
Abstract
Type 2 diabetes (T2D) poses a significant global health burden. We developed a polygenic risk score (PRS) model based on genome-wide association study (GWAS) findings and integrated it with clinical data to predict T2D risk. This study analyzed electronic medical records from a major medical center in Taiwan, comprising 315,424 T2D cases and 141,484 controls. Fourteen genome-wide significant SNPs were identified and used to construct the T2D PRS. The integrated predictive model showed high accuracy (AUROC 0.842) and was validated in the Taiwan Biobank. A risk score ranging from 0 to 19 was established for clinical use. Phenome-wide association study (PheWAS) revealed links between PRSs and T2D-related complications, such as diabetic retinopathy and hypertension. Pathway analysis highlighted biological processes including IL-15 production and WNT/β-catenin signaling. Our findings support the use of PRSs in personalized T2D risk assessment and early prevention strategies.
Similar content being viewed by others


Introduction
As of 2021, approximately 537 million people worldwide suffer from diabetes, and approximately 6.7 million people die from the disease annually1. In Taiwan, diabetes is the sixth leading cause of death, with a mortality rate of 52.7 per 100,000 people in 20222. Type 2 diabetes (T2D) comprises approximately 90% of diabetes cases3. Risk factors for T2D include genetics, obesity, environmental factors, and an unhealthy lifestyle4,5,6. These risk factors lead to insulin resistance, which can result in poor blood sugar control and hyperglycemia7. The complications of T2D include cardiovascular disease, various neuropathies (such as peripheral, autonomic, focal, and proximal neuropathy), heart disease, kidney disease (leading to kidney failure), retinopathy, injuries to limb extremities, skin lesions (which are prone to infection and heal slowly), and sexual dysfunction (such as erectile dysfunction)8,9,10. Additionally, more diseases related to T2D are emerging.
The correlation between polygenic risk scores (PRSs) and T2D has been extensively studied, and advancements in PRS models since 2007 have improved T2D risk assessment. Early PRSs were constructed using a small number of genome-wide significant variants, and although recent studies have explored more complex approaches involving larger sets of variants, the number and selection of SNPs often vary depending on study design, target population, and predictive goals. Advances in GWAS datasets and computational methods have generally improved the predictive accuracy and broader applicability of PRSs across populations. PRSs are crucial in clinical medicine for risk prediction, early diagnosis, and treatment response prediction, leading to better disease outcomes and fewer adverse drug reactions. In T2D research, PRSs have been increasingly applied to predict disease onset, complications, and treatment outcomes, particularly when combined with clinical variables11,12. Despite this, large-scale efforts to establish and apply PRS models for T2D remain limited in Taiwan. Combining PRSs with clinical data allows for more interpretable risk scores that facilitate population-level risk stratification and individualized prevention. Furthermore, constructing risk score models is beneficial to convert the predicted probabilities of outcome events into a simple score. This transformation enhances the readability of the results and facilitates the risk assessment of subjects. Risk scores allow stratification of a population for targeted screening. They use risk factor data to calculate an individual’s total score; a higher score represents a higher risk of disease. Over the past decade, a plethora of risk score methods have been developed and validated for detecting those at risk of T2D13,14,15,16. In this study, we conducted GWAS to identify genetic variants related to T2D and validated them, constructed a risk score model included PRSs and clinical variables to predict the risk of T2D, and used a phenome-wide association study (PheWAS) to identify other diseases associated with T2D, based on a population in Taiwan. Additionally, bioinformatics network analysis was conducted to explore the signaling pathways involved in the pathogenesis of T2D.
Materials
Data source
In this study, electronic medical records (EMRs) from the China Medical University Hospital (CMUH), one of the largest medical centers in Taiwan were utilized. For research purposes, demographic information, laboratory data, International Classification of Diseases codes, and medical procedure data, including personal medical information contained within the CMUH EMRs, were accessed. Additionally, the CMUH database contains genetic information from patients as part of the CMUH Precision Medicine Project (also known as HiGenome database) approved by the Ethics Committee of the Institutional Review Board of CMUH (CMUH107-REC3-058, CMUH110-REC3-005, and CMUH111-REC1-176)17. The current study received approval from the IRB of CMUH (CMUH109-REC1-003).
Identification of T2D cohorts for GWAS and PRS generation
A total of 15,424 T2D cases and 141,484 control subjects were included in the T2D cohort. Cases within this cohort consisted of individuals aged 20 years or older who were diagnosed with T2D at least twice according to the ICD, 9th /10th Revision, Clinical Modification [ICD-9-CM] (ICD-9-CM code = 250.xx, except for 250.x1 and 250.x3; ICD-10-CM = E11.xx). Control subjects were identified as individuals without any diagnosis of diabetes or prescriptions for antidiabetic drugs in their EMR.
Genotyping and GWAS
Genomic DNA was extracted from peripheral blood leukocytes of the participants using a commercial kit (Qiagen, Redwood City, CA, USA), as previously reported18. In CMUH-PMP, a TPMv1 customized single-nucleotide polymorphism (SNP) array (Thermo Fisher Scientific, Santa Clara, CA, USA) was used for genotyping. The Affymetrix Axiom genotyping platform was used according to the standard quality control procedures. Genotype data were imputed using Beagle 5.2. Imputed SNPs with an INFO R2 ≧ 0.3 were kept for further GWAS. Detailed genotyping and imputation procedures have been described earlier19.
