
Abstract
Brain tissue orchestrates neuronal function through biochemical and mechanical cues. Utilizing in vitro modeling, often the dynamics of mechanical aspects in neuronal cell cultures is neglected. However, the growing recognition of the importance of mechanical cues in neural development and healthy brain function necessitates a shift in how we study cultured neurons. Microfluidic platforms, like a Brain-on-Chip (BoC), can take active mechanical stimuli into account. In our BoC design a set of microchannels manufactured in a glass substrate by FEMTOprint technology is assembled with a spin-coated polydimethylsiloxane (PDMS) membrane and a PDMS culture chamber, which was fabricated from a stereolithographically made mold by replication. The membrane can locally deform across the culture chamber by air pressure. This paper describes the design, fabrication and test of such a novel BoC, offering an experimental setting in which we demonstrated mechano-dynamic elevated Calcium signaling in cultured human induced neural stem cell-derived neuronal networks.
Similar content being viewed by others

Introduction
The brain is a mechanically delicate organ that orchestrates brain function through mechanical forces. Additionally, the rigidity of the cranium coupled with its relatively constant volume renders the brain sensitive to mechanical influences1. From this point of view, axonal outgrowth involves applying mechanical force to the surrounding environment2.Cell crawling was first observed in 1675, it was later discovered that this movement is primarily driven by actin filaments within the cell membrane3. Mechanical energy’s impact on living organisms’ brains is pervasive; for instance, when considering how cerebrovascular blood flow, synchronized with each human heartbeat, exerts forces capable of displacing brain tissue by tens of micrometers4. The brain’s intricate control over water and solute transport from the blood into its tissue is crucial for maintaining normal intracranial pressure (ICP) and creating an optimal environment for neuronal function5. Astrocytes play a key role in capillary function, influencing local cerebral blood flow regulation while directly communicating with neurons6. Any significant changes in neuronal activity highlight the critical relationship between neural activity and cerebral blood flow regulation7,8,9.
Mechanical stimulation has been demonstrated to be pivotal in comprehending cerebral function and addressing an array of neurological disorders10. Disruptions to brain homeostasis caused by mechanical forces may result in pathological conditions, underscoring the significance of integrating these stimuli within clinical practices11,12,13,14. Interventions, including whole-body vibration and ultrasound, exhibit promising therapeutic advantages by influencing neuronal activity and enhancing cerebral plasticity15,16,17,18,19. Ultrasound waves can infiltrate biological tissues and alter neuronal activity; these phenomena hold potential answers for the management of chronic pain and to enhance recovery20,21. Overall, mechanical interventions offer promising prospects for both slowing disease progression and modulating neuronal dynamics; however, their full impact on brain function is still not completely understood22,23,24,25.
State-of-the-art in-vitro models provide insights into mechanical stimulation effects on the brain. For example, one approach applies controlled mechanical pulling forces to encourage defined neurite outgrowth, achieving controlled connectivity between neurons26. Another technique involves the use of atomic force microscopy (AFM) to mechanically stimulate neurons through indentation, facilitating the formation of functional neuronal networks27. Additionally, the application of mechanical tension is crucial not only for the development of neuronal networks but also for fine-tuning neural function28. Studies on traumatic brain injury (TBI) show that cellular responses to mechanical stimuli are nonlinear and dependent on the type of force applied, with methods developed to apply these stimuli to cerebral organoids for more accurate modeling29,30. Research shows that neurons are highly sensitive to mechanical forces, with growing evidence of how these forces regulate neuronal dynamics, such as signaling and plasticity1. Mechanical impulses during action potential firing and axonal tension highlight the influence of these forces on axons, impacting initiation22,23, growth22,24, synapse formation via neurotransmitter release31,32. Axonal elongation under mechanical tension suggests an adaptive capacity that holds potential for nerve repair interventions33,34.
In the last couple of decades, the use of various materials for microfluidic devices, such as silicon, glass, polydimethylsiloxane (PDMS)35, SU-8 photoresist36, polycarbonate (PC), polymethyl methacrylate (PMMA), and polystyrene (PS)37, has blurred the distinct advantages and disadvantages of one material over another. Of these, PDMS has demonstrated to contribute to the way cell culture has been innovated due to its permeable nature, which allows for the easy diffusion of O\(_2\) and CO\(_2\)38,39,40,41,42,43. For example, PDMS holds great potential in developing bio-artificial lungs, with its flexibility being a key advantage for lung mimicry, as well as facilitating oxygen transport44. However, high oxygen permeability in PDMS may give rise to hyperoxic conditions within the microenvironments, leading to cell stress45,46. Additionally, scalability of the PDMS devices is a major limitation47. Another drawback of PDMS is related to water vapor permeability, which could lead to evaporation and, consequently, changes in osmolarity, thus affecting cell viability and differentiation in microfluidics48,49,50,51. Finally, PDMS has the tendency to absorb small hydrophobic molecules, that can alter medium composition and subsequently affect cellular growth and the cell-based on chip assay’s outcome41,52. Furthermore, uncrosslinked oligomers in PDMS can leach into solutions and pose a hazard in biological experiments53. The well-documented phenomenon of hydrophobic recovery, in which PDMS reassumes its native hydrophobic state after plasma treatment, further complicates the use of PDMS in microfluidics54,55,56,57,58,59. Methods to overcome this problem, such as full curing or the removal of uncured oligomers, have been suggested, but these methods are often either highly time-consuming or complicated52,60. Despite these challenges, PDMS remains one of the most common materials used in microfluidics due to its ease of prototyping. However, improvements in surface treatment and adsorption control are ongoing53,61,62.
Glass microfluidic devices are increasingly popular, as they effectively prevent the absorption of hydrophobic molecules, improve cell attachment, and offer a more stable environment, making them useful for long-term experiments63,64,65. The properties of reusability and biocompatibility render it especially attractive for biological research concerning cell analysis or drug testing that demands stable and precise outcomes38,54,57. Since the 1980s, there has been significant advancement in microfluidic devices, with glass-based variants emerging prominently in the early 2000s63.
Femtosecond lasers are making it possible to create complex 3D microfluidic structures in glass, offering exciting new possibilities in device design. For example, Italia et al. used this technology to craft a double Y-shaped microfluidic chip with intricate 3D channels59, while Shan et al. developed a 3D helical mixer that shows promise for efficiently mixing larger biological samples66. Calmo et al. advanced this research by incorporating microchannels and resonators within a glass chip designed to quantify bacteria through the analysis of liquid density; this approach has the potential to be extended for the detection of organelles and various small particles67. A significant advancement in this domain is femtoprinting, which enhances the fabrication of these glass devices by facilitating the process and removing the necessity for clean rooms or supplementary procedures such as the application of photoresist67. This technology not only improves surface uniformity and biocompatibility but also enables real-time imaging and has complete integration with electronic devices, thus marking a groundbreaking development for rapid prototyping. In the process, femtoprinting is setting a new standard in the development of complex microfluidic systems by eliminating the burdensome complexities typically involving traditional methods, particularly in biomedical applications.
In this study, the growing recognition of the importance of mechanical cues in neural development and healthy brain function necessitates a shift in how we study cultured neurons. This drives the emergence of innovative BoC concepts, offering a more physiologically relevant setting for studying neural cells in brain research. Often, the dynamics of mechanical aspects in neuronal cell cultures are simply neglected. Our BoC model is capable of incorporating mechanical stimulation, mirroring how cells in the living brain are constantly influenced by forces. Our BoC design features microfluidic, pneumatic pressure-driven channels located beneath a flexible membrane. When pneumatic pressure is employed, the membrane bulges, creating a topographical change that serves as a mechanical stimulus for the cells cultured in the reservoir on top locally. Potentially, this stimulus allows us to study mechanotransduction processes by observing cellular responses to these controlled mechanical inputs. By doing so, our model could offer valuable insights into how mechanical forces regulate brain function.
By utilizing FEMTOprint technology (a combination of femtosecond laser exposure and subsequent chemical etching), we microfabricated a BoC, which consists of a microstructured glass with a PDMS membrane and cell culture reservoir. Applying such type of device in neurobiology studies can enhance our comprehension of the mechanical aspects of the brain microenvironment.
Results and discussion
Microfabrication of the BoC
Channel layer for pneumatic pressure-driven actuation
Femtosecond laser micromachining enables direct writing of 3D features into transparent materials without the need for masks or sacrificial layers as in traditional methods68,69. The fabrication of the BoC channel layer was accomplished by using femtosecond laser micromachining on fused silica glass, which is favored for its optical transparency and chemical durability. A notable outcome of this process is much smaller heat-affected zones compared to ablation driven methods, leading to lesser nonspecific destruction of surrounding areas while maintaining exceptional precision in the changes undergone by materials. During the design step, Alphacam software was used to define the geometry of the microfluidic channels, which consists of inlets and outlets with a 1 mm diameter, with a channel width of 50 \(\upmu\)m, a height of 40 \(\upmu\)m, and a pitch of 1.2 mm between adjacent channels (Fig. 1a). The sketching phase within Alphacam also makes the creation of toolpaths possible. The toolpath defines the sequence of movements undertaken by the laser beam tool and is therefore crucial in translating the design into readable instructions for the FEMTOprint laser system. Optimized toolpaths are provided directly by the software, guiding the laser beam on well-defined trajectories and thus providing a very accurate reconstruction of the intended channel geometry (Fig. 1b). Fig. 1c displays cross-sectional view of the channels and movie of the channel fabrication simulation is embedded into QR code in Fig. 1d.

