
Abstract
This study investigates the mechanisms of Osimertinib resistance in lung adenocarcinoma (LUAD) by identifying prognostic genes associated with SUMOylation. We performed differential expression analysis to identify differentially expressed genes (DEGs) in LUAD samples, Osimertinib-tolerant cell samples and SUMOylation-related genes (SRGs). Utilizing Cox regression and LASSO regression, we developed a prognostic model that highlighted five key prognostic genes—BIRC5, AURKA, BLM, NR3C2, and NDC1. These genes were significantly associated with LUAD progression, revealing their predominant expression in epithelial cells, which play a vital role in tumor development. Furthermore, we explored the biological functions and signaling pathways linked to these prognostic genes, discovering that their expression levels and corresponding risk scores could serve as indicators of CD4 T cell and memory B cell activation. The enriched signaling pathways in LUAD were regulated by ubiquitin-related small modifiers, highlighting the complex interplay between SUMOylation and tumor biology. Our findings suggest the important role of SUMOylation-regulated genes in LUAD progression and Osimertinib resistance, suggesting their potential as valuable biomarkers for prognosis and therapeutic targets to enhance treatment strategies for patients with EGFR-mutant lung adenocarcinoma.
Similar content being viewed by others


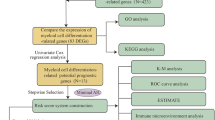
Introduction
Lung cancer remains one of the leading causes of cancer-related mortality worldwide, with non-small cell lung cancer (NSCLC) being the major histological type, accounting for approximately 85% of all lung cancer cases1. Within this category, LUAD is the most prevalent subtype2. Epidemiologically, LUAD has shown a rising incidence, particularly among non-smokers and younger populations, reflecting changing risk factors and environmental influences3. Current treatment options for LUAD include surgical resection, chemotherapy, targeted therapies such as epidermal growth factor receptor (EGFR) inhibitors, anaplastic lymphoma kinase (ALK) inhibitors, and immune checkpoint inhibitors. Among these, EGFR-tyrosine kinase inhibitors (EGFR-TKIs) demonstrate improved clinical efficacy and reduced toxicity compared to traditional chemotherapy, establishing them as the standard first-line treatment for advanced NSCLC with EGFR mutations4. However, despite the significant survival benefits achieved with TKIs, resistance mutations and disease progression remain inevitable challenges. Addressing acquired resistance and developing new therapeutic agents continue to be pivotal areas of research. Therefore, the urgent need to identify novel therapeutic targets is paramount to improve treatment outcomes and enhance the overall survival of patients diagnosed with this aggressive cancer subtype.
Post-translational modifications include ubiquitination, acetylation, glycosylation, and SUMOylation, and they primarily influence tumor growth and progression by regulating tumor cell proliferation, apoptosis, invasion, and metastasis5. These modifications can affect the efficacy of drugs by altering the functionality or stability of target proteins, as well as by modifying the cellular mechanisms of drug perception and response, thus promoting the emergence of drug resistance. While there has been extensive research on ubiquitination, SUMOylation is closely associated with tumor progression and targeted drug resistance6. SUMOylation or SUMO modification, refers to the post-translational modification process in which Small Ubiquitin-like Modifier (SUMO) proteins (such as SUMO1, SUMO2, and SUMO3) are conjugated to target proteins, influencing their activity, stability, localization, and interactions7. SUMOylation is an important post-translational modification of proteins that is involved in regulating various cellular activities, including cell division, signal transduction, DNA repair, and cellular metabolism8,9,10,11. Dysregulation of SUMOylation has been implicated in various diseases, particularly cancer. Abnormal SUMOylation patterns can contribute to tumorigenesis by altering the stability and activity of oncogenes and tumor suppressor proteins, influencing pathways involved in cell proliferation, apoptosis, and metastasis12. Moreover, cancer cells often exploit SUMOylation as a means to adapt to the stress of the tumor microenvironment and to evade therapeutic interventions13,14. Osimertinib is a third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) specifically designed for the common activating mutations such as T790M. It has shown significant clinical efficacy, improving progression-free survival and overall survival compared to earlier generation TKIs15,16,17. However, the development of resistance to Osimertinib remains a significant challenge, often due to secondary mutations in the EGFR gene, such as the C797S mutation, or through the activation of alternative signaling pathways18,19. SUMOylation can influence the stability and functionality of proteins involved in drug response, potentially contributing to the mechanisms of acquired resistance. Therefore, identifying relevant targets within the SUMOylation pathway to enhance Osimertinib sensitivity is crucial.
In this study, we utilized data from public databases to screen and identify prognostic genes associated with Osimertinib resistance and SUMOylation modification in LUAD using bioinformatics methods. Additionally, we established a prognostic model and used regulatory network and single-cell analyses to investigate the regulatory mechanisms and cellular heterogeneity in lung adenocarcinoma. This research aims to explore the roles and prognostic value of these genes in lung adenocarcinoma and Osimertinib treatment resistance, providing a theoretical basis for the treatment of lung adenocarcinoma patients to overcome resistance and improve treatment outcomes for EGFR-mutant lung adenocarcinoma.