The relevant study participants were randomly divided into three groups with an 7:1.5:1.5 ratio, with 70% of subjects as the discovery cohort for GWAS, and 15% and 15% of subjects as the target and validation cohort. The quality control (QC) process was performed as follows: (1) samples were excluded if they had a call rate of < 98%, extreme heterozygosity rate (> 3 standard deviations [SDs] from the mean), and duplicate or related individuals (kinship > 0.354); (2) variants were filtered out if they had a missing rate > 2%, deviated from Hardy–Weinberg equilibrium with P < 1 × 10− 10, and minor allele frequency < 0.01. After the QC process, additive models adjusted for age, sex, and principal components (PCs) 1–10 were used using PLINK 2.020. A Manhattan plot was constructed using the CMplot R package.
Identification of T2D associated SNPs and PRSs
The SNPs identified from GWAS with P value < 0.05 (786,845 SNPs) were included for PRS construction. The PRSs were constructed using the “clumping and thresholding” approach in PRSice-2 v2.3.521. This algorithm iteratively selects a set of SNPs (with a P-value threshold of 0.05) to form clumps around the index SNPs. Each clump was composed of SNPs located within 250 kb of the index SNP and in linkage disequilibrium (LD) with the index SNP, based on a pairwise threshold of r2 = 0.2. A candidate PRS was computed using the resultant index SNPs and the corresponding estimated β-coefficient for its effect allele was computed as weights using the “score” procedure in PLINK v2.020. A total of 14 SNPs with P value < 5 × 10− 8 were included in PRS using PRSice2 (Table 1).
External validation database
For external validation, we used the Taiwan Biobank (TWB) database, which contains genetic information for 95,233 individuals. The TWB included individuals aged 30–70 years from the general Taiwanese population who self-reported as being of Taiwanese Han Chinese ancestry and had no history of cancer. For the TWB, specimens and associated data were collected at enrollment and individuals will receive follow-ups every two to four years via questionnaire. Details of the information contained in the TWB database have been reported previously22. Written informed consent was obtained from the TWB participants in accordance with institutional requirements and the principles of the Declaration of Helsinki. The self-reported T2D disease status and laboratory tests, including fasting glucose value and HbA1c, were used to identify the T2D status of individuals, and external validation was performed with 19,139 subjects (851 T2D cases and 18,288 controls).
Phenotype generation and PheWAS
The PheWAS was based on PRS_T2D and 1,335 health outcomes data obtained from CMUH. We extracted the diagnosis data (ICD-9/10 codes) from the CMUH EMRs and mapped the codes to Phecodes using the R package “PheWAS”23. Logistic regression models were generated to evaluate the association between T2D PRSs and each binary trait. The PheWAS was conducted for phenotypes with more than 50 incidences to ensure sufficient statistical power. In this study, individuals were classified as phenotypic cases for a specific disease phenotype if they had at least two encounters corresponding to the designated Phecode diagnosis. Conversely, phenotypic control subjects included individuals who had never been diagnosed with the corresponding Phecode, as well as those who met the exclusion criteria for the Phecode. Unless otherwise stated, both binary and quantitative traits were adjusted for age, sex, and the first ten principal components (PCs) estimated using EIGENSOFT (version 6).
Biological network and pathway analyses
The significance threshold of PRSs for analyzing biological networks across the genome was determined to be P < 1 × 10–5. Molecular network and genetic correlations between SNPs and diseases were analyzed by using the core analysis module in the IPA software (Qiagen Sciences, Inc.). The statistical significance of the available networks was assessed using Fisher’s exact t-test with a significance level of 0.05.
Statistical analysis
Continuous variables are presented as means with SDs, and categorical variables as proportions. Comparisons between the two groups were conducted using an independent sample t-test for continuous variables and a chi-square test for categorical variables. Baseline BMI was calculated as baseline weight in kilograms divided by height in meters squared (kg/m2) and categorized into four categories: <18.5, 18.5–<25, 25-<30, and 30 + kg/m2. Waist circumference > 80 cm for female and > 90 cm for male was considered as abnormal. For the blood pressure, systolic blood pressure > 130 mmHg or diastolic blood pressure > 85 mmHg was considered as abnormal.
To investigate the association between PRSs and T2D, logistic regression models were used to estimate odds ratios (ORs) and 95% confidence intervals (CIs). The T2D PRS was normalized and analyzed as both a continuous and categorical variable. For categorical analysis, participants were divided into quartiles, with the lowest PRS quartile used as the reference group. Two models were developed to predict the risk of T2D. The clinical model included demographic and clinical variables that were selected based on univariate logistic regression analyses, with variables showing a P value < 0.05 considered for inclusion. The combined model incorporated all variables from the clinical model, along with the T2D PRS, to evaluate the added predictive value of genetic information.
The development of the prediction models followed the framework of the Framingham Heart Study24. Variables with P value < 0.05 in the univariate model were entered simultaneously into a multivariate model with variables with P value < 0.05 retained in the final model. Receiver operating characteristic (ROC) curves were generated to evaluate the predictive accuracy of the models, and the area under the ROC curve (AUCs) was calculated to assess the discriminatory abilities of the models. The DeLong test was applied to determine the significant differences between the AUCs of the correlated ROC curves.
For the PheWAS, only the T2D PRS was used as the primary independent variable. Logistic regression was performed for each phenotype, adjusting for age and sex, to assess its association with the T2D PRS. Bonferroni correction was applied to account for multiple comparisons.
All statistical analyses were performed using SAS (v9.4; SAS Institute, Cary, NC, USA), SPSS (v21.0; IBM, Armonk, NY, USA), and R (v3.4.4, R Core Team, 2018). All tests were 2-sided; P < 0.05 was considered statistically significant.