Channel design overview in Alphacam software. (a) Schematic design of the channels in Alphacam, illustrating the dimensional layout of the microfluidic pneumatic pressure-driven channels. (b) Toolpath view, showing the sequence of movements by the laser beam tool for producing the desired geometry. (c) 3D view of the channel layer in Alphacam with cross-sectional view of the channels. (d) Full movie of the channel fabrication simulation in Alphacam, accessible via embedded QR code.
A wet chemical etching process using 45% KOH was then applied after femtosecond laser micromachining (Fig. 2a) to enable enhanced material removal nd laser micromachining (Fig. 2b). The laser-induced modification of fused silica results in localized structural changes, including variations in refractive index and increased chemical solubility in alkaline solutions like KOH. This laser-induced chemical modification is responsible for achieving selective etching70. The KOH solution preferentially dissolves the laser-modified regions, while largely leaving the unmodified ones intact, thus enabling the realization of high-resolution three-dimensional (3D) microstructures with sharp edge definition nd laser micromachining (Fig. 2c,d). Yielding channel architectures with controlled depth and smooth surfaces71.

Channel fabrication process. (a) Femtosecond laser-machined channels and their inlets/outlets prior to wet etching. (b) Channels (50 \(\upmu\)m) and inlets/outlets (1 mm diameter) after KOH etching, displaying the final structure and confirming successful 3D printing. (c) \(80^{\circ }\) angled view of the femtosecond laser-fabricated 3D channel structure on a fused silica glass captured using the digital microscope. (d) Zoomed-in view of (c).
Membrane layer
The membrane layer was fabricated using a spin-coated PDMS that could be adjusted to a thickness of 10 micrometers by fine tuning the spin-process in three steps using at a PDMS mixture of 1:10 base-to-curing agent to base PDMS material according our previously published work72. This flexible PDMS membrane was selected to enable deformation under mechanical stimuli, as illustrated in (Fig. 5). The deformation height was visualized using a digital microscope. As shown in (Fig. 5b,d), images were captured both before and during membrane actuation. These observations supporting the membrane’s ability to respond to mechanical stimulation, which is its functional role in the chip system.
Reservoir layer
The reservoir layer was fabricated by casting PDMS at a 10:1 ratio onto a 3D-printed mold. The thickness of this layer was sufficiently controlled by adjusting the amount of PDMS poured, resulting in a uniform height of 2.5 mm. As shown in (Fig. 14a), the cast PDMS accurately replicated the mold design, demonstrating smooth surface features. The reservoir openings and contours were clearly defined, indicating that the casting process yielded a well-formed and reproducible structure.
BoC assembly
Assembly was performed manually in sequential steps, starting with bonding the membrane layer to the reservoir layer, followed by alignment and bonding with the channel layer. Oxygen plasma treatment was applied at each bonding interface to ensure strong adhesion between layers. To complete the process, the final assembly was cured in an oven. This procedure ensured the structural integrity and readiness of the final BoC device as shown in (Fig. 7a).
Further details on the fabrication steps and results thereof can already be found in72
Pneumatic pressure-driven BoC characterization
Mechanical stimulation and membrane deformation
In order to investigate dynamic mechanical stimuli and influence the functional responses of the brain cells, compressed air flow was introduced into the chip to produce a controlled mechanical deformation on the flexible PDMS membrane (Fig. 4c,d). When pneumatic pressure is employed to the channels underneath the PDMS membrane, the membrane bulges out and this provides a mechanical stimulus to the cells cultured on top. In this study, neuronal cell cultures were subjected to mechanical stimulation at a pressure of 1 bar. The choice of 1 bar was necessary to achieve observable membrane deformation, as lower pressures did not produce sufficient membrane bulging detectable under a digital microscope. While this pressure level exceeds normal physiological conditions (0.09–0.13 bar), it applies across the entire membrane area and is only applied as a pulse. Hence, the resulting forces do not harm the cells’ viability. Applying pressures beyond the physiological conditions remain relevant for studies exploring extreme conditions such as stroke73, hypertension74. High-pressure environments can provide insights into the impact of abnormal mechanical forces on brain cells. These cellular responses to the mechanical changes represent mechanotransduction processes in which the cell transduces the mechanical forces into biochemical signals. This approach can also provide insight into how mechanical forces might regulate the activities of brain cells in a way relevant to overall cellular function. This model further serves as a platform to study how dynamic mechanical environments influence cellular physiology.
The deformation in the membrane was considered as an arc for the deformed shape. Subsequently, the change of the arc length due to the displacement was calculated through the formulas and it is shown in Fig. 3 (Fig. 4);

Schematic of deformation in the membrane. b: the original width of the channel, b’: the width of deformation, r: radius of the arc, \(\omega\): the height of deformation.

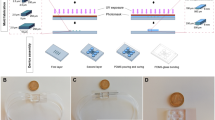
Mechanical stimulation via pneumatic actuation: design, set-up, and visualization. (a) Overview of the mechanical stimulation set-up. (b) BoC and air tubing connection. (c) Air flow on channel 1 and (d) air flow on channel 4. (e) QR code linking to the full video of the mechanical stimulation.
The deformation height was viewed and measured with a digital microscope. Since a perfect cross-sectional view is not possible in the working device, the observation axis was tilted to \(70^{\circ }\) (\(\alpha\)) to view the membrane deformation from this angle (Fig. 5) during operation. To make clear observation of the membrane displacement, the blue colored water was dropped on the edge of the reservoir. \(70^{\circ }\) was considered as the optimum angle to see the deformation height. From this angle, deformation height observed with applying pressure and the pictures were taken before and during the actuation (Fig. 5b,d).