Materials and methods
Data source
Gene expression matrices, clinical characteristics, and mutation information for 585 samples of LUAD from The Cancer Genome Atlas (TCGA) database were obtained from UCSC Xena (https://xena.ucsc.edu/). A subset of 510 case samples (cancer tissue, 01A) and 58 control samples (normal adjacent tissue, 11A) were utilized for analysis. Among the case samples, 497 contained survival information. Gene expression data and phenotype information from the GSE200893 dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200893), GSE50081 dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50081)and single-cell sequencing data GSE131907 were obtained GEO database (https://www.ncbi.nlm.nih.gov/geo/). The GSE200893 included 6 samples using the H1975 cell line, comprising 3 normal cell samples (Control) and 3 Osimertinib-tolerant cell samples (Tolerant), with the chip platform being GPL23159. The GSE50081 consisted of 181 samples using tissue samples, from which 127 LUAD samples were selected for the validation of the prognostic model. The chip platform used was GPL570. The GSE131907 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131907) included 58 samples, from which 11 normal lung tissue samples (nLung) and 11 LUAD lung tissue samples (tLung) were selected for analysis. A total of 189 SUMOylation-related genes (SRGs) were obtained by searching the ‘REACTOME_SUMOYLATION’ gene set from the Msigdb (https://www.gsea-msigdb.org/gsea/msigdb)20.
Differential expression analysis
Differential analysis were analyzed between the Case and Control groups in TCGA-LUAD dataset using the DESeq2 package (v 1.38.3)21. Significant DEGs_LUAD were identified (|log2FC|≥ 1 and P.adj < 0.05). Moreover, in GSE200893 dataset, the limma package (v 3.52.4)22 were used to identify DEGs_Tolerant between Tolerant and Control groups (|log2FC|≥ 1 and P.adj < 0.05).
Identification and analysis of candidate genes
The candidate genes were obtained by taking the intersection of DEGs_LUAD, DEGs_Tolerant, and SRGs. To gain insights into their biological functions and pathways, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were executed using clusterProfiler package (v 4.4.4)23. Finally, data visualization was carried out using the ggplot2 package (v 3.3.6)24.
Construction of prognostic model
The prognostic genes were screened using a multi-step approach. First, the survival package (v 3.4-0)25 was employed to perform univariate Cox regression analysis on each candidate gene individually. Genes significantly linked with survival (P < 0.05) were then subjected to the Proportional Hazards (PH) assumption test, and those that passed were included in least absolute shrinkage and selection operator (LASSO) regression analysis to build a prognostic model. Risk scores were computed based on the model, and all samples in TCGA-LUAD and GSE50081 were divided into high- and low-risk groups based on their median risk score respectively. Expression levels of prognostic genes were compared between two risk groups. Afterwards, K-M analysis was used to compare survival differences between the two risk groups, and 1- to 3-year receiver operating characteristic (ROC) curves were plotted separately for TCGA-LUAD and GSE50081 using the survival ROC package (v 1.0.3.1)26 to assess model accuracy.
Independent prognostic analysis
In TCGA-LUAD, the distribution and significance of risk scores among different groups stratified were examined by various clinical characteristics. For two-group comparisons, we used the Wilcox.test to assess the significance of differences in risk scores. For multiple-group comparisons, the kruskal.test was employed. In addition, in TCGA-LUAD, we performed univariate Cox regression analysis to determine the significance of clinical characteristics and riskScore. Significant characteristics were then subjected to PH assumption tests. Based on the results, a multivariable Cox regression model was constructed to obtain independent prognostic factors. A nomogram was built using the survival (v 3.4-0)25 and rms packages (v 6.3-0)27 based on independent prognostic factors, and was used to predict patients’ 1- to 3-year survival, and calibration curves and decision curve analysis (DCA) curves were drawn to validate the model predictions.
Immune microenvironment analysis
The Seven-Step Tumor Immune Cycle described the interactions between tumors and the immune system and highlighted how the immune system fights tumors. In order to investigate the mechanisms related to the immune microenvironment in LUAD, we assessed the tumor immunophenotypes within the seven-step tumor immune cycle of the TCGA-LUAD samples using the TIP (Tracking Tumor Immunophenotype) website. Subsequently, the seven-step tumor immune cycle was divided into 23 subcategories. A Wilcoxon rank sum test was executed to compare the scores of these 23 subcategories between the high- and low-risk groups. The ssGSEA was executed on the TCGA-LUAD using GSVA package (v 1.44.5)28. The immune related genes were from literature as background gene set29. In TCGA-LUAD, the enrichment scores of 28 immune cells in case samples were calculated and the correlation between immune cells was analyzed. To analyze the differences in enrichment scores of immune cells between two groups, Wilcoxon test was used. Spearman correlation analysis was conducted to explore the relationship between prognostic genes, risk scores, and the differential enrichment scores of immune cells. Finally, the differences between the common immune checkpoints were compared in the high and low risk groups of TCGA-LUAD.
Somatic mutation analysis
Tumor Mutational Burden (TMB) reflected the degree of variation in the genome of tumor cells. In TCGA-LUAD, somatic gene mutations in Case samples were analyzed using the maltols package30.
Gene set enrichment analysis
To elucidate the biological functions and signaling pathways associated with prognostic genes, Spearman analysis was executed between the prognostic genes and other genes within TCGA-LUAD. Subsequently, these genes were ranked based on their correlation strengths. The msigdbr package (v 7.5.1) was utilized to download the C2: KEGG gene set as the background set. Enrichment analysis was then conducted on the ranked genes using the GSEA function (P < 0.05).
Construction of regulatory network
To better understand the regulatory mechanisms of prognostic genes, miRNAs were predicted simultaneously using the Target Prediction and Analysis (PITA) (https://genie.weizmann.ac.il/pubs/mir07/index.html) and microT (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=microT_CDS/index) databases (clipExpNum > = 10). ClipExpNum represents the number of independent CLIP-seq experiments supporting the specific miRNA-target gene interaction31. A threshold of 10 for miRNA screening is used to filter out low-confidence interactions, reduce false positives, and improve specificity. The interactions between lncRNA and miRNA are more tissue-specific or condition-dependent, requiring further experimental validation. Furthermore, miRNA-associated lncRNAs were predicted in starbase database (clipExpNum > = 20), and a ceRNA network was established based on these predictions. Using the cytoHubba plugin in Cytoscape 3.10.1, the top 20 nodes ranked by degree value were selected for display.