Data and resource availability
The data supporting this study’s findings are available from the CMUH, but restrictions apply to the availability of this data which is not publicly available.
Results
Study subjects
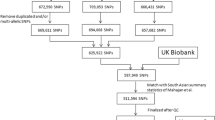
The study design is illustrated in Fig. 1. A total of 140,277 subjects, including 15,363 patients with T2D and 124,914 control subjects without diabetes were identified from the EMR. T2D primarily occurred in men (53.9% vs. 43.4%, P < 0.001). The mean (SD) ages of individuals in the T2D and control groups were 60.14 (12.69) years and 48.17 (16.60) years, respectively (P < 0.001). Baseline BMI was significantly different between the groups (the mean (SD) BMIs were 26.90 (4.65) and 24.35 (4.33) for the case and control groups, respectively; P < 0.001). Higher glucose, HbA1c, and TG values and lower TC, HDL, and LDL values were observed in the T2D group compared with those in the control group (all P values < 0.001) (Table S1).

Flowchart of the study design.
GWAS of Taiwanese patients with T2D
To identify T2D-associated SNPs and calculate PRSs, the dataset was divided into three cohorts: discovery, target, and validation, in a ratio of 7:1.5:1.5. Each group was regarded as an independent sample. GWAS was performed in the discovery cohort, which comprised 10,754 cases and 87,440 controls. The Manhattan and Q-Q plots (Fig. S1A and S1B, respectively) had a genomic inflation factor (λGC) of 1.078. A total of 202 SNPs surpassed the genome-wide significance threshold of 5 × 10− 8 for the Cochran–Armitage trend P-value. After clumping, 14 SNPs remained, including rs1337028053 on Chr5; rs9356744 (CDKAL1, intron) on Chr6, rs1431838560; rs806214 (FSCN3, intron), rs2233580 (PAX4), and rs201976370 (SND1) on Chr7; rs10811661 (CDKN2B-AS1; DMRTA1) on Chr9; rs2283228 and rs2237895 (KCNQ1) on Chr11; rs17676332 (TYRO3), rs76072851 (CORO2B), and rs527631646 (LINC01582; LINC02351) on Chr15; rs551262605 (PSMD3) on Chr17; and rs148612115 (ZNF257) on Chr19. These SNPs were associated with T2D in our study population. The most significant SNP, rs76072851, is located in the intronic region of CORO2B on 15q23 and had a P-value of 8.31 × 10− 74. Novel markers for T2D included the intronic SNP rs551262605 (P = 4.50 × 10− 8) in PSMD3 on 17q21.1, the intronic SNP rs17676332 (P = 1.34 × 10− 12) in TYRO3 on 15q15.1, and the intergenic SNP rs527631646 (P = 1.73 × 10− 8) on 15q26.3 (Table 1). Figure S2A-D show the regional plots for rs76072851 in CORO2B as well as three novel loci for T2D.
PRS models for predicting T2D risk
After the GWAS, 786,845 SNPs with a significance level of P < 0.05 were used to generate PRSs in the target cohort, and 30% of the sample data was used to generate T2D PRSs for each individual. Figure 2A and B show that the mean T2D PRSs were significantly different between two groups in target cohort (mean [SD] PRSs were − 0.3 [0.98] and 0.23 [1.10] for the control and case groups, respectively; P < 0.001) and in validation cohort (mean [SD] PRSs were − 0.3 [0.98] and 0.24 [1.11] for the control and case groups, respectively; P < 0.001). The strata plot demonstrated a positive trend, indicating that higher percentiles were associated with higher OR for T2D risk compared to the reference quantile (Fig. 2C and D).

Distribution of the polygenic risk scores, corresponding strata plot, and the receiver operating characteristic curve of the polygenic risk scores for the prediction of type 2 diabetes in the (A, B, C) target and (D, E, F) validation groups. Clinical variables included T2D_Diag_Age, gender, waist circumference, blood pressure, and diabetic family history.
In the target cohort, the ORs (95% CIs) of T2D in those who were older, male, abnormal waist circumference, and had high blood pressure or a family history of diabetes, and had a higher PRS were 1.04 (1.03, 1.05), 1.46 (1.03, 2.06), 3.75 (2.63, 5.34), 1.83 (1.27, 2.63), 3.16 (1.21, 8.25), and 1.37 (1.18, 1.60), respectively (Table 2). T2D PRSs and family history of diabetes were independent risk factors. A T2D risk model including age, sex, waist, blood pressure, family history of diabetes, and PRS was constructed (Supplemental Table S2). The total risk scores were calculated by adding the scores of all factors, and they ranged from 0 to 19. The estimated risks of T2D of each possible sum of points under the model combining clinical and genetic risk factors in the target cohort are displayed in Supplemental Table S3.
The AUROC values (95% CI) for the T2D prediction model combining the clinical factors and PRS were 0.804 (0.772, 0.835) and 0.842 (0.816, 0.867) for the target and validation cohorts, respectively (Fig. 2). These results demonstrated that our prediction model displayed remarkably good discrimination ability.
External validation
The external validation set consisted of 19,139 subjects from the TWB database, including 851 T2D cases. Among the 14 SNPs identified in the GWAS of the CMUH-TPMI chip, eight SNPs were identical on the TWB chip and six SNPs were replaced with highly linked SNPs located in the same gene. The association between the relevant 14 SNPs and T2D in TWB validation set was shown on Table S4. In the external validation set, the OR (95% CIs) for T2D for those who had higher PRSs was 1.08 (1.01, 1.16) (see Table S5). The AUROC value (95% CI) for the T2D prediction model, which combined clinical factors and wGRS, was 0.733 (0.718, 0.748, P < 0.001) (Fig. S3). The external validation sets demonstrated that the prediction model had good discrimination and calibration.
