
Abstract
Improving anti-inflammatory and analgesic drugs with negligible adverse effects remains a significant challenge for long-term use. In the present study, flurbiprofen-based oxadiazole derivatives were synthesized and evaluated for their in vivo anti-inflammatory activity using the carrageenan-induced paw edema assay in mice. Amongst the tested compounds, 10, 3, and 5 exhibited remarkable anti-inflammatory activity, with edema inhibition rates of 88.33%, 66.66%, and 55.55% respectively, compared to the standard drug flurbiprofen, which showed a 90.01% reduction. Molecular docking studies confirmed stable interactions of these compounds with the COX-2 binding site, similar to those observed with the standard drug flurbiprofen. Moreover, toxicity predictions, ADME profiles, and bioactivity scores were evaluated, along with frontier molecular orbitals (HOMO and LUMO) and the molecular electrostatic potential maps (MEP), to reveal the compounds’ drug-like properties and chemically reactive regions. These findings highlight the potential of these derivatives as promising candidates for further development as anti-inflammatory agents.
Similar content being viewed by others
Introduction
The international association for the study of pain (IASP) defines pain as an unpleasant sensation, which can be both bodily and expressive, and is either directly or indirectly linked to actual or potential tissue damage1. One of the most obvious sources of impairment worldwide is pain, which poses a significant public health concern2,3. The immune system’s response to injured or damaged tissues is inflammation, which involves the transfer of white blood cells from blood to damaged cells, infections, and irritants in order to repair and eliminate them4,5. This process is carried out by neutrophils and macrophages6. Inflammatory mediators, such as reactive oxygen species, prostaglandins, cytokines, and nitrogen species are generated during inflammation, increasing vascular permeability and promoting leukocyte migration to the inflammation sites7,8. Redness, heat, pain, swelling, and loss of function are the primary signs of inflammation9,10. Inflammation can arise from various sources, including chemical and physical irritants, infections by pathogenic organisms, and immunological responses11,12,13.
A variety of biochemical mechanisms, including the immune system, the local vascular system, and various cell types present in the injured tissues, are involved in inflammation, which can be categorised as either chronic or acute14,15. Acute inflammation is the early response and is characterised by an increase in the migration of native immune system cells, such as neutrophils and macrophages from the blood into the injured tissues16,17. Chronic inflammation, on the other hand, is marked by progressive changes in the type of cells that are present at the inflammatory reaction site and is characterised by the simultaneous destruction and curing of the injured tissues18. Reduced severity and signs of inflammation can be achieved by alleviating both inflammation and pain19. A progressive approach is used to manage pain and inflammatory disorders, involving corticosteroids, immunosuppressive medications, selective COX-II inhibitors, and traditional NSAIDs20,21.
NSAIDs (non-steroidal anti-inflammatory drugs) are widely used as a first line of treatment for a number of inflammatory diseases and for pain relief22. However, prolonged use of NSAIDs is associated with serious side effects, including bleeding, platelet dysfunction, hepatic and renal toxicity, aplastic anaemia, impaired bone healing, gastrointestinal ulcerations, and cardiovascular toxicity23,24. Certain selective COX-II inhibitors were introduced to the market with the expectation that they would dramatically reduce the gastrointestinal toxicity associated with both acute and chronic NSAID use25.
Flurbiprofen (FBP), also known as S−2-(4-phenyl-3-fluorophenyl)propanoic acid, a non-selective cyclooxygenase (COX) inhibitor, that targets both COX-1 and COX-226. COX-1 is constitutively expressed in most tissues, playing a vital role in physiological processes such as gastrointestinal protection and platelet aggregation. In contrast, COX-2 is an inducible enzyme that is up regulated in response to inflammatory stimuli, such as carrageenan. Therefore, the anti-inflammatory effects observed in this study were hypothesized to be primarily mediated by COX-2 inhibition under the experimental conditions. Flurbiprofen belongs to the aryl propionic acid derivatives, which constitute the most significant class of non-steroidal anti-inflammatory medications (NSAIDs)27. Since its introduction to the European market in 1977, it has been extensively utilised in the treatment of arthritis. Flurbiprofen, also known as racemic 2-[2-fluoro-4-biphenyllpropionic acid, is an NSAID that Pfizer markets under the brand name ANSAID. It is given as a racemic combination and is a member of the class of NSAIDs known as the “profens” chemical group. The therapeutic activity of flurbiprofen primarily arises from its (S) enantiomer because, like its structural analogues ibuprofen and fenoprofen, only this enantiomer is effective in inhibiting cyclooxygenase28,29. Unlike its structural analogues, flurbiprofen does not undergo metabolic chiral inversion. Flurbiprofen enantiomers may undergo further metabolism via methylation to form 3-hydroxy-4-methoxy flurbiprofen or through oxidation to produce 3-hydroxy-4-dihydroxy flurbiprofen and 4-hydroxy flurbiprofen30. Additionally, it is utilised in the management of keratoconjunctivitis, gingivitis, herpetic stromal keratitis, acute gout, migraine, osteoarthritis, soft tissue injuries such as tendinitis and bursitis, rheumatoid arthritis, post-operative ocular inflammation, and excimer laser photo refractive keratectomy31,32,33.
Numerous pharmacological active compounds have been discovered through evaluation of their biological effects. For example, bis-Schiff bases of ibuprofen have been found as excellent anti-cancer agents34while piroxicam derivatives were successfully tested for their anti-glycation properties35. Similarly, Khan et al. reported albendazole derivatives as anti-glycating agents36 while Alam et al. found the synthesized flurbiprofen derivatives as anti-diabetic and anti-inflammatory agents (Figure 1). In addition, our group previously reported flurbiprofen–oxadiazole derivatives as potent α-amylase and urease inhibitors. The primary objective of the ongoing study is evaluated commercially available flurbiprofen derivatives for their in vivo anti-inflammatory potential.
This study aims to design flurbiprofen-based oxadiazole by-products is to combine the pharmacological aids of the oxadiazole moiety and the parent NSAID, flurbiprofen. There are numerous causes why the oxadiazole group is comprised. Firstly, the oxadiazole ring may enhance the compound’s pharmacokinetic properties, including its solubility, stability, and absorption, may be improved by the oxadiazole ring, which would increase its beneficial effectiveness37,38. Secondly, the selective COX-2 inhibitory activity of oxadiazole derivatives may improve the compound’s safety profile, which may lower the probability of gastrointestinal opposing effects that are commonly linked to NSAIDs39,40. Preclinical data obtained through screening in animal models of inflammation help identify lead compounds for further optimization and potential clinical development. The synthesis and evaluation of flurbiprofen derivatives represent a promising strategy to address unmet medical needs and improve therapeutic outcomes in inflammatory disorders.

Commercially available drugs derivatives with different biological activities.