Single-cell analysis
The original literature data underwent quality control based on the following criteria: (1) mitochondrial genes were limited to ≤ 20%; (2) the number of genes in each cell ranged from 200 to 10,000; and (3) the gene expression UMI fell between 100 and 150,00032. Consequently, the Seurat package (v 4.3.0)33 in R was exclusively employed to import the expression data from 22 samples. Following logarithmic normalization, the vst method was utilized to identify genes with substantial variability across cells, resulting in the identification of 2,000 highly variable genes. Subsequently, Principal Component Analysis (PCA) downscaling was implemented to select principal components (PCs) for subsequent analyses. Unsolicited cluster analysis of the filtered cells was carried out using the Seurat package functions FindNeighbors and FindClusters. Finally, cell types were annotated based on the marker genes provided in the literature32. In addition, the expression of prognostic genes in different cells was verified to identify key cells.
Pseudotime analysis and cellular communication
Cellular communication was assessed using the CellChat package (v 1.6.1)34, which was based on the CellChatDB database (https://www.cellchat.org/db/). This package utilizes cell expression data as input to simulate cell–cell communication in conjunction with ligand-receptor and cofactor interactions. The monocle package (v 2.26.0)35 was used to perform a pseudotime analysis analysis in order to understand the relationship between key cellular transitions at different developmental stages.
Statistical analysis
Bioinformatic analysis were conducted in the R program. Data from different teams were compared via the wilcoxon test.
Results
LUAD progression and EGFRi tolerance were associated with small ubiquitin-related modifier
The TCGA-LUAD case consisted of 510 tumor samples of LUAD sourced from The Cancer Genome Atlas (TCGA) database via UCSC Xena, while the control groups included 58 samples of normal adjacent tissue. Differential analysis revealed a total of 3222 differentially expressed genes (DEGs) between the TCGA-LUAD Case (DEGs_LUAD) and control groups with 1806 genes being up-regulated and 1416 genes down-regulated (Fig. 1A,B). The GSE200893 dataset contained 6 samples utilizing the H1975 cell line, comprising 3 normal cell samples (control groups) and 3 Osimertinib-tolerant cell samples (tolerant groups). From this analysis, 1621 differentially expressed genes in the Tolerant (DEGs_Tolerant) were identified, comprising 852 up-regulated genes and 769 down-regulated genes (Fig. 1C,D). By intersecting DEGs_LUAD, DEGs_Tolerant, and 189 SUMOylation-related genes (SRGs) obtained by searching the ‘REACTOME_SUMOYLATION’ gene set in Msigdb (https://www.gsea-msigdb.org/gsea/msigdb), We identified 13 candidate genes: AURKA, AURKB, BIRC5, BLM, BRCA1, CDCA8, INCENP, NDC1, NR3C2, NUP210, PCNA, PPARG, and TOP2A (Fig. 1E). The enrichment analysis indicated that these candidate genes were associated with Gene Ontology (GO) terms related to the regulation of mitotic cytokinesis and mitotic spindle midzone assembly, which were also linked to small ubiquitin-related modifier (Fig. 1F). Furthermore, these candidate genes exhibited enrichment in KEGG signaling pathways, including those related to the cell cycle and platinum drug resistance. (Fig. 1G).

Screening differentially expressed gene signature associated with small ubiquitin-related modifier in LUAD. (A) Volcano plots of the gene expression signature analysis in LUAD Case based on TCGA-LUAD. (B) Heat map of differential genes in LUAD Case. (C) Volcano plots of the gene expression signature analysis in GSE200893. (D) Heat map of differential genes in GSE200893. (E) Venn diagram of intersection genes. X-axis: number of intersection genes. Y-axis: number of differentially expressed genes in DEGs_LUAD, DEGs_Tolerant, and SRGs. (F) The results of enrichment analysis, the TOP10 functional items of each part (significance ranking) were displayed, the box size indicates the number of enriched genes. (G) The results of enriched KEGG pathways, the TOP5 functional pathway (significance ranking) and its genes were displayed, circle size indicates the number of enriched genes.
SUMOylation associated genes BIRC5, AURKA, BLM, NR3C2, and NDC1 were prognostic genes in LUAD
The candidate genes identified through univariate Cox regression analysis, which included BIRC5, AURKA, BLM, NR3C2, CDCA8, TOP2A, INCENP, NDC1, AURKB, and BRCA1, demonstrated a significant association with patient survival and successfully met the proportional hazards (PH) assumption test (Fig. 2A). Subsequent LASSO analysis further refined this list, confirming BIRC5, AURKA, BLM, NR3C2, and NDC1 as key prognostic genes, which may play a role in osimertinib resistance, but further validation is required. (Fig. 2B). Based on these prognostic genes, a risk score model was developed: RiskScore = BIRC5 * 0.072 + AURKA * 0.050 + BLM * 0.075 + NR3C2 * (−0.122) + NDC1 * 0.021. Among these genes, BIRC5, AURKA, BLM, and NDC1 were risk factors (HR > 1) with higher expression in the high-risk group, while NR3C2 served as a protective factor (HR < 1) showing higher expression in the low-risk group (Fig. 2C,D). K-M results showed poorer survival in the high-risk group than in the low-risk group in TCGA-LUAD and GSE50081 (Fig. 2E). Furthermore, the Area Under the Curve (AUC) values for 1 to 3 years in TCGA-LUAD and GSE50081 were greater than 0.6, indicating that the model was more effective in predicting the survival of the samples (Fig. 2F).