T2D polygenic risk score and phenome-wide association study
We evaluated T2D PRSs across 1,335 EMR-derived phenotypes with at least 50 case and control subjects. The PheWAS plot presented in Fig. 3 displays -log10 (FDR corrected) P values corresponding to each of the association tests for H0: βPRS = 0. Among the top 10 phenotypes related to T2D PRSs, including T2D with renal manifestations (P = 1.68 × 10− 142), diabetic retinopathy (P = 5.99 × 10− 66), and T2D with ophthalmic manifestations (P = 3.22 × 10− 51) (Table S6). Furthermore, we determined associations between T2D PRSs with other endocrine/metabolic disorders such as hyperlipidemia (P = 5.56 × 10− 109), disorders of lipid metabolism (P = 3.28 × 10− 99), and hypercholesterolemia (P = 2.02 × 10− 58). Additionally, we explored associations between PRS and traits beyond endocrine/metabolic disorders and observed a positive association between T2D PRS and abnormal kidney function (P = 4.82 × 10− 62) and essential hypertension (P = 1.18 × 10− 49). Moreover, T2D PRS is associated with different diseases of the circulation, digestive system, skin diseases, sense organs, muscles, and nervous system, and neoplasms. Table S7 illustrates the genetic associations between T2D and the top 10 phenotypes of several diseases, categorized as: (1) the circulatory system; (2) digestive system; and (3) neoplasms. To determine whether the secondary associations of the kidney, hypertension, and neoplasms with T2D PRS were due to patients having both T2D and these phenotypes, we reexamined the T2D PRS-phenotype associations after excluding T2D cases. The results were similar (Fig. S4).

Phenome-wide association study analysis of the correlation between polygenic risk scores and human diseases (SNPs gene loci, denoting a P < 5 × 10–8).
Bioinformatics network analysis of SNPs associated with T2D in PRS models
The significance of 1,566 SNPs was assessed in the context of the entire genome, using a threshold of P < 1 × 10− 5. A comprehensive analysis of 82 SNPs within 72 genes associated with T2D was performed using Bioinformatics IPA software. The gene numbers were cross-analyzed with pathways, resulting in the ranking of various biological processes including apoptosis, cellular immune response, cytokine signaling, cancer progression, disease-specific pathways, organismal growth and development, cell cycle regulation, ingenuity toxicity list pathways, nuclear receptor signaling, xenobiotic metabolism, cellular growth, proliferation and development, cellular stress and injury, and biosynthesis (Fig. S5A). Significant associations were also found between FTO, HNF1A, IDE, KCNQ1, KIF11, NTRK1, PAX4, SLC8A1, TCF7L2, TLE4, TYRO3, CDKAL1, CTSS, DPP6, and FOXP1 in the GWAS (Fig. S5B). Furthermore, our findings demonstrated that T2D characterized by multiple key ingenuity canonical pathways, including IL-15 production, thyroid cancer signaling, osteoblasts in rheumatoid arthritis signaling pathways, sperm motility, aryl hydrocarbon receptor signaling, HOTAIR regulatory pathway, WNT/β-catenin signaling, protein citrullination, granulocyte adhesion and diapedesis, basal cell carcinoma signaling, and pulmonary healing signaling pathway (Fig. S5C and Table 3).
Discussion
Existing T2D risk prediction models/nomograms are based on Asian and non-Asian populations25,26,27,28. As more T2D genetic variants have been identified, the genetic information incorporated in the models has increased the accuracy of models. However, there are few nomograms that combine clinical variables and genetic information. In the current study, we used the CMUH cohort database to conduct GWAS and PRS analysis to validate previous findings and develop new risk prediction formulas for clinical use. We conducted a GWAS to screen for T2D-related polymorphisms and validated them. Based on the associated SNPs identified, we established a nomogram model based on 14 T2D-associated SNPs and five clinical variables, including age, sex, waist circumference, blood pressure, and family history to predict the risk of T2D. This model has promise for aiding early identification and prevention of T2D.
The GWAS results revealed a total of 14 independent gene variants linked to T2D (P < 5 × 10− 8) (Table 1). These SNP loci span 13 genes: CDKAL1, FSCN3, PAX4, SND1, CDKN2B-AS1, DMRTA1, KCNQ1, TYRO3, CORO2B, LINC01582, LINC02351, PSMD3, and ZNF257. Among them, CDKAL1, FSCN3, PAX4, SND1, DMRTA1, KCNQ1, CORO2B, and ZNF257 have been previously reported to be associated with T2D. TYRO3, PSMD3, LINC01582, and LINC02351 were newly identified in our study as associated with T2D. While TYRO3 and PSMD3 have been previously linked to BMI or metabolic syndrome29,30their association with T2D has not been reported before, to the best of our knowledge. The relevant associations of these gene variants with T2D and related metabolic traits, such as HbA1c, insulin, and glucose levels across different ethnic populations, are summarized in Table S831,32,33,34. TYRO3 encodes a receptor tyrosine kinase of the TAM family and is highly expressed in podocytes, specialized epithelial cells essential for glomerular filtration. Studies have demonstrated that TYRO3 activation in podocytes protects against apoptosis and reduces albuminuria, suggesting a role in maintaining glomerular integrity under diabetic conditions35,36,37. These findings support the hypothesis that TYRO3 variants may influence T2D risk through mechanisms involving immune regulation, inflammation, and renal microvascular integrity. PSMD3 encodes a non-ATPase regulatory subunit of the 26 S proteasome, which plays a critical role in ubiquitin-mediated protein degradation and signaling regulation. Genetic variants in PSMD3 have been associated with insulin resistance, as measured by HOMA-IR, particularly in the context of dietary fat and carbohydrate intake38. PSMD3 may influence T2D susceptibility through pathways related to proteostasis, metabolic inflammation, and endocrine signaling. Additional research is warranted to explore the potential roles of TYRO3 and PSMD3 in T2D pathogenesis, including their involvement in gene regulation, metabolic processes, and diabetic complications.
