
Abstract
Intercellular communication signals in the tumor microenvironment are closely related to behaviors such as cancer cell proliferation and immune evasion. However, the specific roles of intercellular signaling pathways in intrahepatic cholangiocarcinoma (ICC) have not yet been fully characterized. In this study, we analyzed publicly available single-cell RNA sequencing (scRNA-seq) data derived from paired samples of two intrahepatic cholangiocarcinoma (ICC) tissues and two adjacent normal tissues, thoroughly examining their cellular composition. InferCNV analysis was employed to compare tumor cells and normal cells, and pseudotime analysis was used to identify the growth and differentiation trajectories of the cells. Additionally, intercellular communication analysis was conducted to elucidate the communication networks between cells. Our analysis delineated the cellular ecosystem of ICC, identifying cell subclusters with shared characteristics between ICC and normal tissues. Notably, we characterized a distinct C7-E-T subcluster that exhibited high expression of CXCR4 and BPTF, markers associated with cancer stem cells (CSCs). Further investigation revealed that the MIF intercellular signaling pathway promotes the progression of ICC by activating intracellular signals in the MYC pathway. This study highlights the dysregulation of intercellular signaling pathways within tumor clusters, which influences the onset and progression of ICC. The cancer stem cell subpopulation (CXCR4hiBPTFhiE-T) exerts a significant influence on ICC progression by secreting relevant signaling molecules via the MIF signaling pathway.
Similar content being viewed by others


Introduction
ICC is a malignant tumor of the liver characterized by high tumor heterogeneity and poor prognosis. However, there is still a limited understanding of the cellular diversity and molecular basis of interactions between tumor cells and niche cells in the surrounding microenvironment, which hinders the development of targeted therapies for this disease1.
ICC arises from the epithelial cells of intrahepatic and extrahepatic bile ducts2. Current studies primarily focus on the genomic and transcriptomic levels, revealing key gene mutations such as TP53, KRAS, C1orf4 (ARID1A), IDH1/2 mutations, and FGFR gene fusions, as well as aberrant signaling pathways driving ICC pathogenesis3. A prominent feature of ICC is the embedding of tumor cells within a dense stroma, with the tumor microenvironment playing a pivotal role in disease progression, invasion, and metastasis4,5.
Single-cell technologies, particularly single-cell RNA sequencing, play a vital role in studying the tumor microenvironment (TME) and uncovering the etiology and potential mechanisms of human diseases, including cancer6,7. Advances in scRNA-seq technology allow us to compare differences between tumor cells and normal cells, thereby gaining deeper insights into complex intercellular communication networks and related biological processes8,9. Aberrant intercellular communication may lead to uncontrolled cell proliferation, driving tumor initiation and progression10. However, the specific mechanisms by which intercellular communication signals promote the growth of ICC tumor cells remain poorly understood.
Intercellular communication is a central mechanism in processes such as cell fate determination, proliferation, migration, and homeostasis11. One of the key challenges in intercellular communication is the depiction of signaling pathways that regulate cancer12. Numerous studies indicate that signaling pathways mutually regulate each other by triggering a series of intracellular signaling events. In certain tumors, there exist subpopulations of tumor stem cells with self-renewal and differentiation abilities, which are considered major drivers of intra-tumor and inter-tumor heterogeneity13,14. Intercellular communication plays a crucial role in maintaining the state of these CSCs and influencing their differentiation fate within the tumor and in interaction with the surrounding environment15.
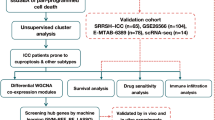
In this study, we utilized an scRNA-seq dataset analyzing a total of 62,944 cells from two ICC tissue samples and two adjacent normal tissue samples. We identified 19 intercellular signaling pathways that were abnormally activated in the tumor cell clusters of ICC. Furthermore, we discovered a cancer stem cell subpopulation capable of secreting aberrant intercellular signaling molecules, providing valuable insights for differentiating cellular mechanisms and developing potential effective immunotherapy strategies.
Methods
scRNA-seq data
The scRNA-seq data from the ICC patients were derived from the GEO database (GSE138709)16. The dataset includes samples from five ICC tissues and three adjacent normal tissues. For further analysis, we selected paired samples of two ICC tissues and two adjacent normal tissues from two patients, extracting high-quality scRNA-seq data.
scRNA-Seq analysis and external validation
We used R software (v.4.4.0) and the Seurat package (v.5.0.3) to read the data from each sample and perform quality control. Cells with gene detection numbers between 200 and 2500 and mitochondrial gene expression below 5% were retained, while low-quality cells were excluded. After quality control, a total of 15,463 high-quality cells from four samples were preserved for subsequent analyses. We normalized the single-cell counts matrix data using the “NormalizeData” function and identified highly variable genes using the “FindVariableFeatures” function. Subsequently, we conducted dimensionality reduction on the top 30 principal components using principal component analysis (PCA), extracting the most significant individual variations in the principal components to uncover key features.