Identification and analysis of candidate genes. (A) Forest plot of Univariate Cox regression analysis. (B) LASSO regression analysis plot. left graph: plot of penalty term parameters, abscissa is log (lambda) value, ordinate is degrees of freedom, representing the error of cross validation. The dashed line on the left represents the position where the cross-validation error is minimal. The corresponding optimal log (Lambda) value is determined at this position (lambda.min), and the number of feature genes is displayed above. The corresponding genes and their coefficients are found on the right plot. Right graph: The abscissa is log(lambda), the ordinate is the coefficient of the gene, and the change of the coefficient of different variables after being penalized with λ. (C) Expression of prognostic genes in TCGA-LUAD (top graph) and GSE50081 (bottom graph). (D) Risk curves according to TCGA-LUAD (top graph) and GSE50081 (bottom graph). The blue line represents the low-risk group, and the red line represents the high-risk group (distinguished by the median). The blue circle represents the survival sample (Alive), and the red dot represents the non-survival sample (Death). (E) Kaplan–Meier survival analysis by different gene expression levels of High-risk group and low-risk group according to TCGA-LUAD (left graph) and GSE50081 (right graph). The abscissa is the survival time, the ordinate is the survival rate, and the Number in the coordinate axis (Number) below the figure is the number of surviving samples in the corresponding survival period. (F) ROC analysis according to TCGA-LUAD (left graph) and GSE50081 (right graph). The abscissa is the false positive rate, the ordinate is the true positive rate, and the area under the curve called Area Under Curve (AUC) represents the prediction accuracy.
Risk score and pathologic_T were independent prognostic factors for the progression of LUAD
In the TCGA-LUAD dataset, we examined the distribution and significance of risk scores across various clinical characteristics, including age, gender, pathologic_M, pathologic_N, pathologic_T, and stage. The analysis of risk score differences among clinical traits revealed significant variations in the between-group distribution of risk scores for all five traits, except for pathologic_M. Notably, risk scores exhibited an upward trend with increasing disease severity in pathologic_N, pathologic_T, and Stage (Fig. 3A). We conducted univariate Cox regression analysis in TCGA-LUAD to assess the significance of clinical characteristics (Age, Gender, pathologic_M, pathologic_N, pathologic_T, Stage) and risk Score. Based on these results, a multivariable Cox regression model was constructed to identify independent prognostic factors. The independent prognostic analysis confirmed that risk Score and pathologic_T are independent prognostic factors (Fig. 3B,C). A nomogram was developed based on these independent prognostic factors and calibration curves along with decision curve analysis (DCA) curves were drawn to validate the model predictions (Fig. 3D). The calibration curve exhibited a slope close to 1, indicating strong concordance between predicted and observed outcomes (Fig. 3E). Furthermore, the decision curve analysis (DCA) demonstrated that the model provided a greater net benefit compared to using a single factor alone, underscoring the enhanced predictive capability of the nomogram (Fig. 3F).

Independent prognostic analysis. (A) Risk scores were distributed across groups for each trait in TCGA-LUAD. (B) Forest plots of independent prognostic analysis: univariate Cox results; (C) Forest plots of independent prognostic analysis: multivariate Cox results. (D) Nomograms curves. (E) Calibration curves. (F) DCA curve of the nomogram.
The expression of prognostic SUMOylation genes was correlated with the activated CD4 T cell and memory B cell
The seven-step tumor immune cycle, first proposed by Chen and Mellman in 2013, includes the following steps: release of antigens by tumor cells (Step 1), presentation of tumor antigens (Step 2), activation of T cells (Step 3), migration of T cells to tumor tissue (Step 4), infiltration of T cells into tumor tissue (Step 5), recognition of tumor cells by T cells (Step 6), and elimination of tumor cells (Step 7).The Seven-Step Tumor Immune Cycle described the interactions between tumors and the immune system emphasizing how the immune system fights tumors. To investigate the mechanisms related to the immune microenvironment in LUAD, we assessed the tumor immunophenotypes within the seven-step tumor immune cycle of the TCGA-LUAD samples using the TIP (Tracking Tumor Immunophenotype) website.
Subsequently, the seven-step tumor immune cycle was divided into 23 subcategories. A Wilcoxon rank sum test was performed to compare the scores of these 23 subcategories between the high- and low-risk groups (according to the median value). The results of the seven-step tumor immune cycle showed significant differences between the 15 tumor immune cycles in the high and low risk groups, such as Step1, Step4, recruiting CD4 T cell etc. (Fig. 4A). Enrichment scores of 22 immune cell subsets exhibited significant differences between groups (P < 0.05), including Activated CD4 T cell, Activated CD8 T cell, and Effector memory CD4 T cell (Fig. 4B,C). Specifically, Activated CD4 T cell and Memory B cell showed positive correlations with prognostic genes (BIRC5, AURKA, BLM, NDC1), and risk score, while displaying negative correlation with prognostic genes NR3C2 (Fig. 4D). These findings suggested that the expression levels of prognostic genes can serve as indicators of the infiltration levels of these two immune cell types to some extent. In addition, Immune checkpoints are a class of immunosuppressive molecules expressed on immune cells that regulate the level of immune activation. Immune checkpoint molecules help maintain the immune system’s activation within a normal range, preventing excessive activation. The use of immune checkpoints in cancer treatment refers to a series of approaches that modulate T cell activity through co-inhibitory or co-stimulatory signals to target and kill tumor cells. In TCGA-LUAD, the expression levels of five immune checkpoints (IDO1, CD274, PDCD1LG2, PDCD1, LAG3) exhibited significant differences between groups (p < 0.05), with all of them being upregulated in the high-risk group compared to the low-risk group (Fig. 4E).

