarget 8, 1. www.impactjournals.com/oncotarget/ (2017).
Feagins, A. R. & Basler, C. F. Amino acid residue at position 79 of Marburg virus VP40 confers interferon antagonism in mouse cells. J. Infect. Dis. 212, S219–S225 (2015).
Valmas, C. & Basler, C. F. Marburg virus VP40 antagonizes interferon signaling in a species-specific manner. J. Virol. 85, 4309–4317 (2011).
Liu, Y. et al. Conserved motifs within Ebola and Marburg virus VP40 proteins are important for stability, localization, and subsequent budding of virus-like particles. J. Virol. 84, 2294–2303 (2010).
Licata, J. M. et al. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol. 77, 1812–1819 (2003).
Urata, S. et al. Interaction of Tsg101 with Marburg virus VP40 depends on the PPPY motif, but not the PT/SAP motif as in the case of Ebola virus, and Tsg101 plays a critical role in the budding of Marburg virus-like particles induced by VP40, NP, and GP. J. Virol. 81, 4895–4899 (2007).
Srivastava, S. et al. Novel antiviral approaches for marburg: a promising therapeutics in the pipeline. Front. Microbiol. 15, 628. https://doi.org/10.3389/fmicb.2024.1387628 (2024).
Tuntland, T. et al. Implementation of Pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of biomedical research. Front. Pharmacol. 5, 174. https://doi.org/10.3389/fphar.2014.00174 (2014).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development q settings. Adv. Drug Deliv. Rev. 46, 1 (2001).
Leeson, P. D. & Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 6, 881–890 (2007).
Veber, D. F. et al. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623 (2002).
Lovering, F., Bikker, J. & Humblet, C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Elmeliegy, M., Vourvahis, M., Guo, C. & Wang, D. D. Effect of P-glycoprotein (P-gp) inducers on exposure of P-gp substrates: review of clinical drug–drug interaction studies. Clin. Pharmacokinet. 59, 699–714. https://doi.org/10.1007/s40262-020-00867-1 (2020).
Seelig, A. & P-Glycoprotein One mechanism, many tasks and the consequences for pharmacotherapy of cancers. Front. Oncol. 10, 59. https://doi.org/10.3389/fonc.2020.576559 (2020).
Yuan, Z. Q. Y. et al. The impact of plasma protein binding characteristics and unbound concentration of voriconazole on its adverse drug reactions. Front. Pharmacol. 11, 1 (2020).
Ryu, J. Y., Lee, M. Y., Lee, J. H., Lee, B. H. & Oh, K. S. DeepHIT: A Deep Learning Framework for Prediction of hERG-Induced Cardiotoxicity. https://doi.org/10.1093/bioinformatics/btaa075/5727757 (2020).
Duijnhoven, R. G. et al. Number of patients studied prior to approval of new medicines: a database analysis. PLoS Med.. 10, 7. https://doi.org/10.1371/journal.pmed.1001407 (2013).
Fourches, D. et al. Cheminformatics analysis of assertions mined from literature that describe drug-induced liver injury in different species. Chem. Res. Toxicol. 23, 171–183 (2010).
Patgiri, A., Jochim, A. L. & Arora, P. S. A hydrogen bond surrogate approach for stabilization of short peptide sequences in α-helical conformation. Acc. Chem. Res. 41, 1289–1300 (2008).
Mane Manohar, M. P. et al. Advancements in Marburg (MARV) virus vaccine research with its recent reemergence in Equatorial Guinea and tanzania: A scoping review. Cureus https://doi.org/10.7759/cureus.42014 (2023).
O’Donnell, K. L. et al. Vaccine platform comparison: protective efficacy against lethal Marburg virus challenge in the hamster model. Int. J. Mol. Sci. 25, 1 (2024).
Das, R., Bhattarai, A., Karn, R. & Tamang, B. Computational investigations of potential inhibitors of Monkeypox virus envelope protein E8 through molecular Docking and molecular dynamics simulations. Sci. Rep. 14, 1 (2024).
Raies, A. B. & Bajic, V. B. In Silico toxicology: computational methods for the prediction of chemical toxicity. Wiley Interdiscip Rev. Comput. Mol. Sci. 1, 147–172. https://doi.org/10.1002/wcms.1240 (2016).
Hassan, A. M. et al. Machine learning-aided identification of natural compounds targeting Marburg virus VP40 with potential antiviral activity. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2024.2435634 (2024).
Abraham, M. J. et al. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
Zhu, Y. & Xie, D. Y. Docking characterization and in vitro inhibitory activity of flavan-3-ols and dimeric proanthocyanidins against the main protease activity of SARS-Cov-2. Front. Plant. Sci. 11, 1 (2020).
Nag, A., Paul, S., Banerjee, R. & Kundu, R. In Silico study of some selective phytochemicals against a hypothetical SARS-CoV-2 Spike RBD using molecular Docking tools. Comput. Biol. Med. 137, 1 (2021).
Ciucx, N. & Peitsrh Urctrophuresis, M. C. SWISS-MODEL and the Swiss-PdbViewer: An Environment for Comparative Protein Modeling. IS, vol. 21. http://www.cxpasy.ch/ (1997).
Government of Sikkim. List of Medicinal and Aromatic Plants Grown and Found in Sikkim (2020).
Kim, S. et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 49, D1388–D1395 (2021).
O’Boyle, N. M. et al. Open babel: an open chemical toolbox. J. Cheminform. 3, 1 (2011).
Hanwell, M. D. et al. SOFTWARE open access avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 1 (2012).
Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F. & Schroeder M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res 43, W443–W447 (2015).
Huang, W., Lin, Z. & Van Gunsteren, W. F. Validation of the GROMOS 54A7 force field with respect to β-peptide folding. J. Chem. Theory Comput. 7, 1237–1243 (2011).
Turner, P. XMGRACE, Version 5.1. 19 (Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology, 2005).
Kumari, R., Kumar, R. & Lynn, A. G-mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 54, 1951–1962 (2014).
David, C. C. & Jacobs, D. J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 1084, 193–226 (2014).
Papaleo, E., Mereghetti, P., Fantucci, P., Grandori, R. & De Gioia, L. Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: the myoglobin case. J. Mol. Graph Model. 27, 889–899 (2009).
Author information
Authors and Affiliations
Contributions
R.D.: Conceptualization, Writing, Editing, and Formal analysis. A.B.: Editing and Formal analysis. B.T.: Conceptualization, Writing, Editing and Formal analysis, N.T.: Editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Das, R., Bhattarai, A., Tamang, B. et al. Identification of potential VP40 inhibitor of Marburg virus through molecular docking, pharmacokinetic analysis and molecular dynamics simulation. Sci Rep 15, 27129 (2025). https://doi.org/10.1038/s41598-025-12917-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-12917-4