Schematic design of the BoC without (a) and with (c) a pneumatic pressure. View of the microfluidic channel with and without pressure applied with the lens tilted at a \(70^{\circ }\) viewing angle, visualized with blue dye (b, d). The white dashed line (b) represents the membrane without pressure, while the white dashed arc (d) demonstrates the membrane under pressure.
The deformation in the membrane was estimated by measuring displacement of the membrane from \(70^{\circ }\) angle by using the digital microscopy and considering an arc for the deformed shape. Subsequently, the change of the arc length due to the displacement was calculated when membrane actuated with 1 bar. The original width of underneath channel is 50 \(\upmu\)m and a membrane actuation of 5.24 \(\upmu\)m high, the membrane is capable of deforming 2.85% of its original width. Hence for the localized deformation within the size of a cell (which is a few micrometers) the membrane would deform at the nanoscale. The deformation experiments were carried out on chips without either PLO+Laminine coating or cells on top of the membrane as a demonstration. The actual scale of the deformation may change during a real cell experiment due to loading with the additional chemical molecules and cells.
Hydrogel stiffness
Hydrogel stiffness can be varied by the concentration of reagents that modulate crosslinking efficiency. Young’s modulus was assessed using a nanoindenter, as detailed in Methods section. For the mechanical characterization experiment, the GelMA precursor solution was cast into a PDMS mold containing a single ring. Photopolymerization was then carried out, resulting in two distinct GelMA hydrogels, referred to as ’stiff’ and ’soft’ (Fig. 6a–d). According to the nanoindentation results, the stiffness of the ’stiff’ GelMA hydrogel was determined to be 2.3 kPa, while the ’soft’ GelMA hydrogel exhibited a stiffness of 1.1 kPa. For the culture experiments we used the soft GelMa to demonstrate the BoC function. The Young’s modulus of GelMA hydrogels was analyzed and plotted using GraphPad Prism and statistical comparison was performed using a two-tailed unpaired t-test (Fig. 6e).

Hydrogel Young’s modulus measurement. (a) Representative image showing the GelMA precursor cast into a PDMS mold with one ring, shaping the gel. (b) GelMA photopolymerization process. Pictures of the (c) stiff and (d) soft GelMA hydrogels after polymerization. (e) Young’s modulus of GelMA hydrogels plotted in GraphPad Prism 10.3 (Two tailed, unpaired t test, **p < 0.05, n = 3).
Cell culture assessment
The cells were introduced to the BoCs by means of two different approaches: 2D and 3D. In 2D, cells were seeded directly onto PLO and biolaminin-coated surfaces within the BoC, while in 3D configuration, GelMA hydrogel was dispensed into PLO and biolaminin-coated BoC reservoirs first up to the level of the first pinning structure of the BoC, 1 mm in height. Next, the hydrogel was polymerized in the chip according to the method described in previous paper72. Following the polymerization process, the hydrogel in both reservoirs was removed using a spatula. The remaining hydrogel in the linker channel was washed three times with cell culture medium and then kept in cell medium at \(4^{\circ }\)C overnight. On the following day, the medium was aspirated. For 2D cell culture, cells were directly seeded onto the glass surface (Fig. 7a), whereas for 3D cell culture, cells were seeded on top of the polymerized GelMA hydrogel (Fig. 7b,c).

The schematic illustration, along with corresponding images, shows (a) the setup of 2D cell culture, and (b, c) the detailed process of GelMA hydrogel integration into the BoC system for 3D cell culture. Following in-chip polymerization of the hydrogel (1.1 kPa), the reservoirs are cleared of the gel, and cells are subsequently seeded on-top of the linker channel.
Calcium imaging analysis
Visualization of the dynamic neuronal activities within a microfluidic chip during mechanical stimulation, calcium imaging was employed using the intracellular \(\text {Ca}^\text {2+}\) indicator Fluo-4, loaded into hiPSC-derived neurons. Our primary objective was to provide a proof-of-concept for the analysis of human stem cell-derived neuronal cell networks subjected to mechanical stimuli via Ca-imaging, under conditions that approximate the in-vivo environment of brain tissue, achieved by incorporating a hydrogel matrix (GelMA).
To establish a comparative baseline, our experimental set-up was contained cultured hiPSC-derived neurons in a 2D and 3D on the chip for three weeks. Following neuronal differentiation, we monitored calcium signaling activity under two conditions: during periods of with mechanical stimulation in both 2D and 3D (Fig. 8), and without mechanical stimulation (Fig. 9a). This setup allowed us to observe baseline calcium signaling patterns and to evaluate how mechanical forces influence neuronal activity relative to their intrinsic signaling behavior. As group with no stimulation, spontaneous calcium fluctuations were recorded for 5 min and the data is shown in Fig. 9b). And recorded movie is accessible via QR code (Fig. 9c). Based on the fluorescence intensity of regions of interest (ROIs) in the control group, neurons were spontaneously active.

Calcium imaging and analyses workflow. (a) Schematic of the experimental strategy for culturing and imaging human induced pluripotent stem cell hiPSC-derived neurons. (b) Time-series images of 2D neuronal cell cultures showing \(\text {Ca}^{2+}\) waves at different time points, extracted from video recordings. The dashed lines indicate the underlying microchannel location. White arrows mark neurons undergoing \(\text {Ca}^{2+}\) flux, indicated by changes in fluorescence intensity under mechanical stimulation. A full movie is accessible via the QR code. (c) \(\text {Ca}^{2+}\) imaging of 2D cultured neuronal cells (DIV 21) loaded with a \(\text {Ca}^{2+}\) indicator. Dashed lines indicate the microchannel position underneath. White circles highlight regions of interest (ROIs) where fluorescence intensity was monitored. (d) \(\text {Ca}^{2+}\) activity profiles of selected cells, corresponding to the ROIs in (c). The red dashed line marks the time point when the stimulus was introduced. (e) Time-series images of a 3D neuronal network (cells extending in 3D via a gel layer), activated by chip stimulation. Images are extracted from video recordings of the observed \(\text {Ca}^{2+}\) wave at different time points. White arrows and dashed lines indicate the underneath microchannel, white arrows displaying neuronal cell firing that were undergoing a \(\text {Ca}^{2+}\) flux, revealed by the change of the fluorescence intensity under mechanical stimulation. The full movie is accessible via the QR code. (f) \(\text {Ca}^{2+}\) imaging of 3D-like neuronal cells, with ROIs marked by white circles. (g) \(\text {Ca}^{2+}\) activity profiles of selected cells, corresponding to the ROIs in (f). The red dashed line marks the time point when the stimulus was introduced. Scale bars: 75 \(\upmu\)m.
As a demonstration of the functioning of the chip, Fig. 8a presents the schematic of the experimental strategy from culturing to imaging of human induced pluripotent stem cell hiPSC-derived neurons. Quantitative analysis of the neuronal response to the mechanical stimulation (Fig. 8c,f) illustrate the selection process for regions of interest (ROIs) within the \(\text {Ca}^{2+}\) imaging datasets, while figure (Fig. 8b,e) present real-time responses of these ROIs, highlighting peaks in fluorescence intensity corresponding to neuronal activation upon stimulation. The analysis reveals that when a mechanical stimulus was introduced, neurons adhered to the free-standing membrane or positioned near the membrane edges experienced mechanical stretching. The pneumatic pressure controller was set to a pressure of 1 bar. The BoC contained five channels, each requiring 10 s of actuation followed by a 1-s delay. A complete run of stimulation for all five channels took a total of 55 min. To facilitate repetitive testing, the pressure controller was configured to perform repetitive runs over a duration of 5 min. During this 5-min period, video recordings were captured to monitor the calcium activity, during which cells in the BoC were stimulated consecutively five times. This mechanical stimuli induced intracellular \(\text {Ca}^{2+}\) fluctuations, which propagated as intercellular \(\text {Ca}^{2+}\) waves across adjacent neurons within the network. Figure 8d depicts the \(\text {Ca}^{2+}\) activity in a 2D neuronal culture, while Fig. 8g illustrates the activity in a 3D culture on the hydrogel. The increased total fluorescence intensity upon stimulation suggests a robust activation of neuronal cells. The \(\text {Ca}^{2+}\) imaging data, which include a series of wavefront propagating from the stimulated region, were analyzed to assess the spatiotemporal dynamics of signal transmission within the network. A time-lapse sequence of these activities, accelerated 100-fold, is accessible via a QR code embedded in Fig. 8b,e.
In summary, we observed distinct differences in calcium signaling between hiPSC-derived neurons with and without mechanical stimulation. When a stimulus was introduced, an average \(\text {Ca}^{2+}\) fluorescence intensity before (pre-) and after (post-) the stimulus was performed using thirty ROIs across two independent 2D BoC samples (Fig. 10). The increase of the total fluorescence intensity implies the activation of the neuronal network due to the stimulation. Neurons cultured without mechanical stimulation exhibited spontaneous calcium signaling activity, characterized by periodic fluctuations intrinsic to their baseline physiology and independent of external stimuli. There is no significant increase in fluorescent intensity observed across all ROIs in control group, while in groups (both 2D and 3D) with mechanical stimulation, after the stimulus is applied, there is a noticeable increase in fluorescence intensity in most ROIs. The increase in fluorescence is sustained over time and may suggest a prolonged calcium response to mechanical stimulation. This observation suggests that mechanical forces can promote more organized and coordinated activity within the neuronal network.
Furthermore, the delayed calcium signaling observed in 3D neuronal cultures compared to 2D might be explained by several interconnected factors. In 3D environments, neurons are embedded in matrix-like materials that act as diffusion barriers, slowing the access of stimuli or signaling molecules. This physical constraint also limits the uniformity and speed of stimulation compared to the flat, easily accessible 2D surfaces75. Additionally, neurons in 3D systems form more spatially complex and less synchronized networks, which can delay calcium signal propagation across the culture. These networks often mature more physiologically, meaning they may express ion channels and synaptic proteins at different rates than 2D cultures, affecting their excitability. The depth of cells within the 3D structure also means that not all neurons are reached by stimuli simultaneously, further contributing to the delay76. Furthermore, light scattering and signal attenuation during calcium imaging in 3D might obscure early responses or make them appear delayed. The calcium dyes used may also diffuse less efficient in 3D matrices, this may reduce response time. Just as drug penetration is slower in 3D, a low drug response occurs77. Overall, while 3D systems offer more in-vivo-like environments, they inherently introduce complexity that affects both neuronal behavior and measurement dynamics (Figs. 9, 10).