Immune microenvironment analysis. (A) Analysis of group differences in tumor immune circulation between high-risk and low-risk groups in TCGA-LUAD. The color of the box indicates the grouping of the samples, and the color of the abscissa indicates the difference between the groups (purple indicates High > Low, green indicates High < Low, and black indicates insignificant). The top is significance, ns represents p > 0.05, * represents p ≤ 0.05, ** represents p ≤ 0.01, *** represents p ≤ 0.001, and **** represents p ≤ 0.0001. (B) Group differences in enrichment scores for immune cells. (C) Correlation between immune cells. (D) Correlation between Prognostic genes, risk scores, and immune-cell. (E) Group differences in immune checkpoints.
The prognostic SUMOylation genes were related to ceRNA networks and enriched signaling pathways in LUAD progression
The somatic mutation response showed a higher percentage of mutations in high-risk samples compared to low-risk samples, with TP53 mutations present in only 25% of low-risk samples but 50% of high-risk samples (Fig. 5A,B). The enrichment analysis revealed that the prognostic genes were significantly enriched in the cell cycle, DNA replication, pyrimidine metabolism, and other signaling pathways. Specifically, the cell cycle pathway was found to be associated with the development of LUAD, while the signaling pathways of DNA replication and pyrimidine metabolism were regulated by small ubiquitin-related modifier. These findings suggested that the obtained prognostic genes align with the expected biological processes (Fig. 5C). To better understand the regulatory mechanisms of prognostic genes, we predicted miRNAs and lncRNAs associated with prognostic genes. We constructed a ceRNA network containing 2 mRNAs, 4 miRNAs, and 14 lncRNAs, with some regulatory relationships such as hsa-miR-330-3p and hsa-miR-330-3p etc. Therefore, ceRNAs, particularly miRNAs and lncRNAs can influence the expression and stability of genes associated with the SUMOylation pathway, thereby contributing to the dynamic changes of the ceRNA network (Fig. 5D). It is important to note that the network is only a predictive result and carries certain uncertainty. Further experimental research is required to validate or refine the findings.


Signaling pathway enrichment and molecular regulatory network analysis. (A) The overall mutational profile of somatic gene mutations in TCGA-LUAD. (B) Somatic mutations in samples from high-risk and low-risk groups. (C) Enrichment gene analysis with GSEA (TOP5 enriched pathways). (D) ceRNA regulatory network, the ellipse represents prognostic genes, the rectangle represents miRNAs, and the pentagon represents lncRNAs.
Epithelial cells express high levels of prognostic SUMOylation genes and regulate cell–cell communication
Single-cell analysis allows for the study of gene expression and biological characteristics at the level of individual cell, revealing the heterogeneity within cell populations. It facilitates the analysis of cellular states and dynamic changes, identifies specialized cell types, and uncovers disease biomarkers. The original literature data underwent quality control based on the following criteria: (1) mitochondrial genes were limited to ≤ 20%; (2) the number of genes in each cell ranged from 200 to 10,000; and (3) the gene expression UMI fell between 100 and 150,000. The Seurat package (v 4.3.0) in R was exclusively employed to import the expression data from 22 samples. Feature RNA quantities and counts before and after QC were demonstrated at (Fig. 6A), and 2000 highly variable genes were selected for subsequent analysis (Fig. 6B). Principal Component Analysis (PCA) downscaling was implemented to select principal components (PCs) for subsequent analyses. The PCs at convergence were selected for cell clustering, so dims = 30 were chosen for subsequent analysis (Fig. 6C). A total of 8 cell types were annotated, including B lymphocytes, Endothelial cells, Epithelial cells, Fibroblasts, MAST cells, Myeloid cells, NK cells, and T lymphocytes (Fig. 6D,E). Gene expression analysis revealed that BIRC5, NR3C2, and NDC1 were mainly expressed in Epithelial cells, whereas AURKA was mainly expressed in MAST cells. Notably, the expression levels of BIRC5 and NDC1 were higher in the Epithelial cells of the tLung samples compared to the nLung samples, and the AURKA expression was also elevated in MAST cells of the tLung samples relative to the nLung samples (Fig. 6F). Conversely, myeloid cells exhibited the strongest intercellular interactions, followed by T lymphocytes. In contrast, disease samples displayed stronger interaction among T lymphocytes, myeloid cells, and epithelial cells. Additionally, disease samples exhibited a slightly stronger communication between epithelial cells and B lymphocytes than control samples (Fig. 6G). Three prognostic genes were expressed in epithelial cells, and the proposed time-series analysis indicated that cells in the control group had a higher percentage of cells in State1, while those in the disease group had a higher percentage of cells in State2 and State3. Specifically, epithelial cells in State2 and State3 exhibited higher expression of the three prognostic genes, suggesting that the epithelial cells continued to differentiate during the development of lung adenocarcinoma (Fig. 6H).

Single cell analysis found cell groups related to the occurrence and development of LUAD. (A) Overview of single-cell analysis of each sample. (B) The degree of variation of each gene in the single-cell data, blue represents the hypervariable genes, black represents the less variable genes, (only the name of the Top10 gene is shown). (C) Inflection point plots (left graph) and cell distribution plots for PCA analysis (right graph). (D) The results of TSNE cluster analysis. (E) Bubble plot results from TSNE cluster analysis. (F) Expression of key genes in single-cell data. (G) Results of cell communication. The right two graphs show the results of the nLung group, and the left two graphs show the results of the tLung group. The size of the circle indicates the number of cells; the larger the circle, the greater the number; The cells emitting the arrow express the ligand, and the cells pointing the arrow express the receptor. The more ligand-receptor pairs, the thicker the lines. The higher the probability/strength of the interaction (the intensity is the sum of the probability values), the thicker the line. (H) Time trajectories and different grouping trajectories were analyzed by pseudo-temporal analysis in epithelial cells. The right panel and he middle panel shows the time trajectory of the proposed time sequence, from blue to red is the sequence of cell differentiation, and the left panel shows the different states of cells.