While numerous PRS models for T2D have been developed, including more than 45 studies involving East Asian populations according to the GWAS Catalog, most are derived from specific Asian or trans-ancestry approaches39,40,41,42. Some recent efforts have focused on improving cross-population applicability or on classifying mechanistic subtypes using multi-ancestry data. However, these models may overlook signals specific to subpopulations such as Taiwanese individuals. Moreover, recent studies have shown that PRS performance can vary depending on the ancestry of the discovery cohort, highlighting the need for population-specific models. To assess the added value of population-specific PRSs, we compared our 14-SNP model with a 287-SNP PRS from prior studies and a 4-SNP model from uniquely Taiwanese variants. All three models, when combined with clinical variables, showed similar predictive performance, suggesting that a concise, locally tailored PRS can match larger models while improving interpretability (Fig. S6).
Currently, there are limited studies utilizing PheWAS to explore SNPs and PRSs in the context of T2D. In our previous work using TWB data, we found that the T2D PRS was inversely associated with age at diagnosis (β = − 0.91 years) and linked to diabetic retinopathy and foot complications43. A PheWAS by Lee et al. identified pleiotropic loci related to glycemic and metabolic traits among 10 diseases and 34 quantitative traits using SNP-level associations44. Expanding on these efforts, we applied the T2D PRS to 1,335 EMR-derived phenotypes and observed strong associations with diabetic complications and metabolic comorbidities, including renal manifestations (P = 1.68 × 10–142), lipid disorders, and hypertension. These associations have been corroborated in previous research on T2D45,46,47. By leveraging real-world EMR data, this analysis offers enhanced clinical relevance and phenotypic resolution. The use of polygenic risk scores enables quantification of cumulative genetic burden and its relationship to a broad spectrum of disease phenotypes in routine healthcare settings. These findings underscore the pleiotropic nature of T2D genetic risk and highlight the potential of PRS as a tool for early risk stratification. Key associations are summarized in Fig. S4 and Table S6.
We conducted bioinformatics network analysis on high-risk gene loci (P < 5 × 10− 8) in the PRS model. The results indicated that numerous signaling pathways are involved in the pathogenesis of T2D, including IL-15 production, aryl hydrocarbon receptor (AHR) signaling, HOTAIR regulatory pathway, and WNT/β-catenin signaling. (Table 3 and Fig. S5(C)). IL-15, a pro-inflammatory cytokine, plays a role in lymphocyte development, survival, proliferation, and activation48. Our results identified genes involved in the IL-15 production pathway, such as EPHA6, NTRK1, PEAK1, and TYRO3 (Table 3) associated with T2D. T2D myotubes produce higher levels of IL-15 than non-diabetic myotubes49. Ye et al.50 suggested that IL-15 may increase energy expenditure, thereby protecting against obesity and T2D. Our study is the first to demonstrate a significant association between the IL15 production pathway and T2D. However, the regulatory role of IL-15 in T2D needs further investigation.
The AHR is a ligand-activated transcription factor, whose transcriptional activity is regulated by small compounds. AHR has been shown to be involved in energy homeostasis and inflammation51. Epidemiological analyses suggest that exposure to xenobiotic AHR ligands contributes to the development of obesity and T2D. Furthermore, increased levels of IL-22 and high frequencies of Th22 cells are associated with insulin resistance in patients with T2D. In patients with T2D, elevated plasma concentrations of IL-22 and IL-17 are associated with increased AHR transcription51. Our findings revealed that, in patients with T2D, the genes involved in AHR signaling include MED1, NFIA, and SMARCA4. Thus, the role of AHR signaling in the regulation of T2D is a new direction for future research.
Homeobox transcript antisense RNA (HOTAIR) an oncogenic long non-coding RNA (lncRNA), as an epigenetic hub may drive T2D pathogenesis52,53. By recruiting the PRC2 complex, HOTAIR can deposit H3K27me3 marks on downstream loci, thereby suppressing HNF1A and modulating TCF7L2, two master regulators of β-cell insulin synthesis and glucose homeostasis. Concurrently, HOTAIR-mediated activation of MMP3 promotes extracellular-matrix remodeling, a precursor to diabetic micro-vascular complications52. These coordinated effects provide a plausible mechanistic link between elevated HOTAIR activity and both the initiation and progression of T2D.
The correlation between WNT/β-catenin signaling and T2D is well-established54. The gene TCF7L2 related to the Wnt signaling pathway has been identified as a risk factor for T2D through GWAS55,56. Two SNPs, rs12255372 and rs7903146, that were also significantly associated with T2D (P values = 2.74 × 10− 7 and 7.35 × 10− 3) in our population, have been shown to exhibit strong correlations with T2D risk57,58. Moreover, certain SNPs of HNF1A increase susceptibility to T2D59. Our results corroborate earlier findings that, the genes involved in the WNT/β-catenin signaling include HNF1A, TCF7L2, and TLE4 are associated with T2D.