Results and discussion
Chemistry
In the current study, we report flurbiprofen-based oxadiazole derivatives as potential anti-inflammatory agents. The combination of the oxadiazole nucleus, a recognized pharmacophore in medicinal/pharmaceutical chemistry, is considered to improve biological activity and decrease the gastrointestinal toxicity related with conservative non-steroidal anti-inflammatory drugs (NSAIDs). This method not only aims to report the restrictions of flurbiprofen but also subsidizes to the continuing growth of more effective, safer anti-inflammatory treatments, tiling the way for new drug candidates with greater therapeutic profiles. These compounds have been reported as potent α-amylase and anti-urease agents by our group (Scheme 1)22,41.

Procedure for the synthesis of oxadiazole derivatives of flurbiprofen.
The structural confirmation for the synthesized derivatives was based on a comprehensive examination of their1H-13, C-NMR and mass spectra. A group wise discussion of the main diagnostic signals is provided below, with spectral interpretation available in the experimental section. Amongst all the derivatives (2–10), the signals for the flurbiprofen ethyl moiety were consistently observed. The methine proton -CH(CH3) seemed as a quartet in the range of δ 4.31–4.59 ppm, and the adjacent methyl protons resonated as a doublet around δ 1.31–1.66 ppm. In the13C-NMR spectra, the two quaternary carbons of 1,3,4-oxadiazole ring were confirmed by characteristic signals in the downfield region of δ ~ 163–165 ppm and δ ~ 158–163 ppm. The structure of the key intermediate, 5-(1-(2-fluoro-[1,1’-biphenyl]−4-yl)ethyl)−1,3,4-oxadiazole-2-thiol (2), was confirmed by its spectral data. The successful cyclization was demonstrated by the characteristic13C-NMR signals for the oxadiazole carbons appeared at δ 163.2 and δ 158.6 ppm. Its mass spectrum showed a clear molecular ion peak (M⁺) at m/z 300, matching its calculated molecular weight (C16H13FN2OS). The presence of the thiol group was confirmed by its successful subsequent S-alkylation to form derivatives (3–5).
S-alkylated oxadiazole derivatives (3–5) characterized by the formation of a thioether linkage. The most vital diagnostic signal in the1H-NMR spectra is a sharp singlet for the methylene protons (-S-CH2-Ar) that appeared among δ 4.44–4.58 ppm. For the most active compound in this group, 3, this singlet was observed at δ 4.58 ppm. The presence of the para nitro benzyl group was confirmed by two distinct doublets observed at δ 8.14 ppm (d, 2H, J = 8.4 Hz) and δ 7.65 ppm (d, 2H, J = 8.8 Hz), characteristic of a 1,4-disubstituted aromatic ring. Its13C-NMR spectrum further supported this, with a signal for the methylene carbon at δ 38.1 ppm and the nitro containing aromatic carbon at δ 146.3 ppm. In direct C-C linked phenyl oxadiazole (6–10) series, a substituted phenyl ring is directly attached to the oxadiazole ring at C-5 position. The1H-NMR spectra were mainly useful for confirming the substitution pattern on this phenyl ring. For example, comparing the isomers 6 (3-nitrophenyl) and 9 (4-nitrophenyl) is illustrative, in the para substituted compound 9, the symmetry of the nitrophenyl ring resulted in a simple AA’BB’ system, showing two clean doublets at δ 8.67 ppm (2H) and δ 7.73 ppm (2H). In contrast, the meta substituted compound 6 lacked this symmetry, displaying four distinct and complex signals for the nitrophenyl protons at δ 8.61, 8.35, 8.14, and 7.80 ppm, clearly confirming the meta orientation. The successful synthesis of all compounds was further confirmed by mass spectrometry, which showed molecular ion peaks consistent with their respective calculated molecular formulas. This detailed, group-specific analysis provides robust and unambiguous evidence for the structures of all synthesized derivatives. Finally, mass spectrometry (EI-MS or HR-ESI-MS) provided definitive confirmation of the molecular weights. The molecular ion peak (M⁺ or [M + H]⁺) was observed for each compound and was consistent with its calculated molecular formula. For example, the thiol precursor 2 showed a molecular ion peak (M⁺) at m/z 300, while the S-alkylated derivative 3 showed an M+ peak at m/z 435. High-resolution mass spectrometry for compounds like 6 provided the exact mass ([M + H]+ at m/z 389.3791), further validating the elemental composition. This combined spectroscopic evidence provides unambiguous proof for the successful synthesis of the target derivatives.”
Biological evaluation
In vivo anti-inflammatory activity
The in vivo anti-inflammatory effect of each synthesised molecule was evaluated by measuring the physiological reactions of animals to both thermal and chemical stressors. The activity of the investigated compounds and a reference standard was assessed prior to the administration of carrageenan. Rats were then given an inflammatory stimulus, and the results were assessed after 30, 60, and 120 min. As shown in Figure 2 the percentage of edema inhibition was computed in comparison to the saline control group. The % edema inhibition was calculated in comparison to the saline control group. In the series, several of the evaluated compounds including 10 (83.33%), 3 (66.66%) and 5 (55.55%) revealed superb activity (decline in edema) (Table-S1). Similar to this, two compounds 9 and 2 exhibited good activity with 33.33 and 30.00% decrease respectively, while four compounds 7, 6, 8, and 4 attributed less to least activity with 26.66 to 3.33% decreases in edema.

Showing the percent inhibition of edema induced by the carrageenan by test compounds.
Structure-Activity relationship (SAR) analysis
The anti-inflammatory activity of the flurbiprofen based oxadiazole derivatives have been assessed by means of a carrageenan-induced mice paw edema assay. The percent decrease in edema after two hours served as the main degree of effectiveness. The investigation highlights perfect trends connecting structural features to the detected anti-inflammatory potential.
In the series, compound 10 revealed the highest activity, with an 83.33% decrease in edema. This brilliant potency is credited to the presence of a nitro group (electron-withdrawing) at position 2 on phenyl ring attached to the oxadiazole ring that is likely improves binding interfaces with the COX-2 active site. Similarly, compound 3 exhibited strong activity (66.66%), with a nitro group positioned at 4 on the phenyl attached to the oxadiazole ring, contributing to its effectiveness, while compound 5 (55.55%) comprises an unsubstituted phenyl ring. Though potent, its somewhat lower efficacy compared to compounds 10 and 3 suggests that the lack of an electron-withdrawing group decreases binding affinity to some extent.