Discussion
In this study, we conducted functional enrichment analysis to identify 13 differentially expressed genes and 11 functional pathways associated with SUMOylation in lung adenocarcinoma, particularly in Osimertinib-resistant cases. Based on univariate Cox analysis and LASSO regression analysis, we ultimately selected five genes (AURKA, NDC1, BLM, BIRC5, NR3C2) that are significantly associated with the survival and prognosis of lung adenocarcinoma patients. We constructed a prognostic assessment model for lung adenocarcinoma by calculating the correlation between the prognostic genes and other genes. Through model calculations and gene set enrichment analysis, we identified functional pathways related to the prognostic genes, where signaling pathways such as the cell cycle, DNA replication, and metabolism are regulated by SUMOylation. Additionally, we observed the immune microenvironment in lung adenocarcinoma and found significant differences in 15 tumor immune cycles, there were significant differences in the enrichment scores of 22 immune cell types between groups (p < 0.05), and five immune checkpoints (IDO1, CD274, PDCD1LG2, PDCD1, LAG3) also exhibited significant differences (p < 0.05). In the single-cell dataset, we performed dimensionality reduction and cell clustering using highly variable genes and found that BIRC5 and NDC1 are primarily expressed in epithelial cells, while AURKA is predominantly expressed in mast cells, indicating that epithelial cells undergo continuous differentiation during the progression of lung adenocarcinoma.
We identified five genes significantly associated with the survival and prognosis of lung adenocarcinoma patients. Among these, BIRC5 is a protein essential for regulating mitosis and inhibiting apoptosis, while also participating in pathological processes36. Research indicates that BIRC5 is upregulated in tumor tissues; however, high expression of BIRC5 is indicative of poor clinical prognosis37. SUMOylation may influence the function of BIRC5/Survivin, thereby affecting its localization and stability within cells. Additionally, SUMOylation might also play a role in regulating the transcriptional activity of Survivin, impacting the expression of its downstream target genes38,39. AURKA (Aurora kinase A) is believed to be involved in the regulation of the cell cycle and centrosome function40. Studies have shown that in EGFR-mutant LUAD, AURKA is associated with resistance to EGFR inhibitors, and high expression of AURKA correlates with poor prognosis in non-small cell lung cancer (NSCLC) patients41. SUMOylation may affect AURKA’s intracellular localization and regulate its interactions with other proteins, thereby influencing AURKA’s function in cell cycle regulation and mitosis42. BLM is a DNA helicase involved in DNA damage repair. Research has shown that BLM can undergo SUMOylation, which facilitates the formation of RAD51 foci during replication stress, thereby impacting the homologous recombination (HR) repair process. Furthermore, SUMOylation of BLM may also regulate its role in DNA damage repair43. NDC1 is a transmembrane nucleoporin that plays a critical role in the assembly of the nuclear pore complex and nucleocytoplasmic transport. Studies have indicated that NDC1 is overexpressed in lung cancer samples, and its elevated expression is associated with poor prognosis. The SUMOylation of NDC1 may play a significant role in its localization and function within the nucleus, particularly during the formation of the nuclear pore complex and spindle poles44. NR3C2 is a nuclear receptor involved in intracellular signal transduction regulation. Abnormal expression of the NR3C2 gene is closely related to the development and progression of various malignancies, potentially inhibiting tumor progression by suppressing tumor cell proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT)45. The protein encoded by the NR3C2 gene is the mineralocorticoid receptor (MR). Multiple SUMOylation sites have been identified within MR, and SUMOylation of MR affects its ligand-dependent nucleocytoplasmic shuttling, as well as protein–protein interactions and subsequent transcriptional regulation46,47.
We found that there are differences in immune responses between the high-risk and low-risk groups in lung adenocarcinoma, potentially involving variations in the levels of immune cell infiltration and the expression of immune checkpoints, such as activated CD4 T cells and memory B cells. Immunotherapy has shown certain efficacy in LUAD, including the use of immune checkpoint inhibitors. Patients in the high-risk group may benefit from immunotherapy strategies. Further investigation of the immune differences between the high-risk and low-risk groups could aid in optimizing immunotherapy approaches and developing personalized treatment regimens48.
Cell clustering analysis has revealed the importance of epithelial cells in the progression of LUAD. The abnormal proliferation and malignant transformation of alveolar and bronchial epithelial cells lead to the development of lung adenocarcinoma. Dysregulation of epithelial cell proliferation is a key feature in the progression of LUAD. Interactions between epithelial cells and other cells in the tumor microenvironment, such as immune cells and stromal cells, contribute to the process of epithelial-mesenchymal transition (EMT)20. This transition endows epithelial cells with enhanced migratory and invasive capabilities, facilitating the infiltration of tumor cells into surrounding tissues and distant metastasis. Through cell clustering analysis, a deeper understanding of the heterogeneity, structural relationships, and biological markers of the epithelial cell population in lung adenocarcinoma is achieved. These analytical results provide new insights and targets for further research into the development and treatment of LUAD.
This study has several limitations. First, the relatively small sample size of the osimertinib resistance dataset resulted in low statistical power, potentially amplifying the impact of batch effects and stochastic biological variability. This may introduce noise and compromise the reliability of our conclusions. Second, the lack of detailed pharmacotherapy information in the clinical data could bias the results. Third, as a preliminary exploratory study, our work remains primarily at the bioinformatic analysis level without experimental validation; the specific functional roles of these genes remain unconfirmed.
Future research should prioritize enhancing the robustness and generalizability of findings by expanding sample sizes or incorporating multi-center clinical trials. Collecting and integrating more comprehensive patient pharmacotherapy history data would facilitate investigating the potential influence of drug treatments on candidate genes and their associated mechanisms, thereby improving the accuracy and clinical translatability of the study.