Calcium imaging and analysis workflow without stimulation. (a) \(\text {Ca}^{2+}\) imaging of 2D cultured neuronal cells at DIV 21, loaded with a calcium indicator, and observed without mechanical stimulation. Image is extracted from video recording of the observed \(\text {Ca}^{2+}\) wave. White circles highlight regions of interest (ROIs) where fluorescence intensity was monitored. (b) \(\text {Ca}^{2+}\) activity profiles of selected cells, corresponding to the ROIs in (a). (c) The full movie is accessible via the QR code. Scale bar: 75 \(\upmu\)m.

Comparison of average Ca2+ fluorescence in 2D BoC before (pre-) and after (post-) stimulus application at DIV21. Statistical analysis was performed using an unpaired two-tailed t-test (*p < 0.0001, n = 30) in GraphPad Prism.
Immunostaining analysis (neuronal morphology and synapse formation)
In 2D model, neurons were directly cultured on pre-coated BoCs (Fig. 11a), while in 3D model, cells were plated onto hydrogel layer in BoC (Fig. 11f) providing structural support. Neuronal morphology and synapse formation were characterized in both systems using immunostaining techniques. Neurons were identified through the expression of specific neuronal markers, MAP2 (soma-dendritic marker, green) and SMI312 (axonal marker, orange), to distinguish dendritic and axonal compartments in the cultures (Fig. 11b,g). For synapse assessment, synaptic puncta were observed, defined by the colocalization of the presynaptic marker SYN1/2 (synaptophysin, red) and the postsynaptic marker HOMER-1 (blue), as seen in Fig. 11c–e,h–j.
Synaptic density was quantified using Image J, taking into consideration only those images that were acquired from the linker channel of the chips. Synaptic density represents one of the most important factors which could explain how different environments influence the overall connectivity and functional status of neural networks. Synaptic density in 2D BoC vs. 3D BoC was statistically compared using an unpaired, two-tailed t-test with GraphPad Prism software. Literature has shown that 3D cultures typically better recapitulate the in-vivo condition than 2D cultures. In 3D, neurons produce more mature and complicated synapses, while maintaining higher synaptic connectivity69. That agrees with the findings in this analysis when synaptic density was statically higher in the 3D BoC group (Fig. 11l)
Noteworthy to mention that cells in 2D formed small spheroid-like aggregates (Fig. 11b–e). Conversely, in 3D, individual cells remained more dispersed on the hydrogel (Fig. 11g–j), instead of clustering since hydrogel provides cells with more spatial freedom. However, in 3D BoC model, cells demonstrated similar clustering behavior in 2D regions such as left and right reservoirs (Fig. 11d).

Overview of hiPSC-derived neuron cultures in 2D (a) and 3D (f) BoC models on day 21. Hydrogels were removed from the reservoirs in the 3D BoC cultures. Co-localization of the post-synaptic marker HOMER-1 (c, h) and the pre-synaptic marker SYN1/2 (d, i) on axons. (e, j) All neuronal markers visualized together in panels (e) and (j). Neurons are labeled with SMI312 (axonal marker, orange), MAP2 (soma-dendritic marker, green), SYN1/2 (pre-synaptic marker, red), and HOMER-1 (post-synaptic marker, blue). Scale bars: 100 \(\upmu\)m. (k) Schematic illustration of synapse (created with Biorender). (l) Statistical comparison between 2D BoC and 3D BoC groups was performed using an unpaired two-tailed t-test (*p < 0.05, n=17) by GraphPad Prism.
SMI312 was performed to visualize axonal structures and axonal cytoskeleton. We, therefore, used SMI312 in the identification of sprouting axons, and axonal organization in different types of neuronal microenvironments, such as 2D and 3D. SMI312 staining result displays explicit visualization of axonal patterns in Fig. 12b,e, MAP2 staining illustrates soma-dendritic organization in Fig. 12c,f. Neurons cultured in the 2D BoC (Fig. 12a) exhibit linear axonal patterns extending from one end to the other. Neurons near the edges, where the cells not only pull themselves but also force adjacent neurons. With this collective movement, the cells at the edges gradually shift toward the center, forming connections with neighboring neurons along the way. The entire neuronal population eventually becomes interconnected through linear and thicker axonal projections. In the 3D BoC model (Fig. 12d), axonal growth exhibited a more complex and highly branched pattern. Within the linker channel of the chip, where a hydrogel layer was positioned beneath the cells, fewer tightly packed cell clusters were seen. The hydrogel provided an overall structural support to the cells such that they would not move around and instead, it would encourage a more organized network of connections. Supplementary Figure 1 presents the 3D visualization of BoC cultures using the Volume Viewer plug-in in ImageJ. The images were reconstructed from z-stack acquisitions taken at multiple angles to enhance the detailed visualization of synaptic structures (Homer-1: Blue, SYN1/2: Red) as well as neuronal features, marked by MAP2 (green) and SMI312 (Supplementary Figure 1c,d).