We downloaded and analyzed bulk RNA sequencing data from The Cancer Genome Atlas cholangiocarcinoma (TCGA-CHOL) cohort to validate the expression levels of candidate genes in tumor versus normal samples. Comparative analyses were further performed using the GEPIA web server (http://gepia.cancer-pku.cn/), which integrates transcriptomic data from both the TCGA and GTEx projects.
Identification of signature genes
We utilized the “FindAllMarkers” function and the Wilcoxon rank-sum test (lnFC > 0.25, p < 0.05, and min.pct > 0.1) to calculate differentially expressed genes between cell clusters and other cells.
inferCNV analysis
To detect large-scale copy number variations in tumor cells, we utilized the inferCNV package (v.1.20.0) to compare the gene expression levels of each tumor cell with a set of normal adjacent cells, inferring the chromosomal variation of tumor cell clusters by analyzing gene expression intensities at different genomic locations to explore potential chromosomal alterations17.
Single-cell trajectory analysis
To understand gene expression changes during the process of cell differentiation, we constructed differentiation trajectories of epithelial cells using Monocle 2 (v.2.20.0)18. We performed pseudotime analysis to order individual cells based on biological processes such as differentiation and transition, employing default parameters and the DDRTree algorithm for trajectory construction. Additionally, we analyzed branching fate-determining genes using the Branch Expression Analysis Model (BEAM).
Cell–cell interaction analysis
We used CellChat (v.1.6.1) to analyze intercellular interactions based on the single-cell gene expression matrix19. CellChat integrates prior knowledge of ligands, receptors, and their cofactors to calculate communication probabilities between cells. We aggregated ligand-receptor interactions for each signaling pathway, calculated communication probabilities at the pathway level, and visualized the intercellular communication network. CellChat provides ligand-receptor databases for both humans and mice; this study utilized the "CellChatDB.human" for data import.
Single-cell transcription factor analysis
Through transcription factor analysis (SCENIC)20, the regulatory strength of key transcription factors (TFs) within each candidate cell cluster was evaluated, and potential core regulatory TFs were identified. During the analysis, the SCENIC R package was used to calculate the activity of genes regulated by each TF, and the area under the recovery curve (AUC) values were used to quantify the functions of these TFs. To further compare TF activity differences across cell clusters, differential analysis was performed using the limma R package.
Cell culture
The human intrahepatic cholangiocarcinoma (ICC) cell line HUCCT1 was purchased from Procell Life Science & Technology Co., Ltd. (China). Cells were cultured in RPMI-1640 medium (Procell, China) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. All cultures were maintained at 37 °C in a humidified incubator with 5% CO2.
Cell transfection and quantitative real-time PCR (qRT-PCR)
To achieve BPTF knockdown, HUCCT1 cells were transfected with BPTF-specific small interfering RNAs (siRNAs). The siRNAs, control sequences, and transfection vectors were designed and synthesized by GenePharma Co., Ltd. (Suzhou, China). Transfection was performed using Lipofectamine 3000 reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. Cells were harvested 48 h post-transfection for further analysis. Total RNA was extracted from siRNA-transfected and non-transfected HUCCT1 cells using the HiPure Total RNA Mini Kit (Magen, China), and complementary DNA (cDNA) was synthesized using the PrimeScript™ RT Master Mix (Perfect Real Time) kit (TaKaRa, Japan). mRNA expression was analyzed by quantitative PCR using the GS AntiQ qPCR SYBR Green Fast Mix (Universal) kit (Genesand, China).
In vitro functional assays
Cell viability was evaluated using the Cell Counting Kit-8 (CCK-8) assay. Briefly, 0.5–1.0 × 104 cells suspended in 100 μL complete medium were seeded into 96-well plates. After incubation, 10 μL of CCK-8 solution was added to each well and incubated at 37 °C. Absorbance was measured at 450 nm using a microplate reader. The impact of BPTF knockdown on cell migration was preliminarily assessed using a wound-healing assay. In brief, a scratch was created using a sterile 200-μL pipette tip across a confluent monolayer of cells in a 6-well plate. Detached cells were removed by washing with sterile PBS, and the remaining adherent cells were cultured in serum-free medium. Cell migration into the wound area was imaged and recorded at 24 h.