On the other hand, compound 9 presented a 33.33% reduction in edema. Its structure is like to that of compound 3 but lacks a sulfur atom that is likely contributes to the observed decline in activity. Interestingly, compound 2 showed a 30.00% decrease in edema. Its structure is different with no phenyl ring and only an SH group is present. The lack of a wider hydrophobic moiety or electron-withdrawing substituent probably restricts its efficiency. Both the compounds propose that the existence of a nitro group or sulfur stabilizes important interactions within the COX-2 binding pocket, contributing to moderate activity. Besides, compound 7 demonstrated some activity, with a 26.66% reduction in edema, while compounds 6, 8, and 4 showed minimal activity, with reductions of 13.33%, 10.00% and 3.33%, respectively. The low effectiveness of these derivatives is probably because of the existence of electron-donating groups or the absence of sulfur in their structures, which may weaken interactions with the COX-2 active site. Lastly, the combination of the flurbiprofen nucleus is indispensable for activity, as established by the standard anti-inflammatory activity of flurbiprofen itself that accomplished a 90.01% decrease in edema. Differences in the activity of the synthetic compounds propose that oxadiazole substituents effect the contribution of the flurbiprofen moiety by modifying its contact with the COX-2 enzyme.
Molecular docking
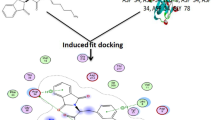
The in vivo activity of the most active oxadiazoles 10, 3, 5, 9, 2, and 7 based on flurbiprofen could be explained and corroborated by performing docking studies. Since the crystal structure of COX-2 is available with pdb code 3RR342. This would allow us to gain more insights into the molecular interactions and binding modes of these compounds with the oxidoreductase enzyme. The docking protocol was tested by re-docking flurbiprofen ligand that was co-crystallized with COX-2 with 3RR3 and comparing its conformation with the reference ligand43,44. The superimposed view of the docked and reference ligands is shown in Figure 3.

Re-docking the most active anti-inflammatory compounds 10, 3, 5, 9, 2, and 7 poses compared with flurbiprofen at active COX-2 site.
The RMSD (Root Mean Square Distance) of the docked ligands 10, 3, 5, 9, 2, and 7 was within the acceptable range of 0.6 Å to 2.2 Å, as shown in table-S2. The docking protocol confirmed that the indomethacin could bind to the crystal structure with 3RR3 in a similar way as reported the co-crystallized flurbiprofen. The docking studies were performed using Glide’s module. The initial inhibitor’s installation into the binding site to achieve crystal configurations indicates a preparatory step for structural analysis. The use of RMSD and scoring functions to evaluate the stability of the inhibitor within the binding pocket. These metrics help in ranking the docked poses, thereby selecting the most stable conformation for further analysis. Redocking the active analogues and comparing them to a stander inhibitor, such as indomethacin, provides a comparative framework to assess the efficacy of compounds 10, 3, 5, 9, 2, and 7. The Glide ΔG score is particularly useful to estimates the free energy of binding, which is crucial for understanding the binding mechanism and stability of the docked compounds 10, 3, 5, 9, 2, and 7. The Oples3e molecular-mechanics force field assists in generating the poses, with the selection based on the lowest ΔG and RMSD, to ensure the most favorable binding affinities. The interactions observed, including polar bonds, hydrogen bonding, and π-π interactions, are indicative of strong ligand-3RR3 oxidoreductase binding, which is essential for the potential efficacy of the compounds being investigated. Flurbiprofen make hydrophobic interactions, via the oxadiazole moiety, which is embedded in a hydrophilic area His388, His386 and Gln203 (Figure 3). Additional validation of the docking protocol involved overlaying the co-crystallized docked poses and reference inhibitor onto the 3RR3 oxidoreductase binding site. The resulting conformation occupied the identical binding position (Figure 3). The most active compounds 10, 3, 5, 9, 2, and 7 showed higher binding efficiency compared to flurbiprofen with ΔG= −7.54 Kcal/mol., as represented in table-S2. Docking results showed that they can occupy the same binding site as a co-crystallized ligand (flurbiprofen) and shared similar binding mode with it (Figure 3). The most active compound 10 with 83.33% decreasing inflammation compound exhibits a significant binding affinity, as indicated by the Gibbs free energy change (ΔG) of −7.59 Kcal/mol. This suggests a strong and spontaneous interaction with the binding pocket. The stabilization is achieved through a series of non-covalent interactions, which are crucial for the binding specificity and strength. The π-π stacking interactions, particularly between the phenyl group of the flurbiprofen fragment and the imidazole rings of HIS386 and HIS207, as well as between the oxadiazole ring and the imidazole of HIS388, contribute to the overall stability of the compound within the binding site. Additionally, the n-π interactions between the flurbiprofen and ASN382, and between the oxadiazole and LEU391, further reinforce the compound’s position in the binding pocket. The second most active compounds 3 (ΔG = 7.401 Kcal/mol) and 5 (ΔG = 8.304 Kcal/mol) with their decreasing percentage 66.6% and 55.55%, can be attributed to their inhibitory effects on the COX-2 enzyme. The inhibition of COX-2 by compounds 3 and 5 can be attributed to the formation of π-π stacking and n-π interactions, which are crucial in drug design due to their role in molecular recognition and binding stability. The π-π stacking interaction, particularly between the nitrophenyl group of compound 3 and the imidazole rings of HIS386, is significant in the stabilization of molecular structures. Similarly, the n-π interaction, such as that between the nitrophenyl ring of compound 3 and the CH of HIS388, involves the delocalization of lone pair electrons to π* orbitals, influencing the stabilization of peptides, proteins, and drugs, as well as interactions between flurbiprofen and the CH3 of Val477 (Figure 3). In contrast, compound 5 exhibits a significant hydrogen bond interaction between the phenyl substituent and HIS388, providing directionality and strength to the binding of molecules, and other n-π interactions with HIS214 and Gln203. These molecular interactions collectively are crucial for the compounds’ decreasing inflammation and highlight the specificity of their binding sites within the COX-2 enzyme. Compounds 9, 2, and 7 exhibit decreasing inflammation properties with Gibbs free energy (ΔG) values of −8.33.8 Kcal/mol, −7.386 Kcal/mol, and − 7.300 Kcal/mol, respectively. These compounds demonstrate their inhibitory action on COX-2 through various interactions. Compound 9 forms n-π interactions with HIS386 and ASN382, and π-π interactions with oxadiazole and HIS386, Phe210. Compound 2 engages the binding pocket via π-π interactions with flurbiprofen and HIS386, Phe210, and n-π interactions with ALA202, GLN203. Meanwhile, compound 7 interacts through π-π bonding with HIS 388, HIS 386, HIS 214, and HIS207. These interactions are crucial for the compounds’ ability to inhibit the COX-2 enzyme, which plays a key role in the process of inflammation. Understanding these interactions can lead to the development of more effective anti-inflammatory drugs with fewer side effects.