Illustration of immunofluorescence images of both 2D (a) and 3D (d) neuronal cultures to show differences in axonal sprouting and organization. (b, c) Zoomed image of a and (f, g) Zoomed image of (d). (SMI312: yellow, MAP2: green). Scale bars: 100 \(\upmu\)m.
Conclusion
Although mechanical forces play a crucial role in brain function and pathology, they are often overlooked in traditional neuronal cell culture models as an important parameter influencing cell and network functions. BoC platforms address this gap by integrating mechanical stimuli to more accurately replicate in-vivo dynamics in brain, enhancing the study of neuronal function and disease mechanisms by providing a more accurate simulation of the brain’s mechanical microenvironment.
In this article, we propose a new approach for BoC fabrication to study brain dynamic microenvironment. BoC comprises five independent microfluidic pneumatic pressure driven channels in a fused silica glass substrate, a spin-coated PDMS membrane, and a PDMS reservoir layer made by replica molding from a 3D printed mold to host the cells and medium. The three parts were oxygen plasma bonded. Air pulses can be applied to bulge the membrane out and deliver localized and time-dependent dynamic mechanical stimuli to the brain cells in the vicinity of a channel. Our chip provides a relatively mild mechanical stimulus by means of a 2.85% elongation of the membrane’s width during the application of a pressure pulse underneath the membrane using a microfluidic channel design. Providing insights into synapse development and function in dynamic neuronal network cultures, we cultured hiPSCs to grow neurons in BoC, focusing on neuronal maturation and synapse formation within 21 days. Immunostaining revealed evidence of synapse formation, with co localization of SYN-1 and Homer-1 signals along MAP2-positive dendrites and SMI312-positive axons. Next, we operated the BoC under dynamic conditions using calcium live imaging to assess neuronal activity. The \(\text {Ca}^{2+}\) responses observed upon mechanical stimulation in both 2D and 3D in-vivo-like neuronal cell networks, using GelMA as a scaffold on the membrane. Neuronal activity monitored and quantified, results revealed significant changes in fluorescence intensity, correlating with \(\text {Ca}^{2+}\) flux and neuronal firing under mechanical stimuli.
In addition to its ability to simulate dynamic mechanical environments, the BoC platform is also designed to mimic the static conditions of brain tissue. Our chip enables the creation of vertical gel stacks which can replicate brain layered architecture and stiffness variation. Moreover, the BoC platform offers potential for co-culturing neurons with glial cells, such as astrocytes, which play a crucial role in neurogenesis and neural network formation though this aspect requires further investigation. An additional feature of this chip design is the integration of feeder channels within each layer, which supply essential nutrients to cells and supporting continuous flow, which helps to replicate in-vivo-like physiological conditions. Although this setup has not yet been tested with cells encapsulated in hydrogel stacks, future experiments could refine and validate these design features for enhanced performance.
The novelty of our BoC platform, it brings two mechanical aspects static and dynamic mechanical forces in native brain tissue together in a single chip. Such a combination in one chip provides an extended model for the simulating of native brain tissue and thus is an invaluable tool in drug discovery and testing concerning neurological disorders.
Our BoC platform holds promises for studying and testing therapies for a range of neurological diseases where mechanical cues play a significant role. As one example, Alzheimer’s disease involves disrupted synaptic function and altered neuronal connectivity. Our BoC model, which incorporates both static and dynamic mechanical forces, allows for investigation of how mechanical microenvironments influence synaptic density and network stability which are the key elements in Alzheimer’s pathology. Another example might be Traumatic brain injury (TBI) and chronic traumatic encephalopathy (CTE) are directly linked to abnormal mechanical forces. Our system’s ability to replicate mechanical strain makes it highly relevant for modeling such conditions. Moreover, our chip system can be further adapted to test fluid pressure instead of air pressure. This might help with Hydrocephalus, conditions involving intracranial pressure fluctuations could also benefit from this model, as the dynamic mechanical component can simulate fluid pressure and tissue deformation effects on brain function. Overall, the combination of static and dynamic mechanical cues in our BoC platform might enable us to simulate disease-specific microenvironments more realistically.
Methods
Microfluidic BoC fabrication
A BoC was fabricated in three separate layers, as channel layer, membrane layer and reservoir layer respectively.
Channel layer
Microfluidic channels were fabricated on fused silica glass slides (75 mm \(\times\) 25 mm \(\times\) 1 mm, Siegert wafer) by using femtosecond laser micromachining, which was performed on the FEMTOprinter F200 aHead (FEMTOprint SA, Switzerland) as demonstrated (Fig. 14c). The fabrication process mainly consisted of three steps: (1) designing the channels with Alphacam software, (2) exposing the fused silica glass to ultrashort-pulsed femtosecond laser radiation, and (3) performing selective chemical etching to remove the exposed material. The laser beam, when focused within the glass, alters its refractive index and chemical properties in specific regions, enabling the creation of intricate, high-precision three-dimensional structures with a high aspect ratio through chemical etching. The initial step was done by placing five independent microfluidic air-flow channels with spacing from center-to-center set at 1.2 mm. All the channels had a depth of 40 \(\upmu\)m and a width of 50 \(\upmu\)m, with air-tube insertion areas (in-outlets) at both ends, having a diameter of 1 mm. The tool path for the laser scanning process was generated using Alphacam, then forwarded to the FEMTO printer F200 for the exposure. Pulse energy, repetition rate, and polarization of the laser were set as 230 nJ, 1000 kHz, and \(90^{\circ }\), respectively, during the femtosecond laser exposure. The laser beam was focused through a 20x microscope objective (Thorlabs) which had a NA of 0.4. After the end of the laser micromachining process, the glass slides were immersed into a 45% potassium hydroxide (KOH, Sigma-Aldrich) solution, which had been diluted in water. The etching process was conducted in an ultrasonic bath at 85 \(^{\circ }\)C for 9 h to remove the laser-exposed material selectively. After etching, cleaning of the glass slides was performed using acetone and deionized (DI) water three times in each. Additionally, an ultrasonic bath in ethanol was given for an hour. Finally, the glass slides were air-dried using an air-gun in preparation for the bonding process. Figure 13 presents the microfluidic channel patterned on the surface of the glass.

Schematic representation of the BOC fabrication process. The reservoir layer, designed to contain the cellular medium, is formed using SLA-based 3D printing for mold creation, followed by PDMS replica molding. A 10 \(\upmu\)m-thick membrane layer is deposited via precision spin coating. The channel layer is fabricated on a fused silica glass substrate through femtoprinting technology, incorporating femtosecond laser micromachining and subsequent wet etching to define the microchannel structures.
The mixture was then degassed in a vacuum desiccator to remove any air bubbles. The degassed PDMS mixture was then poured onto the silicon wafer substrate, filling the entire wafer. The spin coating process was then completed according to the parameters in Table 1. After the spin coating process, the PDMS layer was allowed to rest for 5 minutes before being placed in an oven set to 80 \(^{\circ }\)C, where it underwent curing overnight to finalize the membrane formation.
Spin coat parameters has been set as below.
Membrane layer
The membrane layer fabricated in the BoC was a 10 \(\upmu\)m layer of polydimethylsiloxane (PDMS). The fabrication started with the preparation of a 100 mm silicon wafer substrate (SiMat), which was cleaned initially with pressurized nitrogen. Then the wafer surface was activated with a 20 W air plasma for 30 s in a plasma asher (Emitech, K1050X). Silanization was carried out to form an anti-stiction layer to promote the PDMS being released after curing. The silicon wafer was introduced into a vacuum desiccator along with a clean glass vial containing 20 \(\upmu\)l of Chlorotrimethylsilane (CTMS, Sigma). The desiccator was then evacuated, and the wafer was maintained under vacuum for approximately 15 min, followed by an extended period under vacuum conditions overnight to ensure thorough silanization. After completing the silanization treatment, the wafer was retrieved from the vacuum desiccator and transferred to a spin coater (Laurell Technologies, WS-650-23B). For the preparation of the PDMS membrane, the PDMS base (\(\text {Sylgard}^\text {TM}\) 184, Dow Corning) was mixed with the curing agent in a 10:1 (w/w) ratio (base: curing agent).
Reservoir layer
The reservoir layer was fabricated by using SLA-based 3D Printing and PDMS replica molding combination as indicated in Fig. 13. Fabrication protocol is explained in detail in our previous article72. Briefly, the reservoir layer features two wells, linked by a straight channel. The sidewalls are designed with a sawtooth-shaped structure with a rectangular microfluidic feeder channel (integrated into the sidewalls), which surrounds the reservoir for fluid distribution. This sawtooth configuration facilitates the precise formation of two hydrogel layers stacked at a certain height as previously demonstrated in our paper72.
Chip assembly
Initially, reservoir layer (Fig. 14a) and PDMS membrane (Fig. 14b) were assembled. PDMS membrane, which was formed on a silicon wafer substrate, along with the PDMS reservoir layer, underwent air plasma treatment at a power setting of 20 W for a duration of 30 s using a plasma asher. This plasma exposure facilitated surface activation, thereby enabling covalent bonding. Subsequent to the plasma activation process, the reservoir layer was aligned and placed into contact with the PDMS membrane situated on the silicon wafer, thereby facilitating consistent adhesion. The membrane-reservoir unit was cut off the silicon wafer using a razor and then gently peeled away to prevent any damage to the membrane. A laser-cut PMMA stencil (Fig. 14d) is employed to create precise holes for in- and outlets (Fig. 14e) using a puncher (UniCore Punches, Qiagen), and an air gun was used to blow the removal of any PDMS residues. In the following step, the membrane-reservoir unit was attached to the channel layer by using oxygen plasma activation. Both surfaces were exposed to 20 W oxygen plasma for 30 s. The second step was essentially about the exact positioning of the punched inlets and outlets on the membrane-reservoir unit with regards to the respective access points on the glass channel layer. Inverted and aligned with great care, the channel layer was laid atop the membrane-reservoir unit a task necessary for ensuring good bonding without trapping air pockets between the layers. To enhance the bonding strength and ensure long-term stability, the assembled device was kept in an oven at 80 \(^{\circ }\)C for an h. Hence resulting in a completely assembled device (Fig. 14f–h), ready for use in microfluidic applications.