Our study has some strengths and limitations. We utilized a large EMR database with longitudinal information on various diseases and conducted a large-scale GWAS using a Taiwanese population. This provided a valuable opportunity to understand the correlation between T2D-related genes and various diseases. For external validation, we used the TWB database, which contains data from the general population, helping to reduce selection bias. However, the PRS construction approach used in this study was based on the “clumping and thresholding” method, which may not fully account for the effects of LD. Alternative analytical methods such as PRS-CS43 or machine learning approaches60 could potentially identify additional significant SNPs and enhance the models’ accuracy. Additionally, we developed a risk score model that may be useful for clinical screening in Taiwanese populations. However, the accuracy of this model requires validation using an independent database. The generalizability of our study is limited, as our study population included only Taiwanese individuals. Due to genetic variations and clinical differences across ethnicities, future studies should incorporate more genetic and environmental variables related to T2D. A more comprehensive nomogram model should also be established to predict T2D risk, and its diagnostic efficacy should be verified in different populations. Moreover, while the sample size of this study is large, particularly in the context of PheWAS, only phenotypes with sufficient occurrences were analyzed. This may lead to a lack of power for subgroup analyses or for specific phenotypes. Additionally, the causative relationships among these observed associations could be explored in future research.
Conclusion
Our predictive risk model based on key genetic and clinical factors can effectively distinguish individuals with different risk levels of T2D. Individualized risk estimation for T2D is important to help inform decision-making when considering measures for primary prevention of T2D.
Data availability
Data supporting the findings of this study are available from the corresponding author upon request.
References
Liu, J. et al. Polygenic risk score, lifestyles, and type 2 diabetes risk: A prospective Chinese cohort study. Nutrients 15, 1253. https://doi.org/10.3390/nu15092144 (2023).
The Ministry of Health and & Welfare, T. Statistics on causes of death in Taiwan in 2022 (2023). https://www.mohw.gov.tw/cp-16-74869-1.html.
Shitomi-Jones, L. M., Akam, L., Hunter, D., Singh, P. & Mastana, S. Genetic risk scores for the determination of type 2 diabetes mellitus (T2DM) in North India. Int. J. Environ. Res. Public. Health. 20, 12. https://doi.org/10.3390/ijerph20043729 (2023).
Cheng, C. F. et al. Genetic risk score constructed from common genetic variants is associated with cardiovascular disease risk in type 2 diabetes mellitus. J. Gene Med. 23, e3305. https://doi.org/10.1002/jgm.3305 (2021).
Thornton, J. M. et al. Multigenerational diabetes mellitus. Front. Endocrinol. (Lausanne). 14, 1245899. https://doi.org/10.3389/fendo.2023.1245899 (2023).
Zahalka, S. J. et al. Endotext (eds. Feingold, K. R.) (2000).
Yang, J. S. et al. Autophagy and its link to type II diabetes mellitus. Biomed. (Taipei). 7, 8. https://doi.org/10.1051/bmdcn/2017070201 (2017).
Soheilipour, F., Abbasi Kasbi, N., Imankhan, M. & Eskandari, D. Complications and treatment of Early-Onset type 2 diabetes. Int. J. Endocrinol. Metab. 21, e135004. https://doi.org/10.5812/ijem-135004 (2023).
Mannar, V. et al. Epigenetics of the pathogenesis and complications of type 2 diabetes mellitus. TouchREV Endocrinol. 19, 46–53. https://doi.org/10.17925/EE.2023.19.1.46 (2023).
Yang, J. S. et al. Genome–wide association study and polygenic risk scores predict psoriasis and its shared phenotypes in Taiwan. Mol. Med. Rep. 30, 523. https://doi.org/10.3892/mmr.2024.13239 (2024).
Irvin, M. R. et al. Polygenic risk for type 2 diabetes in African Americans. Diabetes https://doi.org/10.2337/db23-0232 (2024).
Jiang, W. et al. Maternal smoking, nutritional factors at different life stage, and the risk of incident type 2 diabetes: a prospective study of the UK biobank. BMC Med. 22, 50. https://doi.org/10.1186/s12916-024-03256-8 (2024).
Pesaro, A. E. et al. The Finnish diabetes risk score (FINDRISC), incident diabetes and low-grade inflammation. Diabetes Res. Clin. Pract. 171, 108558. https://doi.org/10.1016/j.diabres.2020.108558 (2021).
Hippisley-Cox, J. & Coupland, C. Development and validation of QDiabetes-2018 risk prediction algorithm to estimate future risk of type 2 diabetes: cohort study. BMJ 359, j5019. https://doi.org/10.1136/bmj.j5019 (2017).
Pantalone, K. M. et al. The probability of A1C goal attainment in patients with uncontrolled type 2 diabetes in a large integrated delivery system: a prediction model. Diabetes Care. 43, 1910–1919. https://doi.org/10.2337/dc19-0968 (2020).
Lin, Z., Guo, D., Chen, J. & Zheng, B. A nomogram for predicting 5-year incidence of type 2 diabetes in a Chinese population. Endocrine 67, 561–568. https://doi.org/10.1007/s12020-019-02154-x (2020).
Liu, T. Y. et al. Diversity and longitudinal records: genetic architecture of disease associations and polygenic risk in the Taiwanese Han population. Sci. Adv. 11, eadt0539. https://doi.org/10.1126/sciadv.adt0539 (2025).
Liao, W. L. et al. Analysis of HLA variants and graves’ disease and its comorbidities using a high resolution imputation system to examine electronic medical health records. Front. Endocrinol. (Lausanne). 13, 842673. https://doi.org/10.3389/fendo.2022.842673 (2022).