In Silico Pharmacokinetic prediction
The physicochemical properties and drug-likeness of the investigated derivatives were calculated using SwissADME and compared with standard inhibitor indomethacin45. The oral bioavailability for these active analogues 10, 3, 5, 9, 2, and 7 and flurbiprofen as standard inhibitor were calculated and introduced in Rader. Figure 4 is a comprehensive illustration of the pharmacokinetic properties of various compounds. Part A of the figure, the bioavailability radar, provides a visual representation of how well each compound meets the optimal criteria for drug-like properties. These criteria include lipophilicity (−0.7 ≤ XLOGP3 ≤ + 5.0), size (150 ≤ MW ≤ 500 g/mol), polarity (TPSA ≤ 140 Ų), solubility (log S ≤ 6), saturation fraction of carbons in hybridization (sp³ ≥ 0.25), and flexibility (rotatable bonds ≤ 9). The red zone highlights the range within which a compound is considered to have favorable attributes for bioavailability. Part B, the BOILED-Egg plot, further categorizes compounds based on their likelihood of gastrointestinal absorption and blood-brain barrier permeation. The color-coded regions and points allow for quick assessment of a compound’s potential as a P-glycoprotein substrate (PGP+) and P-gp non-substrate (PGP−), which is crucial for understanding drug distribution and potential interactions within the body. This dual approach provides a multidimensional view of compound characteristics, aiding in the selection and optimization of drug candidates.

In silico pharmacokinetic study of active oxadiazole derivatives (2, 3, 5, 7, 9, and 10) and flurbiprofen through SwissADME. (A) Bioavailability radar plot viewing the drug-likeness profile of each compound based on flexibility, saturation, solubility, polarity, size and lipophilicity. Compounds falling within the red zone are measured to have promising oral bioavailability. (B) BOILED-Egg model showing blood brain barrier penetration and gastrointestinal absorption potential. The plot also expects P-glycoprotein substrate status (PGP+ or PGP−), aiding in the assessment of pharmacokinetic distribution.
In Figure 4, we calculated the ADME properties for the most active compounds 10, 3, 5, 9, 2, 7 and flurbiprofen. The investigation revealed that the oxadiazole compounds exhibit desirable ADME characteristics. These properties are crucial for identifying potential drug candidates and should align with various rule-based filters. Notably, all oxadiazoles and flurbiprofen comply with drug similarity guidelines (including PAINS, Lipinski, Golden Triangle, Ghose violations, Veber violations, Egan violations, and Muegge violations). Based on these findings, they are approved as unique drug candidates.
To ensure proper systemic circulation, a new drug must pass through intestinal cell membranes. We evaluated drug permeability using human colon adenocarcinoma cell lines (Caco-2). According to the rule, a target compound with a projected value > −5.15 log cm/s is considered to have suitable Caco-2 permeability. Applying this criterion, we found that the investigated oxadiazoles and flurbiprofen fall within the range of −5.05 to −5.15 log cm/s, indicating nearly proper Caco-2 permeability (Table 1).
In our study, the investigated these oxadiazoles exhibited a bioavailability score of 0.55, which is lower than that of flurbiprofen (0.85). This categorizes them as drug-like compounds. Importantly, these oxadiazoles lack undesirable functional groups that could lead to carcinogenic, mutagenic, or hepatotoxic effects46. To predict gastrointestinal absorption and brain permeability, we employed the Brain or Intestinal Estimated Permeability approach (BOILED-Egg). This model relies on small molecule lipophilicity and polarity calculations. It offers a rapid, simple, and reproducible method for assessing the pharmacokinetic properties of compounds relevant to drug development. The white part of the egg (the yolk) represents the physicochemical zone associated with substances likely to be absorbed by the gastrointestinal tract. In contrast, the yellow region symbolizes the Blood-Brain Barrier a critical boundary that regulates the entry of chemicals into the brain. Compounds 10, 3, 5, 9, 2, and 7 flurbiprofen have High GI absorption and high permeability against BBB. None of the most active compounds and flurbiprofen can be transported out of the cells by P-glycoprotein, which is a protein that pumps foreign substances out of cells. This implies that the compounds can stay longer in the cells and have more effects on the receptors47. In recent times, metabolism prediction models have focused on the interaction between target compounds and cytochrome P450 mono-oxygenase enzymes (specifically CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4). These enzymes play a critical role in the phase one metabolism of pharmaceuticals, primarily within the liver. Inhibition of cytochrome enzymes is a key mechanism underlying pharmacokinetic drug-drug interactions48. Table 1 reveals that skin permeability correlates linearly with molecular size and lipophilicity. Notably, the more negative the log Kp (cm/s), the less skin permeation occurs49. Consequently, the investigated compounds exhibit promising skin penetration values, making them suitable for topical application, such as through skin massage (Table 1). Additionally, the low synthetic accessibility score (ranging from 4.95 to 5.11) for oxadiazoles, compared to a value of 4.93 for flurbiprofen, is crucial in selecting the most promising virtually tested molecules for production and subsequent use in biological experiments (Table 1).
We focused on estimating the uptake efficiency of target compounds 10, 3, 5, 9, 2 and 7 using the apparent permeability coefficient. To achieve this, we developed Madin-Darby Canine Kidney cells (MDCK) as a model for permeability screening. Additionally, we evaluated the impact of the blood-brain barrier (BBB) by analyzing Papp values from MDCK cell lines. A target compound is considered to have high passive MDCK permeability when its Papp exceeds 20 × 10−6 cm/s. notably; the most active oxadiazoles 10, 3, 5, 9, 2, and 7 exhibit such high permeability, ranging from 1.9 × 10−5 to 2.8 × 10−5 cm/s, whereas flurbiprofen shows a lower value of 2.90 × 10−6 cm/s. Furthermore, we assessed the plasma protein binding (PPB) and the percentage unbound in plasma (FU) of the drugs. If the expected PPB and FU values are greater than or equal to 5% and less than 90%, respectively, the target drug is considered to have appropriate plasma protein binding affinity. Based on our findings, the investigated compounds exhibit high binding with plasma proteins (predicted values ranging from 98.95 to 100%) compared to flurbiprofen (98.29%). However, their predicted values of FU are less than 5%. These compounds may have a low therapeutic index, indicating slight efficiency in crossing membranes.
The Ames test was used to predict the compounds 10, 3, 5, 9, 2, and 7 and flurbiprofen compounds induced toxicity and to evaluate the safety of these compounds. Table 1 shows that compounds were non-mutagenic, that showed positive Ames test results. Moreover, the brain/blood partition coefficient values were low for all compounds, indicating that they have a potential to cross the blood-brain barrier and causes CNS related toxicity, compared to flurbiprofen value (0.41). The oral bioavailability of the compounds depends on their intestinal absorption. The results indicate that all compounds have a good calculated intestinal absorption, which means that they can be easily absorbed by the intestine. The new compounds which synthesized have desirable properties, such as high intestinal absorption, large volume of distribution, good BBB penetration and low toxicity. They also have strong COX-2 inhibition and could be useful for further research.