Overview of the BoC fabrication process in real time/world. (a) Reservoir Layer, (b) Reservoir and membrane layer assembly. (c) femtoprinted underneath channels in fused silica glass. (d, e) laser cut PMMA sheet with in-outlets with a 1mm puncher to make hole for in-outlets in the chip. (f–h) Final BoC from different angles.
Mechanical stimulation
Mechanical stimulation was performed by connecting home-made gas flow control device to the BoC through air tubing, as shown in Fig. 3. The pressurized air was connected to the homemade gas flow control device, the pressure regulator was set to 1 bar, and chip was actuated channel 1 up to channel 5 subsequently for 4 times. Each channel was actuated for 10 s, following was a delay for 1 s. Altogether, the cell culture was actuated for 5 min (2D) and 10 min (3D). A stimulus with 10 s duration and an applied pressure of 1 bar was used throughout the experiments presented in this paper as a demonstration.
Membrane deformation
Compressed air flow was introduced into the chip to produce a deformation in the flexible membrane. The deformation was confirmed with a digital microscope (Keyence, VHX-5000) (Fig. 4a,b). Schematic illustrations of membrane deformation without and with applied pressure are presented in Fig. 5a,c, respectively. The deformation height was observed with colored liquid. The liquid was dropped in the edge of the reservoir. The microscope was bent over \(70^{\circ }\), this was optimized by changing different angles. \(70^{\circ }\) was considered as the optimum angle to see the deformation height. From an angle, deformation heigh observed with applying pressure and taking pictures before and during the actuation. The experiments were carried out on chips without PLO+Laminine coating or cells on top of the membrane. Hence, the scale of deformation may change according to the cell amount and cell-membrane attachment during cell experiment.
3D mimicry of extracellular matrix
To mimic the 3D in vivo tissue microenvironment and to investigate how extracellular matrix material interactions stimulate neuronal cell behavior, we applied two types of GelMA hydrogels were characterized: ”soft” and ”stiff” GelMA. The only difference between the two hydrogel types is their composition, specifically the concentration of triethanolamine (TEA) used in their preparation. To prepare the soft GelMA hydrogel, 5% (w/v) GelMA was dissolved in a base solution comprising cell medium, 0.01 mM Eosin Y, 0.1% (w/v) TEA, and 37 nM N-vinyl-2-pyrrolidinone (NVP). Then the mixture was kept in a waterbath at \(37 ^{\circ }\)C for 30 min to get a homogeneous precursor solution. In contrast, the stiff GelMA hydrogel was prepared similarly except that the concentration of TEA was increased from 0.1% (w/v) to 1% (w/v). This modification was set to observe the mechanical properties of the resulting hydrogel. Subsequent to preparation, both GelMA precursor solutions were cast into BoCs. Thereafter, the hydrogels were then photo-polymerized a light source (KL 2500 LCD with a light intensity of 4E, Schott) at room temperature for 10 min. Afterwards, the gels remained for another 5 min without light exposure to ensure the completion of polymerization. Upon successful polymerization, the GelMA hydrogels were carefully scooped from the reservoirs and let the gel remained in the linker channels of the microfluidic chips.
Hydrogel (GelMA) stiffness was determined by means of a nanoindentor (PIUMA, Optics11 Life). A 0.51 N/m stiffness probe with a tip radius in 28 \(\upmu\)m was used for measurement tests. Hydrogel samples were tested in water. Hydrogels were created in a PDMS ring in 1 mm height as cylindircal shaped. Placed into the petri dish and added water ontop till cover the gel.
Cell culture
Chip sterilization
Prior to cell culture experiments, BoCs underwent a sterilization procedure. For proper surface decontamination of BoCs, treatment with 70% ethanol was done for 30 min. Following ethanol treatment, the chips were rinsed three times with Milli-Q (MQ) water to eliminate any traces of ethanol. The chips were then air-dried for 1 h in a biosafety cabinet, protecting them from environmental microbial contamination. As a final step, the chips were exposed to ultraviolet (UV) light for 30 min to further eradicate any remaining microorganisms. After completing these steps, these BoCs were deemed sterile and ready for subsequent cell culture studies. ñ
Cell culture protocol
hiPSCs (Human WTC-11, Ngn2+ overexpressing line; kindly provided by Prof. Nael Nadif Kasri, Radboud University) were expanded and differentiated towards cortical excitatory neurons as previously described78. Briefly, hiPSCs were thawed on Matrigel coated 6-well plates in C8 medium containing E8 flex basal medium, E8 flex basal supplement, Primocin, RevitaCell, G418 and Puromycin. Medium was changed daily. To initiate differentiation, once hiPSC colonies reached 80% of confluency, hiPSC colonies were dissociated with accutase, thereafter hiPSCs were plated with a density of \(12 \times 10^4\) \(\text {cells/cm}^2\) in BoCs coated with 50 \(\mu\)g/mL poly-L-ornithine (PLO; Merck, P4957) and 20 \(\mu\)g/mL biolaminin (Biolamina, LN521). Cells received a half medium change and were then fed every other day with differentiation medium which are indicated for each day separately in the supplementary information. All materials and suppliers indicated in supplementary information.
Calcium imaging
Calcium imaging was conducted to detect cytosolic calcium ions (\(\text {Ca}^{2+}\)) in iNeurons using the Fluo-4 Calcium Imaging Kit according to the instructions of the manufacturer (Invitrogen, F10489). Neuronal cultures were incubated at 37 \(^{\circ }\)C for 20 min with a PowerLoad solution and probenecid in the presence of Fluo-4 dye. Following this initial incubation, further incubation at room temperature was performed for 15 min in the dark to ensure the best possible dye loading. After incubation, the cells were washed once in cell medium to remove excess dye. An imaging solution, consisting of a background suppressor suspended in cell medium, was added to each chip and helped enhance the clarity of the image. Transfer the chips to the pre-heated microscope stage, set to 37 \(^{\circ }\)C, for live-cell image acquisition. Next, green fluorescence movies were recorded using a fluorescence microscope (DMI8, Leica), for tracing calcium fluxes in the neurons. The intracellular calcium was monitored for 5-minute period, capturing images at a rate of 4 frames per second with on stage incubation.
Immunostaining