For experimental validation, subsequent studies should employ gene overexpression or knockdown/knockout techniques to investigate the effects of key genes on LUAD cell behaviors (e.g., proliferation, migration, invasion, apoptosis). Functional assays should further verify whether these genes operate through specific molecular pathways, potentially offering novel insights for LUAD therapeutic strategies. Additionally, investigating the interactions within the CeRNA network (lncRNA-miRNA-mRNA) could reveal their potential roles in LUAD pathogenesis. More detailed functional experiments are essential to elucidate the molecular mechanisms of the CeRNA network and advance precision medicine research.
Finally, to comprehensively validate the role of genetic variations in LUAD development, future studies should incorporate and comparatively analyze additional variant types beyond SNPs (e.g., insertions/deletions, copy number variations, structural variants). This multidimensional validation approach would strengthen the support for our findings.
Data availability
The datasets presented in this study can be found in online repositories. The original contributions presented in the study are included in this article. Further inquiries can be directed to the corresponding authors.
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263. https://doi.org/10.3322/caac.21834 (2024).
Ettinger, D. S. et al. NCCN guidelines insights: Non-small cell lung cancer, version 2.2023. J. Natl. Comp. Cancer Netw. 21, 340–350. https://doi.org/10.6004/jnccn.2023.0020 (2023).
Shi, J. F. et al. Clinical characteristics and medical service utilization of lung cancer in China, 2005–2014: Overall design and results from a multicenter retrospective epidemiologic survey. Lung Cancer 128, 91–100. https://doi.org/10.1016/j.lungcan.2018.11.031 (2019).
Ramalingam, S. S. et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl. J. Med. 382, 41–50. https://doi.org/10.1056/NEJMoa1913662 (2020).
Han, Z. J., Feng, Y. H., Gu, B. H., Li, Y. M. & Chen, H. The post-translational modification, SUMOylation, and cancer (review). Int. J. Oncol. 52, 1081–1094. https://doi.org/10.3892/ijo.2018.4280 (2018).
Qin, Y. et al. SUMOylation alterations are associated with multidrug resistance in hepatocellular carcinoma. Mol. Med. Rep. 9, 877–881. https://doi.org/10.3892/mmr.2014.1882 (2014).
Johnson, E. S. Protein modification by SUMO. Annu. Rev. Biochem. 73, 355–382. https://doi.org/10.1146/annurev.biochem.73.011303.074118 (2004).
Wang, J. & Schwartz, R. J. Sumoylation and regulation of cardiac gene expression. Circ. Res. 107, 19–29. https://doi.org/10.1161/circresaha.110.220491 (2010).
Liebelt, F. & Vertegaal, A. C. Ubiquitin-dependent and independent roles of SUMO in proteostasis. Am. J. Physiol. Cell Physiol. 311, C284-296. https://doi.org/10.1152/ajpcell.00091.2016 (2016).
Bettermann, K., Benesch, M., Weis, S. & Haybaeck, J. SUMOylation in carcinogenesis. Cancer Lett. 316, 113–125. https://doi.org/10.1016/j.canlet.2011.10.036 (2012).
Eifler, K. & Vertegaal, A. C. O. SUMOylation-mediated regulation of cell cycle progression and cancer. Trends Biochem. Sci. 40, 779–793. https://doi.org/10.1016/j.tibs.2015.09.006 (2015).
Kessler, J. D. et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335, 348–353. https://doi.org/10.1126/science.1212728 (2012).
Bogachek, M. V. et al. Inhibiting the SUMO pathway represses the cancer stem cell population in breast and colorectal carcinomas. Stem Cell Rep. 7, 1140–1151. https://doi.org/10.1016/j.stemcr.2016.11.001 (2016).
Du, L. et al. Role of SUMO activating enzyme in cancer stem cell maintenance and self-renewal. Nat. Commun. 7, 12326. https://doi.org/10.1038/ncomms12326 (2016).
Park, K. et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 17, 577–589. https://doi.org/10.1016/s1470-2045(16)30033-x (2016).
Hsu, W. H., Yang, J. C., Mok, T. S. & Loong, H. H. Overview of current systemic management of EGFR-mutant NSCLC. Ann. Oncol. 29, i3–i9. https://doi.org/10.1093/annonc/mdx702 (2018).
Remon, J., Steuer, C. E., Ramalingam, S. S. & Felip, E. Osimertinib and other third-generation EGFR TKI in EGFR-mutant NSCLC patients. Ann. Oncol. 29, i20–i27. https://doi.org/10.1093/annonc/mdx704 (2018).
Thress, K. S. et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 21, 560–562. https://doi.org/10.1038/nm.3854 (2015).
Du, X. et al. Acquired resistance to third-generation EGFR-TKIs and emerging next-generation EGFR inhibitors. Innovation 2, 100103. https://doi.org/10.1016/j.xinn.2021.100103 (2021).
Alwarsamy, M., Gooneratne, R. & Ravichandran, R. Effect of fucoidan from Turbinaria conoides on human lung adenocarcinoma epithelial (A549) cells. Carbohydr. Polym. 152, 207–213. https://doi.org/10.1016/j.carbpol.2016.06.112 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141. https://doi.org/10.1016/j.xinn.2021.100141 (2021).
Gustavsson, E. K., Zhang, D., Reynolds, R. H., Garcia-Ruiz, S. & Ryten, M. ggtranscript: An R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 38, 3844–3846. https://doi.org/10.1093/bioinformatics/btac409 (2022).
Ramsay, I. S. et al. Model selection and prediction of outcomes in recent onset schizophrenia patients who undergo cognitive training. Schizophr. Res. Cogn. 11, 1–5. https://doi.org/10.1016/j.scog.2017.10.001 (2018).
Heagerty, P. J., Lumley, T. & Pepe, M. S. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 56, 337–344. https://doi.org/10.1111/j.0006-341x.2000.00337.x (2000).