Our findings are supported by the bioavailability radar in Figure 4, which indicates that the oxadiazole derivatives meet optimal criteria for drug-like properties. Additionally, we suggest that oxadiazole moieties can introduce hydrogen bonding capabilities, which may further enhance absorption and distribution. Furthermore, the oxadiazole moiety enhances absorption by optimizing the lipophilicity and hydrophobicity balance of the compounds. Our Caco-2 permeability data shows that the oxadiazole derivatives exhibit permeability values close to the threshold for good intestinal absorption, suggesting that this moiety facilitates membrane crossing. This balance is crucial for effective absorption in the gastrointestinal tract. The oxadiazole group contributes to high plasma protein binding, as evidenced by the predicted values ranging from 98.95 to 100.97%, which is comparable to flurbiprofen. However, the low percentage unbound in plasma (FU < 5%) indicates a potential low therapeutic index, suggesting that the oxadiazole moiety may enhance tissue penetration and distribution, although this requires further investigation. The oxadiazole moiety appears to confer metabolic stability by minimizing interactions with cytochrome P450 enzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4). This reduces the likelihood of significant drug-drug interactions and suggests that the compounds have a prolonged half-life in the body, as they are less prone to metabolic degradation.
Frontier molecular orbitals (FMOs) of oxadiazole derivatives 10, 3, 5, 9, 2, and 7
The terms HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) describe specific electronic states within molecules50. These orbitals play a crucial role in chemical reactions and reactive regions51. By analyzing the frontier molecular orbitals (FMOs), we efficiently assess internal charge transfer and reveal the kinetic instability or reactivity the compounds 10, 3, 5, 9, 2, and 7 compared with flurbiprofen52. To investigate these electronic characteristics, we employed TD-DFT (wb97x-d3), a method known for its accurate predictions of molecular properties and condensed phase geometries, making it suitable for diverse chemical applications53,54. To achieve accurate results aligning well with experimental data for the synthesized oxadiazoles, the 6-311 + + G** basis set, has been employed for its ability to provide precise outcomes.

FMOs for oxadiazoles 10, 3, 5, 9, 2, and 7 based on flurbiprofen.
The FMO analysis indicates that the distribution of electron density within the HOMO is mainly focused over oxadiazoles for six most active investigated flurbiprofen derivatives 10, 3, 5, 9, 2, 7 and on the π-conjugated system for the aromatic substituents ring with (2-NO2-C6H4) for 10, (4-NO2-C6H4-S) for 3, (4-NO2-C6H4) for 9, (C6H4) for 7. Besides, this orbital is inverted for compounds 5 and 2, which shifted to concentrate above the aromatic system of flurbiprofen (Figure 5). Furthermore, the Lowest Unoccupied Molecular Orbital (LUMO) levels in compounds 10, 3, 9 and 7 are primarily associated with the flurbiprofen fragments, indicating potential sites for chemical reactivity. In contrast, compounds 5 and 2 exhibit a more dispersed electron density for HOMO and LUMO orbitals across the entire flurbiprofen fragment, suggesting a different reactivity profile (Figure 5). The results of reactivity indices are presented in figures S1-S3.
Molecular electrostatic potential studies
The molecular electrostatic potential (MEP) is a visual way of showing how the charge is distributed on the surface of a molecule, which affects how it reacts with other molecules55. The color blue indicates a positive charge area (a nucleophilic site), the color red indicates a negative charge area (an electrophilic site), and the colors green and cyan indicate neutral charge areas. The negative areas localized over the oxygen and the nitrogen atoms of the oxadiazole for all most active investigated compounds (Figure 6), which make them more likely to be attacked by electrophiles. Positive areas are found around the hydrogen atoms the methyl group, which make them more likely to interact with nucleophiles. The nitrogen of and oxygen atom showed high nucleophilic tendencies.

Molecular electrostatic potential of oxadiazole 10, 3, 5, 9, 2, and 7 based on flurbiprofen.
In contrast, the carbon atoms of the phenyl rings are more prone to electrophilic attacks, which could be due to their lower electron density. The dispersed electron density distribution in the most active compounds, such as 10 and 3, compared to other compounds 5, 9, 2, and 7, suggests they have distinct electronic characteristics that may contribute to their biological activities, including the potential to inhibit inflammation (Figure-6). This understanding of electron density and reactivity is essential for the design and synthesis of new pharmaceuticals with targeted biological effects.
Molecular dynamic (MD) simulations
The Molecular Mechanics-Poisson-Boltzmann Surface Area (MM-PBSA) approach was used to conduct MD simulation at the nano scale (ns) in order to validate and gain a clear understanding of the ligand-protein interactions that emerged from the molecular docking results. The binding free energy changes between the majority of active compounds 10, 3, 5 and the protein complexes were computed using that. The MD simulations for ligand-protein interactions are carried out over a time period of 100 ns. The interactions between ligands and proteins are analyzed every 2 ns (Figure-S4, supplementary materials), illustrating the variations in binding strength between the compounds 10, 3, and 5 and the COX-2 protein complexes. The binding free energy changes of the most active ligand molecules for COX-2. Negative values for MM-PBSA free energies indicate stronger binding interactions. The small divergence for MM-PBSA for investigated complexes with oligopeptidase over the entire time-scale indicated a robust contact between Ligand and amino acid backbone, indicating a firm H-bonding pattern. The consistent MM-PBSA values over the simulation period suggest a stable interaction, likely due to a strong hydrogen-bonding network between the ligand and the protein’s amino acid backbone.
We analyzed the structural changes of target protein during MD simulation by calculating the root means square deviation (RMSD) of the protein backbone and root mean square fluctuation (RMSF) of each residue. The RMSD of the protein backbone of each residue indicated the structural changes of the protein over the 100 ns simulation. The results revealed that the protein backbone of 10, 3, and 5 complexes had RMSD values between 05 nm and 1.2 nm after 5 ns of simulation. This is a relatively low value, which implies that the drug candidates interacted stably with the protein and did not modify their protein backbone structure. The low RMSD values also support the hypothesis that the drug candidates are suitable for binding to the protein in a stable and specific manner that does not disrupt its structure. The assessment of the spatial variations at the residual level (RMSF) provided the details about the residues that are more sensitive to drug binding. Residue numbers 120 to 350 of protein backbone were located inside of the ligand-binding site, while other outside amino acid residues deviated more than other residues upon binding of compounds 10, 3, and 5 (Figure-S4, supplementary materials). The 3-complex had a lower mean RMSD than the other complexes, indicating that formed a stronger and more stable complex with the protein’s active site. The backbone of the 3-complex and stabilized between 5 ns and 15 ns after 43 ns of simulation, while the other complexes required more time to stabilize. The compound 5 had a higher RMSD deviation until the 50th ns, but then the graphs converged. This implies that compound 5 also formed a stable complex with the protein, but with a longer time span. After 78 ns, the deviation of compound 10 was higher than the other compounds 3 and 5, and the overall deviation of compounds 15 and 14 is relatively lower than the compound 3. This suggests that the compound 3 is able to form the most stable complex with the protein, followed by compounds 5 and 10.