3-week-old iNeurons in the BoCs were washed with Dubecco’s phosphate buffered saline (DPBS, calcium, magnesium, Gibco, 14040133) and fixated for 15 min at room temperature with a solution of 4% formaldehyde (Thermo Scientific Alfa Aesar, 043368) and 4% w/v sucrose (Thermofisher, J65148.36). Subsequently, the cells were washed three times for 5 min at room temperature with DPBS again and permeabilized with 0.2% Triton X-100 (SigmaAldrich, 9036-19-5) solution for 15 min at room temperature. Thereafter cells were incubated with blocking buffer solution of DPBS with 5% normal goat serum (Invitrogen, 31873) for 1 h at room temperature. Primary antibodies (MAP2, Rabbit polyclonal, Abcam ab32454, 1:500; SMI-312, Biolegend, 837904,1:500; Homer-1, Chicken polyclonal, Synaptic systems, 160 006, 1:500; Synapsin 1/2, Guinea pig polyclonal, Synaptic systems, 106 004, 1:1000) were diluted in 1% blocking buffer were applied to the cells and incubated overnight at 4\(^{\circ }\)C. Then, cells were washed three times for 5 min with DPBS followed by incubation with secondary antibodies (Goat anti-Rabbit IgG Alexa Fluor 488, Termofisher, 10236882, 1:2000; Goat anti-Mouse IgG Alexa Fluor 555, Termofisher, 10433982, 1:500; Goat antiGuinea pig IgG Alexa Fluor 647, Termofisher 10624773, 1:2000; Goat anti-Chicken IgG Alexa Fluor 405, Termofisher 17121839, 1:2000) diluted in 1% blocking buffer for 4 h at room temperature. Finally, cells were washed 3 times for 5 min with DPBS followed by submerging the chips in fluorescent mounting medium (Mowiol 4-88, polysciences, 25213-24-5). Then, cells were ready to be imaged using a fluorescence microscopy (DMI8, Leica).
Data availability
The data that support the findings of this study are available within the article or its supplementary material.
References
Guo, T., Ren, P., Hao, S. & Wang, B. The underestimated role of mechanical stimuli in brain diseases and the related in vitro models. Curr. Pharmaceutical Design. 23 (2016).
Koser, D. E. et al. Mechanosensing is critical for axon growth in the developing brain. Nat. Neurosci. 19, 1592–1598 (2016).
Chicurel, M. Cell migration research is on the move. Science 295, 606–609 (2002).
Tyler, W. J. The mechanobiology of brain function. Nat. Rev. Neurosci. 13, 867–878 (2012).
Cipolla, M. J. Control of cerebral blood flow. in The Cerebral Circulation. (Morgan & Claypool Life Sciences, 2009).
Rennels, M. L. & Capillary, E. Capillary innervation in the mammalian central nervous system: An electron microscopic demonstration. Am. J. Anat. 144, 233–241 (1975).
Cohen, Z., Bonventot, G., Lacombe, P. & Hamel, E. Serotonin in the regulation of brain microcirculation. Progress Neurobiol. 50, 335–362 (1996).
Hamel, E. Regulation of the cerebral circulation: Perivascular nerves and the regulation of cerebrovascular tone. J. Appl. Physiol. 100, 1059–1064 (2024).
Lok, J. et al. Cell cell signaling in the neurovascular unit. Neurochem. Res. 32, 2032–2045 (2007).
Tufail, Y., Yoshihiro, A., Pati, S., Li, M.-M. & Tyler, W. J. Ultrasonic neuromodulation by brain stimulation with transcranial ultrasound. Nat. Protocols 6, 1453–1470 (2011).
Rezayat, E. & Toostani, I. G. A review on brain stimulation using low intensity focused ultrasound. Basic Clin. Neurosci. 7, 187–194 (2016).
Liu, X., Qiu, F., Hou, L. & Wang, X. Review of noninvasive or minimally invasive deep brain stimulation. Front. Behav. Neurosci. 15, 1–13 (2022).
Szotek, S., Kobielarz, M. & Maksymowicz, K. Mechanical response of brain to mechanical stimuli animal model investigation. Neurologia i Neurochirurgia Polska 41, 525 (2007).
Zusman, M. The meaning of mechanically produced responses. Aust. J. Physiother. 40, 35–39 (1994).
Lam, F. M. H., Liao, L. R., Kwok, T. C. Y. & Pang, M. Y. C. Effects of adding whole-body vibration to routine day activity program on physical functioning in elderly with mild or moderate dementia: a randomized controlled trial. Int. J. Geriatr. Psychiatry 33, 21–30 (2018).
Furness, T. P. & Maschette, W. E. Influence of whole body vibration platform frequency on neuromuscular performance of community-dwelling older adults. J. Strength Conditioning Res. 23, 1508–1513 (2009).
lvarez Barbosa, F. et al. Effects of supervised whole body vibration exercise on fall risk factors, functional dependence and health-related quality of life in nursing home residents aged 80+. Maturitas 79, 456–463 (2014).
Mueller, J. K. Advancing transcranial focused ultrasound for noninvasive neuromodulation of human cortex. Thesis (2015).
Fini, M. & Tyler, W. J. Transcranial focused ultrasound: A new tool for non-invasive neuromodulation. Int. Rev. Psychiatry 29, 168–177 (2017).
Hodkinson, D. J., Drabek, M. M., Jung, J. Y., Lankappa, S. T. & Auer, D. P. Theta burst stimulation of the human motor cortex modulates secondary hyperalgesia to punctate mechanical stimuli. Neuromodulation 27, 812–823 (2024).
Mansour, Z. M. et al. Brain response to non-painful mechanical stimulus to lumbar spine. Brain Sci. 8, 1–10 (2018).
Bray, D. Axonal growth in response to experimentally applied mechanical tension. Develop. Biol. 102, 379–389 (1984).
Zheng, J. et al. Tensile regulation of axonal elongation and initiation. J. Neurosci. 11, 1117–1125 (1991).
Breau, M. A. et al. Extrinsic mechanical forces mediate retrograde axon extension in a developing neuronal circuit. Nat. Commun. 8 (2017).
Akcay, G. & Luttge, R. Microenvironments matter: Advances in brain-on-chip. Biosensors 13, 1–28 (2023).
Baldi, A., Fass, J. N., De Silva, M. N., Odde, D. J. & Ziaie, B. A micro-tool for mechanical manipulation of in vitro cell arrays. Biomed. Microdevices 5, 291–295 (2003).
Gaub, B. M. et al. Neurons differentiate magnitude and location of mechanical stimuli. Proc. Natl. Acad. Sci. 117, 848–856 (2020).
Joy, M. S. H. et al. Synapses without tension fail to fire in an in vitro network of hippocampal neurons. Proc. Natl. Acad. Sci. 120, e2311995120 (2023).
Maneshi, M. & Z., S. H. Early cell response to mechanical stimuli during tbi. Recent Adv. Biomech. https://doi.org/10.5772/intechopen.93295 (2020).
Beltrn, S. M. et al. Characterization of neural mechanotransduction response in human traumatic brain injury organoid model. Sci. Rep. 13, 1–11 (2023).
Siechen, S., Yang, S., Chiba, A. & Saif, M. T. Mechanical tension contributes to clustering of neurotransmitter vesicles at presynaptic terminals. Proc. Natl. Acad. Sci. 106, 12611–12616 (2009).
Minegishi, T. et al. Mechanical regulation of synapse formation and plasticity. Seminars Cell Develop. Biol. 140, 82–89 (2023).
Pfister, B. J. et al. Extreme stretch growth of integrated axons. J. Neurosci. 24, 7978–7983 (2004).
Smith, D. H., Wolf, J. A. & Meaney, D. F. A new strategy to produce sustained growth of central nervous system axons: Continuous mechanical tension. Tissue Eng. 7, 131–139 (2001).
Whitesides, G. M., Ostuni, E., Takayama, S., Jiang, X. & Ingber, D. E. Soft lithography in biology and biochemistry. Annu. Rev. Biomed. Eng. 3, 335–373 (2001).
Arscott, S. Su-8 as a material for lab-on-a-chip-based mass spectrometry. Lab Chip 14, 3668–3689 (2014).
Berthier, E., Young, E. W. & Beebe, D. Engineers are from pdms-land, biologists are from polystyrenia. Lab Chip 12, 1224–1237 (2012).
Roy, E. et al. Overview of materials for microfluidic applications. In Advances in Microfluidics–New Applications in Biology, Energy, and Materials Science. https://doi.org/10.5772/65773 (IntechOpen, 2016).
Hughes, M. P. & Hoettges, K. F. The Application of Microfluidics in Biology, vol. 19 (2010).