Sachs, M. C. plotROC: A tool for plotting ROC curves. J. Stat. Softw. https://doi.org/10.18637/jss.v079.c02 (2017).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Charoentong, P. et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18, 248–262. https://doi.org/10.1016/j.celrep.2016.12.019 (2017).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756. https://doi.org/10.1101/gr.239244.118 (2018).
Zhang, Y. J., Huang, C., Zu, X. G., Liu, J. M. & Li, Y. J. Use of machine learning for the identification and validation of immunogenic cell death biomarkers and immunophenotypes in coronary artery disease. J. Inflamm. Res. 17, 223–249. https://doi.org/10.2147/jir.S439315 (2024).
Kim, N. et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 11, 2285. https://doi.org/10.1038/s41467-020-16164-1 (2020).
Tan, Z. et al. Comprehensive analysis of scRNA-Seq and bulk RNA-Seq reveals dynamic changes in the tumor immune microenvironment of bladder cancer and establishes a prognostic model. J. Transl. Med. 21, 223. https://doi.org/10.1186/s12967-023-04056-z (2023).
Liu, Z., Sun, D. & Wang, C. Evaluation of cell-cell interaction methods by integrating single-cell RNA sequencing data with spatial information. Genome Biol. 23, 218. https://doi.org/10.1186/s13059-022-02783-y (2022).
Rao, Y. et al. Single-cell transcriptome profiling identifies phagocytosis-related dual-feature cells in a model of acute otitis media in rats. Front. Immunol. 12, 760954. https://doi.org/10.3389/fimmu.2021.760954 (2021).
Altieri, D. C. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 22, 8581–8589. https://doi.org/10.1038/sj.onc.1207113 (2003).
Xu, L., Yu, W., Xiao, H. & Lin, K. BIRC5 is a prognostic biomarker associated with tumor immune cell infiltration. Sci. Rep. 11, 390. https://doi.org/10.1038/s41598-020-79736-7 (2021).
Hamy, A. S. et al. BIRC5 (survivin): A pejorative prognostic marker in stage II/III breast cancer with no response to neoadjuvant chemotherapy. Breast Cancer Res. Treat. 159, 499–511. https://doi.org/10.1007/s10549-016-3961-2 (2016).
Zhou, L. et al. High nuclear survivin expression as a poor prognostic marker in pancreatic ductal adenocarcinoma. J. Surg. Oncol. 118, 1115–1121. https://doi.org/10.1002/jso.25253 (2018).
Taylor, N. J. et al. Genetic variation in cell cycle regulatory gene AURKA and association with intrinsic breast cancer subtype. Mol. Carcinog. 54, 1668–1677. https://doi.org/10.1002/mc.22238 (2015).
Shah, K. N. et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 25, 111–118. https://doi.org/10.1038/s41591-018-0264-7 (2019).
Deng, B. et al. AURKA emerges as a vulnerable target for KEAP1-deficient non-small cell lung cancer by activation of asparagine synthesis. Cell Death Dis. 15, 233. https://doi.org/10.1038/s41419-024-06577-x (2024).
Cohen, S. et al. A POLD3/BLM dependent pathway handles DSBs in transcribed chromatin upon excessive RNA:DNA hybrid accumulation. Nat. Commun. 13, 2012. https://doi.org/10.1038/s41467-022-29629-2 (2022).
Chen, J., Smoyer, C. J., Slaughter, B. D., Unruh, J. R. & Jaspersen, S. L. The SUN protein Mps3 controls Ndc1 distribution and function on the nuclear membrane. J. Cell Biol. 204, 523–539. https://doi.org/10.1083/jcb.201307043 (2014).
Li, X. et al. Nuclear receptor subfamily 3 group c member 2 (NR3C2) is downregulated due to hypermethylation and plays a tumor-suppressive role in colon cancer. Mol. Cell. Biochem. 477, 2669–2679. https://doi.org/10.1007/s11010-022-04449-6 (2022).
Walters, T. S. et al. SUMO-modification of human Nrf2 at K(110) and K(533) regulates its nucleocytoplasmic localization, stability and transcriptional activity. Cell. Physiol. Biochem. 55, 141–159. https://doi.org/10.33594/000000351 (2021).
Sun, Y. Y. et al. Identification of NR3C2 as a functional diagnostic and prognostic biomarker and potential therapeutic target in non-small cell lung cancer. Cancer Innov. 3, e122. https://doi.org/10.1002/cai2.122 (2024).
Chen, K., Liu, S., Lu, C. & Gu, X. A prognostic and therapeutic hallmark developed by the integrated profile of basement membrane and immune infiltrative landscape in lung adenocarcinoma. Front. Immunol. 13, 1058493. https://doi.org/10.3389/fimmu.2022.1058493 (2022).
Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 82374288), Chinese Medicine Science and Technology Project of Shandong Province in 2020 (Nos. 2020M108).
Author information
Authors and Affiliations
Contributions
Xiaoping Yang: Methodology, Investigation, Formal analysis, Validation, Writing – original draft. Yongji Liu and Wen Jiang: Validation, Visualization, Formal analysis, Writing – original draft. Xiaochun Liu and Xiaonan Zhang: Methodology, Validation. Huiying Liu: Investigation, Methodology, Validation. Daijun Xing and Keer Wang: Project administration, Resources. Xin Zheng: Project administration, Supervision, Writing—review & editing. Wenqing Jiang: Funding acquisition, Project administration, Supervision, Writing—review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, X., Liu, Y., Jiang, W. et al. Identification of SUMOylation modifiers involved in lung adenocarcinoma progression and Osimertinib resistance by integrated bioinformatics analysis. Sci Rep 15, 31130 (2025). https://doi.org/10.1038/s41598-025-16615-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-16615-z