Multiplex immunofluorescence (mIF) staining
The tumor tissue sections used in this study were provided by the Department of Pathology at our institution. To assess the expression levels of target proteins (e.g., CXCR4, BPTF), tissue samples were processed using multiplex immunofluorescence (mIF) staining. Paraffin-embedded sections were first dewaxed and subjected to antigen retrieval. To minimize nonspecific binding, the sections were blocked with serum. Primary antibodies targeting the genes of interest were then applied at 4 °C and incubated overnight. The following day, after washing, the sections were incubated at room temperature for 1–2 h with fluorescence-labeled secondary antibodies and counterstained with DAPI to label the cell nuclei. Finally, the sections were mounted and imaged using either a fluorescence microscope or a confocal microscope to observe the expression and spatial distribution of the target proteins.
Statistical analyses
In this study, all data were analyzed and visualized using R (v. 4.4.0). The t-test and Wilcoxon test were used to assess the differences between cell clusters, while the AUCell algorithm was employed to calculate transcription factor activity. A p-value of < 0.05 was considered statistically significant in all statistical tests.
Declaration
All methods described in this study were conducted in accordance with the guidelines and regulations of your journal.
Results
Analysis of the transcriptional states of epithelial and tumor cells in ICC and cell type annotation
To investigate the transcriptional states of individual cells in ICC and normal bile duct tissues, we performed a comprehensive analysis of the scRNA-seq data obtained from the GEO database16. We extracted scRNA-seq data from two paired ICC samples and adjacent non-tumor tissue samples for analysis. After integrating the scRNA-seq datasets and performing quality control, we constructed a cellular map consisting of 15,463 individual cells.
Through analysis, these cells were divided into 16 distinct cell clusters (Fig. 1A). We annotated the 16 cell clusters by analyzing the expression patterns of classical cell marker genes21 (Fig. 1B). Based on this marker gene expression, we further classified these 16 clusters into seven distinct cell types, offering a clearer understanding of the cellular composition within the ICC samples (Fig. 1C). In clusters 7, 14, and 15, we observed that tumor cells and epithelial cells in ICC tissues exhibited overlapping marker gene expressions, which created challenges in differentiating between the two cell types (Fig. 1B). Consequently, we temporarily designated clusters 7, 14, and 15 in ICC samples as "epithelial/tumor" cells, while in normal bile duct tissues, clusters 7, 14, and 15 were labeled as epithelial cells (Fig. 1C). Further analysis demonstrated a marked increase in the proportion of "epithelial/tumor" cells in ICC samples when compared to adjacent non-tumor tissue samples (Fig. 1D). These findings suggest a complex relationship between epithelial cells and tumor cells during the progression of ICC.

The expression profiles of 15,463 single cells from paired ICC and adjacent non-tumor tissues. (A) UMAP plots derived from single-cell analysis show the distribution of cells from ICC and normal sample groups (left). Different cell clusters from ICC and normal sample groups are shown (right). (B) Bubble plots of the marker genes expressed in the major cell types. Dot color reflects expression level and dot size represents the percent of cells expressing marker genes in different cell types. (C) UMAP plot of 15,463 cells profiled in the present study colored by major cell types. (D) The proportion of major cell types in the four samples.
Identification of epithelial/tumor cells and chromosomal copy number variation analysis in ICC
To precisely differentiate "epithelial/tumor" cells in ICC tissues, we carried out an in-depth analysis involving 800 "epithelial/tumor" cells from ICC samples and 272 epithelial cells from adjacent normal tissues, totaling 1072 cells. We performed dimensionality reduction analysis using Uniform Manifold Approximation and Projection (UMAP), dividing the cells into 12 distinct subpopulations (Fig. 2A). In normal samples, the cells were categorized into 11 cell clusters (Fig. 2B), while the cells in ICC samples were divided into 10 cell clusters (Fig. 2C). Among these cell clusters, we observed that clusters 0, 1, 2, 3, 5, 6, 7, 9, and 11 were present in both normal and ICC samples. Notably, clusters 8 and 10 were exclusive to normal samples, while cluster 4 was specific to ICC samples. Therefore, we annotated clusters 0, 1, 2, 3, 5, 6, 7, 9, and 11 from normal samples, as well as clusters 8 and 10, as epithelial cells.

Identification of the characteristics of epithelial and tumor cells. (A) UMAP plots from single-cell analysis show the different “epithelial/tumor” cell clusters in the ICC and normal sample groups. (B) UMAP plots from single-cell analysis show the different epithelial cell clusters in the normal sample group. (C) UMAP plots from single-cell analysis show the different "epithelial/tumor" cell clusters in the ICC group. (D) Violin plots depicting the expression of ICC tumor marker genes KRAS and MET across different cell clusters. (E) Violin plots showing the expression of KRAS and MET in the ICC and normal sample groups. (F) UMAP plots illustrating the expression of ICC tumor marker genes KRAS and MET across different cell clusters. (G) Violin plots depicting the expression of ICC tumor marker genes KRAS and MET in different cell clusters. (H) Heatmap showing the results of CNV analysis. The upper heatmap represents the CNV profile of reference cells, while the lower heatmap represents the CNV profile of target cells. Red indicates CNV amplification, blue indicates CNV deletion and the depth of color represents the magnitude of CNV variation. (I) Box plot illustrating the CNV levels across distinct cell clusters (Mann–Whitney U test, *** p < 0.001).