Shakeri, A., Khan, S. & Didar, T. F. Conventional and emerging strategies for the fabrication and functionalization of pdms-based microfluidic devices. Lab Chip 21, 3053–3075 (2021).
Raj, K. M. & Chakraborty, S. Pdms microfluidics: A mini review. J. Appl. Polym. Sci. 137, 1–14 (2020).
Miranda, I. et al. Properties and applications of pdms for biomedical engineering: A review. J. Funct. Biomater. 13, 1–31 (2022).
Banik, S. et al. The revolution of pdms microfluidics in cellular biology. Crit. Rev. Biotechnol. 43, 465–483 (2023).
Vickers, J. A., Caulum, M. M. & Henry, C. S. Generation of hydrophilic poly(dimethylsiloxane) for high-performance microchip electrophoresis. Analyt. Chem. 78, 7446–7452 (2006).
Gewandter, J. S., Staversky, R. J. & O’Reilly, M. A. Hyperoxia augments er-stress-induced cell death independent of bip loss. Free Radical Biol. Med. 47, 1742–1752 (2009).
Tang, Y. et al. Cyp1b1 and endothelial nitric oxide synthase combine to sustain proangiogenic functions of endothelial cells under hyperoxic stress. Am. J. Physiol.-Cell Physiol. 298 (2010).
Campbell, S. B. et al. Beyond polydimethylsiloxane: Alternative materials for fabrication of organ-on-a-chip devices and microphysiological systems. ACS Biomater. Sci. Eng. 7, 2880–2899 (2020).
Yun, S. H. et al. Characterization and resolution of evaporation-mediated osmolality shifts that constrain microfluidic cell culture in poly (dimethylsiloxane) devices. Analyt. Chem. 79, 1126–1134 (2007).
Zhang, Y., Ishida, M., Kazoe, Y., Sato, Y. & Miki, N. Water-vapor permeability control of pdms by the dispersion of collagen powder. IEEJ Trans. Electr. Electron. Eng. 4, 442–449 (2009).
Scott Lynn, N., Henry, C. S. & Dandy, D. S. Evaporation from microreservoirs. Lab Chip 9, 1780–1788 (2009).
Millet, L. J. & Gillette, M. U. Over a century of neuron culture: From the hanging drop to microfluidic devices. Yale J. Biol. Med. 85, 501–521 (2012).
Lee, H. S. et al. Effect of hydrophilicity on electrically driven flow in microchannels. Colloids Surfaces A Physicochem. Eng. Aspects 311, 77–82 (2007).
Almutairi, Z., Ren, C. L. & Simon, L. Evaluation of polydimethylsiloxane (pdms) surface modification approaches for microfluidic applications. Colloids Surfaces A Physicochem. Eng. Aspects 415, 406–412 (2012).
Nielsen, J. B. et al. Microfluidics: Innovations in materials and their fabrication and functionalization. Analyt. Chem. 92, 150–168 (2020).
Devices, G. M. et al. A rapid and low-cost procedure for fabrication of. J. Microelectromech. Syst. 16, 1193–1200 (2007).
Annabestani, M., Dokht, P. & Fardmanesh, M. A novel, low cost, and accessible method for rapid fabrication of the modifiable microfluidic devices. Sci. Rep. 10, 1–9 (2020).
Hirama, H. et al. Glass-based organ-on-a-chip device for restricting small molecular absorption. J. Biosci. Bioeng. 127, 641–646 (2019).
Berlanda, S. F. et al. Recent advances in microfluidic technology for bioanalysis and diagnostics. Analyt. Chem. 93, 311–331 (2020).
Italia, V. et al. Laser-inscribed glass microfluidic device for non-mixing flow of miscible solvents. Micromachines 10, 1–12 (2018).
Toepke, M. W. & Beebe, D. J. Pdms absorption of small molecules and consequences in microfluidic applications. Lab Chip 6, 1484–1486 (2006).
Feng, C., Takahashi, K. & Zhu, J. Simple one-step and rapid patterning of pdms microfluidic device wettability for pdms shell production. Front. Bioeng. Biotechnol. 10, 1–10 (2022).
Chen, I.-J. & Lindner, E. The stability of radio-frequency plasma-treated polydimethylsiloxane surfaces. Langmuir 23, 3118–3122 (2007).
Aralekallu, S., Boddula, R. & Singh, V. Development of glass-based microfluidic devices: A review on its fabrication and biologic applications. Mater. Design 225, 111517 (2023).
Tourovskaia, A., Figueroa-Masot, X. & Folch, A. Differentiation-on-a-chip: A microfluidic platform for long-term cell culture studies. Lab Chip 5, 14–19 (2005).
Ofner, A. et al. High-throughput step emulsification for the production of functional materials using a glass microfluidic device. Macromol. Chem. Phys. 218, 1600472 (2017).
Shan, C., Chen, F., Yang, Q., Jiang, Z. & Hou, X. 3d multi-microchannel helical mixer fabricated by femtosecond laser inside fused silica. Micromachines 9, 29 (2018).
Calmo, R. et al. Monolithic glass suspended microchannel resonators for enhanced mass sensing of liquids. Sensors Actuators B Chem. 283, 298–303 (2019).
Zhu, D. et al. Fabrication and applications of surface micro/nanostructures by femtosecond laser. Colloids Interface Sci. Commun. 59, 100770 (2024).
Butkutė, A. & Jonu auskas, L. 3d manufacturing of glass microstructures using femtosecond laser. Micromachines. 12 (2021).
Suthisomboon, T. et al. Design and simulation of xz mems micropositioning with 3d-complex structure. In 2020 Symposium on Design, Test, Integration and Packaging of MEMS and MOEMS (DTIP), 1–5 (IEEE, 2020).
Steimle, A. 3d micro-printing: A new era for med-tech applications. Laser Technik J. 15, 32–34 (2018).
Akcay, G., van Venrooij, J. & Luttge, R. Stacked hydrogel-based brain-on-chips utilizing capillary force flow pinning. J. Vacuum Sci. Technol. B. 42 (2024).
Procès, A., Luciano, M., Kalukula, Y., Ris, L. & Gabriele, S. Multiscale mechanobiology in brain physiology and diseases. Front. Cell Develop. Biol. 10, 823857 (2022).
Huang, C. Hypertension, mechanical force, and renal disease. Ann. Clin. Exp. Hypertens. 2, 1009 (2014).
LaPlaca, M. C., Vernekar, V. N., Shoemaker, J. T., Cullen, D. K. & Coulter, W. Three-dimensional neuronal cultures. Methods Bioeng. 3D Tissue Eng. 187–204 (2010).
Frega, M., Tedesco, M., Massobrio, P., Pesce, M. & Martinoia, S. Network dynamics of 3d engineered neuronal cultures: A new experimental model for in-vitro electrophysiology. Sci. Rep. 4, 5489 (2014).
Xu, F. X., Sun, R., Owens, R., Hu, K. & Fu, D. Assessing drug uptake and response differences in 2d and 3d cellular environments using stimulated Raman scattering microscopy. Analyt. Chem. 96, 14480–14489 (2024).
Frega, M. et al. Rapid neuronal differentiation of induced pluripotent stem cells for measuring network activity on micro-electrode arrays. J. Visualized Exp. 2017, 1–10 (2017).
Acknowledgements
The research tasks of Regina Luttge are partially funded by the European Union’s Horizon 2020 research and innovation program H2020-FETPROACT-2018-01 under Grant (Grant no. 824070). We extend our sincere gratitude to the Nael Nadif Kasri Lab at Radboud University for generously providing us with Ngn2+ hiPSCs.
Author information
Authors and Affiliations
Contributions
G.A: Conceptualization (equal); Investigation (lead); Visualization (lead); Writing – original draft (lead); Writing – review and editing (equal). R.L: Conceptualization (equal); Funding acquisition (lead); Project administration (lead); Supervision (lead); Writing – review and editing (equal). All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Akcay, G., Luttge, R. Mechanodynamic brain on chip for studying human stem cell derived neuronal networks. Sci Rep 15, 29631 (2025). https://doi.org/10.1038/s41598-025-14187-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14187-6