Liu, T. Y. et al. Comparison of multiple imputation algorithms and verification using whole-genome sequencing in the CMUH genetic biobank. Biomed. (Taipei). 11, 57–65. https://doi.org/10.37796/2211-8039.1302 (2021).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7. https://doi.org/10.1186/s13742-015-0047-8 (2015).
Choi, S. W. & O’Reilly, P. F. PRSice-2: polygenic risk score software for biobank-scale data. GigaScience 8, 452. https://doi.org/10.1093/gigascience/giz082 (2019).
Fan, C. T., Lin, J. C. & Lee, C. H. Taiwan biobank: a project aiming to aid taiwan’s transition into a biomedical Island. Pharmacogenomics 9, 235–246. https://doi.org/10.2217/14622416.9.2.235 (2008).
Carroll, R. J., Bastarache, L. & Denny, J. C. R phewas: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics 30, 2375–2376. https://doi.org/10.1093/bioinformatics/btu197 (2014).
Sullivan, L. M., Massaro, J. M. & D’Agostmo, R. B. Presentation of multivariate data for clinical use: the Framingham study risk score functions. Stat. Med. 23, 1631–1660. https://doi.org/10.1002/sim.1742 (2004).
Xu, S. et al. Risk prediction models for incident type 2 diabetes in Chinese people with intermediate hyperglycemia: a systematic literature review and external validation study. Cardiovasc. Diabetol. 21, 182. https://doi.org/10.1186/s12933-022-01622-5 (2022).
Lindstrom, J. & Tuomilehto, J. The diabetes risk score: a practical tool to predict type 2 diabetes risk. Diabetes Care. 26, 725–731. https://doi.org/10.2337/diacare.26.3.725 (2003).
Wilson, P. W. et al. Prediction of incident diabetes mellitus in middle-aged adults: the Framingham offspring study. Arch. Intern. Med. 167, 1068–1074. https://doi.org/10.1001/archinte.167.10.1068 (2007).
Xu, T., Yu, D., Zhou, W. & Yu, L. A nomogram model for the risk prediction of type 2 diabetes in healthy Eastern China residents: a 14-year retrospective cohort study from 15,166 participants. EPMA J. 13, 397–405. https://doi.org/10.1007/s13167-022-00295-0 (2022).
Zhu, Z. et al. Shared genetic and experimental links between obesity-related traits and asthma subtypes in UK biobank. J. Allergy Clin. Immunol. 145, 537–549. https://doi.org/10.1016/j.jaci.2019.09.035 (2020).
Huang, J. et al. Genomics and phenomics of body mass index reveals a complex disease network. Nat. Commun. 13, 7973. https://doi.org/10.1038/s41467-022-35553-2 (2022).
Steinthorsdottir, V. et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet. 39, 770–775. https://doi.org/10.1038/ng2043 (2007).
Kwak, S. H. et al. Nonsynonymous variants in PAX4 and GLP1R are associated with type 2 diabetes in an East Asian population. Diabetes 67, 1892–1902. https://doi.org/10.2337/db18-0361 (2018).
Imamura, M. et al. Genome-wide association studies in the Japanese population identify seven novel loci for type 2 diabetes. Nat. Commun. 7, 10531. https://doi.org/10.1038/ncomms10531 (2016).
Sun, Q., Song, K., Shen, X. & Cai, Y. The association between KCNQ1 gene polymorphism and type 2 diabetes risk: a meta-analysis. PLoS One. 7, e48578. https://doi.org/10.1371/journal.pone.0048578 (2012).
Zhong, F. et al. Tyro3 is a podocyte protective factor in glomerular disease. JCI Insight. 3, 123. https://doi.org/10.1172/jci.insight.123482 (2018).
Zhong, F. et al. TYRO3 agonist as therapy for glomerular disease. JCI Insight. 8, 856. https://doi.org/10.1172/jci.insight.165207 (2023).
Curtis, J. L., Todt, J. C., Hu, B., Osterholzer, J. J. & Freeman, C. M. Tyro3 receptor tyrosine kinases in the heterogeneity of apoptotic cell uptake. Front. Biosci. (Landmark Ed). 14, 2631–2646. https://doi.org/10.2741/3401 (2009).
Zheng, J. S. et al. Genetic variants at PSMD3 interact with dietary fat and carbohydrate to modulate insulin resistance. J. Nutr. 143, 354–361. https://doi.org/10.3945/jn.112.168401 (2013).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424. https://doi.org/10.1038/s41588-021-00931-x (2021).
Jung, H. U. et al. Assessment of polygenic risk score performance in East Asian populations for ten common diseases. Commun. Biol. 8, 374. https://doi.org/10.1038/s42003-025-07767-9 (2025).
Ge, T. et al. Development and validation of a trans-ancestry polygenic risk score for type 2 diabetes in diverse populations. Genome Med. 14, 70. https://doi.org/10.1186/s13073-022-01074-2 (2022).
He, Y. et al. Comparisons of polyexposure, polygenic, and clinical risk scores in risk prediction of type 2 diabetes. Diabetes Care. 44, 935–943. https://doi.org/10.2337/dc20-2049 (2021).
Wang, S. H., Huang, Y. C., Cheng, C. W., Chang, Y. W. & Liao, W. L. Impact of the trans-ancestry polygenic risk score on type 2 diabetes risk, onset age and progression among population in Taiwan. Am. J. Physiol. Endocrinol. Metab. 326, E547–E554. https://doi.org/10.1152/ajpendo.00252.2023 (2024).