Methods
General
Each of the chemicals, solvents and reagents were acquired from Sigma-Aldrich, TCI, Merck and BDH and was of analytical quality. For thin layer chromatography (TLC), aluminium plates covered with silica gel (Kieselgel 60, 254, E. Merck, Germany) were utilised. Spots were seen using UV light with two wavelengths (254 and 365 nm). The MAT 312 and MAT 113D mass spectrometers were used to record the electron impact mass experiment. Operating at 300 and 400 MHz, Bruker AM machines were used to record the1H-NMR spectra. The coupling constant (J) is given in Hz, and the chemical shift values are given in ppm (δ) with respect to tetramethylsilane (TMS) as an internal reference. Using the Stuart SMP10 melting point instrument, the compounds’ melting points were ascertained. On an Italian Carlo Erba Strumentazione-Mod-1106, CHN analyses were performed.
Experimental animals
All the experiments performed on animals were approved by the Ethical Research Committee, Department of Pharmacy, CUI, Abbottabad Campus (PHM.Eth/CS-M01-03-0323) and conformed with the rulings of the Institute of Laboratory Animal, Resources, Commission (on Life Sciences,) National Research Council (NRC, 2010). Additionally, all the procedures performed on animals were according to the updated guidelines reported in accordance with the ARRIVE guidelines 2.056. Balb/C mice (20–22 g) of either sex were obtained from the National Institute of Health (NIH), Islamabad, Pakistan, and housed in the animal house of CUI, Abbottabad. The temperature was maintained at 25 ± 2 °C. All the animals were fed with standard rat diet and tape water ad libitum and a 12 h light-dark cycle was maintained in the animal house.
Carrageenan-Induced acute inflammatory model
Using the carrageenan-induced mice paw edema experiment, anti-inflammatory activity was assessed as previously reported57. Thirty minutes before the carrageenan injection, test compounds were given intraperitoneally. Edema was created in the right hind paws of each group of mice by sub plantar injection of 50 µL of a newly made 1% carrageenan solution in distilled water. Paw thickness was measured at “0 hour,” right before the carrageenan injection, and then at 0.5, 1, and 2 h following the injection. The difference between the paw thickness at “0 hour” and the paw thickness at the corresponding hours was used to measure the increase in paw thickness.
Molecular modeling
Time-dependent density functional theory (TD-DFT) is a pivotal quantum mechanical method used to analyze the electronic structure and reactivity of molecules under time-varying conditions. The use of hybrid functional like wb97x-d3 in conjunction with a 6-311 + + G** basis set al.lows for a detailed understanding of molecular properties58,59,60. Full geometry optimizations using single-point TD-DFT calculations enable the computation of harmonic vibrational frequencies, using first singlet excited state, ensuring the stability of the optimized structures as indicated by the absence of imaginary frequencies. The calculation of frontier molecular orbitals (FMOs), such as the highest occupied molecular orbital (EHOMO) and the lowest unoccupied molecular orbital (ELUMO), provides insight into a molecule’s reactivity and interaction with other species. Additional parameters like the energy gap, electron affinity, hardness, electronegativity, ionization energy, softness, and electrophilicity index are crucial for a comprehensive understanding of molecular behavior. The molecular electrostatic potential (MEP) map, complemented by Laplacian analysis, offers a visual representation of charge distribution and potential reactive sites. TD-DFT also facilitates the study of electronic transitions, which is essential for performed the topology analysis further aids in examining non-covalent interactions, which are significant in the stability and function of molecular systems. Visualization tools such as the Materials Science Suite202461, along with computational packages like jaguar package62are indispensable for interpreting and presenting the complex data derived from these simulations.
Molecular Docking method
The Glide was implemented to conduct a docking investigation for the target al.ongside its employment in the discovery studio63. After attaining the crystallized COX-2, water and inhibitor molecules were removed, and the H atoms were added. The examined ligands were redocked into the vacant active site after the standard inhibitor was removed from it. The scoring function was generated for measuring the binding affinity, and the charges were assigned with the CHARMM force field. The structure with the lowest RMSD score was utilized to generate different ligand poses. For calculating the binding affinity, the scoring function was integrated, and the CHARMM force field was used to assign charges.
Synthesis of the compounds (2–10)
These compounds have been published earlier by our group as potent α-amylase and urease inhibitors22,41.
Conclusion
Some oxadiazole derivatives bearing flurbiprofen moiety have been successfully synthesized and screened for their anti-inflammatory activity using carrageenan induced mice paw edema assay. In the series, three compounds that includes 10 (83.33%), 3 (66.66%) and 5 (55.55%) exposed outstanding activity (decline in edema). Moreover, two compounds 9 and 2 exhibited good activity with 33.33 and 30.00% decrease in edema respectively, while four compounds 7, 6, 8, and 4 attributed less to least activity with 26.66 to 3.33% decreases in edema. Furthermore, the in-silico study of most active amides 10, 3, 5, 9, 2, 7 and flurbiprofen were performed against human COX-2 oxidoreductase and these compounds showed more stable docking energies than flurbiprofen and confirming their plausible mode of action. Also, the drug-likeness prediction showed that all the prepared oxadiazole agree with drug likeness rules and these derivatives are accepted as new drug candidates. Using TD-DFT, we expanded active oxadiazoles and calculated their FMO, which revealed their stability, bioactivity and charge transfer properties. We visualized the reactive regions of molecules with MEP, and found that they were near the Nitrogen atoms, which were more electrophilic and could interact with protein receptors. We performed AIM-topological analysis of investigated molecules showed that the H-bond energy was minimal and that the interactions were non-covalent. The hopeful consequences inspire more in vivo studies to evaluate toxicity, bioavailability, and pharmacokinetics profiles of the potent derivatives. Optimization of the structure-activity relationship (SAR) may possibly improve their selectivity and efficacy. Moreover, discovering their potential against other inflammation-related corridors and enzymes might extend their therapeutic usefulness. These efforts might pave the technique for the expansion of safer as well as effective anti-inflammatory drugs.
Data availability
All data generated or analyzed during this study are included in this published article and Supplementary Materials.
References
Alam, A. et al. Synthesis and characterization of biologically active flurbiprofen amide derivatives as selective prostaglandin-endoperoxide synthase II inhibitors: in vivo anti-inflammatory activity and molecular Docking. Int. J. Biol. Macromol. 228, 659–670 (2023).