Lee, C. J. et al. Phenome-wide analysis of Taiwan biobank reveals novel glycemia-related loci and genetic risks for diabetes. Commun. Biol. 5, 1175. https://doi.org/10.1038/s42003-022-04168-0 (2022).
Kember, R. L. et al. Polygenic risk scores for cardiometabolic traits demonstrate importance of ancestry for predictive precision medicine. Pac. Symp. Biocomput. 29, 611–626 (2024).
Chung, R. H. et al. Evaluating polygenic risk scores for predicting cardiometabolic traits and disease risks in the Taiwan biobank. HGG Adv. 5, 100260. https://doi.org/10.1016/j.xhgg.2023.100260 (2024).
Suzuki, K. et al. Genetic drivers of heterogeneity in type 2 diabetes pathophysiology. Nature 627, 347–357. https://doi.org/10.1038/s41586-024-07019-6 (2024).
Mishra, A., Sullivan, L. & Caligiuri, M. A. Molecular pathways: interleukin-15 signaling in health and in cancer. Clin. Cancer Res. 20, 2044–2050. https://doi.org/10.1158/1078-0432.CCR-12-3603 (2014).
Amir Levy, Y., R, R. H. & T, P. C., S, R. M., S, A. P. & Adipose tissue from subjects with type 2 diabetes exhibits impaired capillary formation in response to groalpha: involvement of MMPs-2 and – 9. Adipocyte 11, 276–286. https://doi.org/10.1080/21623945.2022.2070949 (2022).
Ye, J. Beneficial metabolic activities of inflammatory cytokine Interleukin 15 in obesity and type 2 diabetes. Front. Med. 9, 139–145. https://doi.org/10.1007/s11684-015-0377-z (2015).
Zhao, R. X. et al. Increased AHR transcripts correlate with Pro-inflammatory T-Helper lymphocytes polarization in both metabolically healthy obesity and type 2 diabetic patients. Front. Immunol. 11, 1644. https://doi.org/10.3389/fimmu.2020.01644 (2020).
Akbari Dilmaghnai, N. et al. Non-coding RNAs modulate function of extracellular matrix proteins. Biomed. Pharmacother. 136, 111240. https://doi.org/10.1016/j.biopha.2021.111240 (2021).
Yang, W., Lyu, Y., Xiang, R. & Yang, J. Long noncoding RNAs in the pathogenesis of insulin resistance. Int. J. Mol. Sci. 23, 5263. https://doi.org/10.3390/ijms232416054 (2022).
Kang, X. et al. Exendin-4 ameliorates Tau hyperphosphorylation and cognitive impairment in type 2 diabetes through acting on Wnt/beta-catenin/NeuroD1 pathway. Mol. Med. 29, 118. https://doi.org/10.1186/s10020-023-00718-2 (2023).
Del Bosque-Plata, L., Hernandez-Cortes, E. P. & Gragnoli, C. The broad pathogenetic role of TCF7L2 in human diseases beyond type 2 diabetes. J. Cell. Physiol. 237, 301–312. https://doi.org/10.1002/jcp.30581 (2022).
Khan, S. M. et al. Association between type 2 diabetes mellitus & TCF7L2 gene variants in the Emirati population: genetics of diabetes in the united Arab Emirates. Am. J. Hum. Biol. 33, e23434. https://doi.org/10.1002/ajhb.23434 (2021).
Jin, T. Current Understanding and dispute on the function of the Wnt signaling pathway effector TCF7L2 in hepatic gluconeogenesis. Genes Dis. 3, 48–55. https://doi.org/10.1016/j.gendis.2015.10.002 (2016).
Florez, J. C. et al. TCF7L2 polymorphisms and progression to diabetes in the diabetes prevention program. N Engl. J. Med. 355, 241–250. https://doi.org/10.1056/NEJMoa062418 (2006).
Li, L. M., Jiang, B. G. & Sun, L. L. HNF1A: from Monogenic diabetes to type 2 diabetes and gestational diabetes mellitus. Front. Endocrinol. (Lausanne). 13, 829565. https://doi.org/10.3389/fendo.2022.829565 (2022).
Huang, Y. J., Chen, C. H. & Yang, H. C. AI-enhanced integration of genetic and medical imaging data for risk assessment of type 2 diabetes. Nat. Commun. 15, 4230. https://doi.org/10.1038/s41467-024-48618-1 (2024).
Acknowledgements
We thank the iHGenome Platform from the China Medical University Hospital for data exploration and administrative support. We also thank the Office of Research and Development, China Medical University (Taiwan), for providing Medical Research Core Facilities to perform the experiments and data analysis.
Funding
This study was supported by the China Medical University and China Medical University Hospital (DMR-111-137, 111-2813-C-039-060-B, and DMR-113-092) and the National Science and Technology Council, Taiwan (NSTC112-2314-B-039-041-MY2). The sponsors had no role in the study design, in the collection, analysis, and interpretation of data, in the writing of the report, or in the decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
W.L.L. conceived of the project, designed experiments, interpreted results, and was a major contributor in writing and revised the manuscript. J.S.Y. conducted the pathway analyses. T.Y.L. and H.F.L. contributed to data curation and methodology. Y.W.C. conducted the statistical analyses. F.J.T. conceived of the project. All authors review the manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liao, WL., Yang, JS., Liu, TY. et al. Pathway insights and predictive modeling for type 2 diabetes using polygenic risk scores. Sci Rep 15, 28956 (2025). https://doi.org/10.1038/s41598-025-13391-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-13391-8