Imkamp, M. S., Theunissen, M., Viechtbauer, W. & van Kuijk, S. M. & Van Den Beuken–van, M. H. Shifting views on cancer pain management: a systematic review and network meta-analysis. Journal Pain Symptom Management 68 (2024).
Inchingolo, A. M. et al. Advancing postoperative pain management in oral cancer patients: A systematic review. Pharmaceuticals 17, 542 (2024).
Nickels, K. et al. Cancer pain management in patients with opioid use disorder. Current Addict. Reports 11, 1–17 (2024).
Tayeb, E. A. A. et al. The role of nursing in pain management for cancer patients: strategies and challenges. Journal Int. Crisis Risk Communication Research 7, 1641–1655 (2024).
Scott, I. C. et al. Pain management in people with inflammatory arthritis: British society for rheumatology guideline scope. Rheumatol. Adv. Pract. 8, rkae128 (2024).
Huette, P. et al. Effect of non-steroidal anti-inflammatory drugs on the management of postoperative pain after cardiac surgery: a multicenter, randomized, controlled, double-blind trial (KETOPAIN Study). Trials 25, 613 (2024).
Shkodina, A. D. et al. Pharmacological and Non-pharmacological approaches for the management of neuropathic pain in multiple sclerosis. CNS Drugs. 38, 205–224 (2024).
Gu, Q. et al. Distinct patterns of vital sign and inflammatory marker responses in adults with suspected bloodstream infection. J. Infect. 88, 106156 (2024).
Cederholm, T. et al. Guidance for assessment of the inflammation etiologic criterion for the GLIM diagnosis of malnutrition: A modified Delphi approach. Clin. Nutr. 43, 1025–1032 (2024).
Li, H. et al. Research trends and hotspots on global influenza and inflammatory response based on bibliometrics. Virol. J. 21, 1–13 (2024).
Bhol, N. K. et al. The interplay between cytokines, inflammation, and antioxidants: mechanistic insights and therapeutic potentials of various antioxidants and anti-cytokine compounds. Biomed. Pharmacother. 178, 117177 (2024).
Chadda, K. R., Blakey, E. E., Davies, T. W. & Puthucheary, Z. Risk factors, biomarkers, and mechanisms for persistent inflammation, immunosuppression, and catabolism syndrome (PICS): a systematic review and meta-analysis. British J. Anaesthesia 133 (2024).
Yao, J., Zhang, S., Zhou, F., Zhuang, M. & Fei, S. The relationship between inflammatory cytokines and in-hospital complications of acute pancreatitis. Immun. Inflamm. Dis. 12, e1203 (2024).
Zhao, X. et al. Macrophages in the inflammatory response to endotoxic shock. Immun. Inflamm. Dis. 12, e70027 (2024).
van der Stouwe, J. G. et al. Body temperature, systemic inflammation and risk of adverse events in patients with acute coronary syndromes. Eur. J. Clin. Invest. 54, e14314 (2024).
Wang, S. & Zhang, G. Association between systemic Immune-inflammation index and adverse outcomes in patients with acute coronary syndrome: a Meta-analysis. Angiology, 00033197241263399 (2024). https://doi.org/10.1177/0003319724126339.
Weiss, M. et al. IRF5 controls both acute and chronic inflammation. Proc. Natl. Acad. Sci. 112, 11001–11006 (2015).
Jensen, G. L. et al. Guidance for assessment of the inflammation etiologic criterion for the GLIM diagnosis of malnutrition: A modified Delphi approach. J. Parenter. Enter. Nutr. 48, 145–154 (2024).
Klein, A. & Eliakim, R. Non steroidal anti-inflammatory drugs and inflammatory bowel disease. Pharmaceuticals 3, 1084–1092 (2010).
Brune, K. & Patrignani, P. New insights into the use of currently available non-steroidal anti-inflammatory drugs. Journal Pain Research 8, 105–118 (2015).
Ahmad, S. et al. Novel flurbiprofen clubbed oxadiazole derivatives as potential urease inhibitors and their molecular Docking study. RSC Adv. 13, 25717–25728 (2023).
Alam, A. et al. Bio-oriented synthesis of novel (S)-flurbiprofen clubbed hydrazone schiff’s bases for diabetic management: in vitro and in Silico studies. Pharmaceuticals 15, 672 (2022).
Alam, A. et al. Flurbiprofen clubbed schiff’s base derivatives as potent anticancer agents: in vitro and in Silico approach towards breast cancer. J. Mol. Struct. 1321, 139743 (2025).
Khan, M. et al. Flurbiprofen derivatives as potential DPPH and ABTS radical scavengers. Russ. J. Org. Chem. 59, 1577–1582 (2023).
Geisslinger, G. & Schaible, H. G. New insights into the site and mode of antinociceptive action of flurbiprofen enantiomers. J. Clin. Pharmacol. 36, 513–520 (1996).
Hmood, K. S., Ammar, A., Al-Bayati, R. I. & Saleh, A. M. Synthesis, and anti-tumor evaluation of some new flurbiprofen derivatives against MCF-7 and WRL-68 cell lines. Indonesian J. Pharm. 32, 17–34 (2021).
Tang, K., Song, L., Liu, Y., Pan, Y. & Jiang, X. Separation of flurbiprofen enantiomers by biphasic recognition chiral extraction. Chem. Eng. J. 158, 411–417 (2010).
Lv, J., Xu, Y., Xu, L. & Nie, L. Quantitative functional evaluation of liver fibrosis in mice with dynamic contrast-enhanced photoacoustic imaging. Radiology 300, 89–97 (2021).
Gu, L. et al. Direct distinction of ibuprofen and flurbiprofen enantiomers by ion mobility mass spectrometry of their ternary complexes with metal cations and cyclodextrins in the gas phase. J. Sep. Sci. 44, 2474–2482 (2021).
Brogden, R., Heel, R., Speight, T. & Avery, G. Flurbiprofen: a review of its Pharmacological properties and therapeutic use in rheumatic diseases. Drugs 18, 417–438 (1979).
Richy, F., Rabenda, V., Mawet, A. & Reginster, J. Y. Flurbiprofen in the symptomatic management of rheumatoid arthritis: a valuable alternative. Int. J. Clin. Pract. 61, 1396–1406 (2007).
He, X., Jiang, Z., Akakuru, O. U., Li, J. & Wu, A. Nanoscale covalent organic frameworks: from controlled synthesis to cancer therapy. Chem. Commun. 57, 12417–12435 (2021).
Ayaz, M. et al. Biooriented synthesis of ibuprofen-clubbed novel bis-schiff base derivatives as potential hits for malignant glioma: in vitro anticancer activity and in Silico approach. ACS Omega. 8, 49228–49243 (2023).
Ullah, S. et al. Synthesis and characterization of novel piroxicam derivatives and their antiglycation activity. J. Mol. Struct. 1239, 130470 (2021).
Khan, M. et al. Biology-oriented drug synthesis (BIODS), structural characterization and bioactivities of novel albendazole derivatives. Lett. Drug Des. Discovery. 16, 1329–1338 (2019).
Rajak, H., Veerasamy, R., Kharya, M. & Mishra, P. Design, synthesis, and Pharmacological evaluation of novel oxadiazole and Oxadiazoline analogs as anti-inflammatory agents. J. Enzyme Inhib. Med. Chem. 25, 492–501 (2010).
Abd-Ellah, H. S. et al. Novel 1, 3, 4-oxadiazole/oxime hybrids: synthesis, Docking studies and investigation of anti-inflammatory, ulcerogenic liability and analgesic activities. Bioorg. Chem. 69, 48–63 (2016).
Alam, M. M. et al. Naproxen based 1, 3, 4-oxadiazole derivatives as EGFR inhibitors: design, synthesis, anticancer, and computational studies. Pharmaceuticals 14, 870 (2021).
Chawla, G., Naaz, B., Siddiqui, A. A. & Exploring 1, 3, 4-oxadiazole scaffold for anti-inflammatory and analgesic activities: a review of literature from 2005–2016. Mini reviews in medicinal chemistry 18, 216–233 (2018).
Khan, M. et al. Flurbiprofen derivatives as novel α-amylase inhibitors: Biology-oriented drug synthesis (BIODS), in vitro, and in Silico evaluation. Bioorg. Chem. 81, 157–167 (2018).
Duggan, K. C. et al. R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat. Chem. Biol. 7, 803–809 (2011).
Elhenawy, A. A., Al-Harbi, L., El-Gazzar, M., Khowdiary, M. M. & Alosaimi, A. M. Naproxenylamino acid derivatives: design, synthesis, docking, QSAR and anti-inflammatory and analgesic activity. Biomed. Pharmacother. 116, 109024 (2019).
Elhenawy, A. A. et al. Synthesis, comparative docking, and Pharmacological activity of Naproxen amino acid derivatives as possible anti-inflammatory and analgesic agents. Drug Des. Dev. Therapy 13, 1773–1790 (2019).
Ezzat, A. et al. Synthesis, spectral characterization, antimicrobial evaluation and molecular Docking studies of new Cu (II), Zn (II) thiosemicarbazone based on sulfonyl Isatin. J. Mol. Struct. 1251, 132004 (2022).
Sharma, S. et al. Insight into the synthesis approaches of oxadiazole and its derivatives, focuses on their Pharmacological activities. Indian J. Pharm. Educ. Res. 59, s25–s45 (2025).
Montanari, F. & Ecker, G. F. Prediction of drug–ABC-transporter interaction—Recent advances and future challenges. Adv. Drug Deliv. Rev. 86, 17–26 (2015).
Deodhar, M. et al. Mechanisms of CYP450 Inhibition: Understanding drug-drug interactions due to mechanism-based Inhibition in clinical practice. Pharmaceutics 12, 846 (2020).
Chen, C. P., Chen, C. C., Huang, C. W. & Chang, Y. C. Evaluating molecular properties involved in transport of small molecules in stratum corneum: A quantitative structure-activity relationship for skin permeability. Molecules 23, 911 (2018).
Mumit, M. A. et al. DFT studies on vibrational and electronic spectra, HOMO–LUMO, MEP, HOMA, NBO and molecular Docking analysis of benzyl-3-N-(2, 4, 5-trimethoxyphenylmethylene) hydrazinecarbodithioate. J. Mol. Struct. 1220, 128715 (2020).
Tsuneda, T. Chemical reaction analyses based on orbitals and orbital energies. Int. J. Quantum Chem. 115, 270–282 (2015).
Sengupta, A., Li, B., Svatunek, D., Liu, F. & Houk, K. Cycloaddition reactivities analyzed by energy decomposition analyses and the frontier molecular orbital model. Acc. Chem. Res. 55, 2467–2479 (2022).
Snigdha, K. et al. Green synthesis of pyrazole derivatives via one-pot three component Knoevenagel–Michael addition utilizing TiO2/RuO2/CuO as a ternary nanocatalytic system: characterization, DFT and molecular Docking studies. Research Chem. Intermediates 51 , 1–27 (2025).
Fedunov, R. G. et al. Photophysics and photochemistry of (n-Bu4N) 2 [Pt (NO3) 6] in acetonitrile: ultrafast pump-probe spectroscopy and quantum chemical insight. Photochem. Photobiol. Sci. 23, 1957–1970 (2024).
Gul, S. et al. Exploring the synthesis, molecular structure and biological activities of novel Bis-Schiff base derivatives: A combined theoretical and experimental approach. J. Mol. Struct. 1306, 137828 (2024).
Percie du Sert. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. BMC Vet. Res. 16 https://doi.org/10.1186/s12917-020-02451-y (2020).
Alam, A. et al. Discovery of (S)-flurbiprofen-based novel Azine derivatives as prostaglandin endoperoxide synthase-II inhibitors: synthesis, in-vivo analgesic, anti-inflammatory activities, and their molecular Docking. Bioorg. Chem. 141, 106847 (2023).
Jacquemin, D., Perpete, E. A., Ciofini, I. & Adamo, C. Assessment of the ωB97 family for excited-state calculations. Theor. Chem. Acc. 128, 127–136 (2011).
Chai, J. D. & Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008).
Adamo, C. & Barone, V. Toward reliable density functional methods without adjustable parameters: the PBE0 model. J. Chem. Phys. 110, 6158–6170 (1999).
Release, S. 4: Materials Science Suite. Schrödinger, LLC, New York, NY 2015 (2015).
Bochevarov, A. D. et al. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 113, 2110–2142 (2013).
Accelrys Accelrys Software Inc,. (2018).
Acknowledgements
This work was supported by the Ongoing Research Funding Program (ORF-2025-235), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
K.S., and A.A., performed experimentations, while A.A.E, M.K and F.U.R., wrote the original draft of the manuscript. I. A, K and M.U.R performed the anti-inflammatory inhibitory activity of the compounds. A.A.E and A.A. performed molecular docking study of the compounds. M. K A.A. and F.A supervised the project, assisted in the writing, the reviewing, and the editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
All the experiments performed on animals were approved by the Ethical Research Committee, Department of Pharmacy, CUI, Abbottabad Campus (PHM.Eth/CS-M01-03-0323) and conformed with the rulings of the Institute of Laboratory Animal, Resources, Commission (on Life Sciences,) National Research Council (NRC, 2010).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shaheen, K., Alam, A., Elhenawy, A.A. et al. In vivo anti-inflammatory evaluation of oxadiazole derivatives bearing flurbiprofen moiety using experimental and computational approaches. Sci Rep 15, 29144 (2025). https://doi.org/10.1038/s41598-025-13351-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-13351-2