Further analysis revealed that clusters 0, 1, 2, 3, 4, 5, 6, 7, 9, 11, and 12 exhibited elevated expression levels of KRAS and MET (Fig. 2D–F), KRAS and MET are among the most frequently mutated oncogenes in cancer22,23, indicating that the cells in these clusters may possess tumor characteristics. In contrast, clusters 8 and 10 showed typical expressions of epithelial cell markers while lacking any tumor-specific markers, confirming that these clusters correspond to normal epithelial cells (Fig. 2G).
Using InferCNV analysis, we explored the chromosomal copy number variations (CNV) in these cells. By comparing with the 272 epithelial cells from normal bile duct tissues, we inferred chromosomal variations at different genomic locations. The results showed significant levels of CNV elevation in clusters 0, 1, 2, 3, 4, 5, 6, 7, 9, and 11 in ICC samples (Fig. 2H and I). Thus, based on these findings, we annotated clusters 0, 1, 2, 3, 5, 6, 7, 9, and 11, as well as cluster 4, in ICC samples as tumor cells.
Pseudotime analysis of tumor cell differentiation trajectories and activation of key genes
To further explore the differentiation trajectories among different cell subpopulations, we performed pseudotime analysis using Monocle 2, identifying six distinct cell states (Fig. 3A and B). By analyzing fate 1 and fate 2 of the cells, we could observe the developmental trajectory of epithelial cells toward malignant cells. In these trajectories, two cell clusters (8 and 10) represent the differentiation trajectory of epithelial cells, primarily associated with cell states 5 and 6. The differentiation trajectories of the other 10 cell clusters are mainly associated with the differentiation of tumor cells, reflecting the trajectories of cell states 1, 2, 3, and 4. From the pseudotime plot, we can see that cells in normal tissues, especially clusters 8 and 10, are at the starting stage of the differentiation trajectory, while cells in tumor tissues, particularly cluster 4, are at the terminal stage of the differentiation trajectory (Fig. 3C–E). This indicates that the differentiation process of tumor cells drives their malignant transformation. Moreover, gene expression analysis revealed that multiple genes were significantly activated in tumor cells, which are closely related to specific behaviors of tumor cells. For instance, KRAS and MET are associated with cell proliferation and display dynamic expression along the differentiation trajectory from epithelial to malignant cells (Fig. 3F). PIK3CA contributes to anti-apoptotic mechanisms in tumor cells, whereas CDKN2A inhibits cell proliferation and promotes apoptosis (Fig. 3G). The activation of these genes further supports our findings regarding the biological functional changes of tumor cells during the differentiation process.

Pseudotime trajectory of all epithelial cells. (A) Displaying the beginnings and endings of pseudo-time trajectories. The colors from dark to light represent the order of pseudo-time. (B) Differentiation trajectory of epithelial cells, with each color representing a different cell state. (C) Differentiation trajectory of epithelial cells, with each color representing a sample type. (D) Differentiation trajectory of epithelial cells, with each color representing a cluster. (E) Differentiation trajectories for each individual cell cluster. (F) Differentiation trajectories of MET and KRAS. (G) Heatmap showing the expression of genes with significantly increased activity.
Analysis of communication between epithelial tumor cell (E-T) clusters and identification of key signaling pathways
ICC originates from epithelial cells and undergoes complex genomic and epigenetic changes during its transformation into tumor cells. Some early-stage cells retain their original epithelial characteristics24. Based on this, we defined the specific cell populations shared by both normal and tumor samples (clusters 0, 1, 2, 3, 5, 6, 7, 9, and 11) as epithelial tumor (E-T) cells and labeled them sequentially as C0-E-T, C1-E-T, C2-E-T, C3-E-T, C5-E-T, C6-E-T, C7-E-T, C9-E-T, and C11-E-T. These coexisting E-T cell populations may play a pivotal role during the early stages of tumorigenesis and are of significant importance for elucidating the oncogenic mechanisms underlying ICC.
To further investigate the intercellular communication among these E-T clusters, we constructed a communication map between the 12 cell clusters using the CellChat R package25. Through the analysis of these clusters, we have identified signaling pathways that are significantly enriched in both outgoing and incoming signals (Fig. 4A). Among them, the MIF, MK, and SPP1 signaling pathways were the top three contributors (Fig. 4B). Using heatmap analysis, we visualized the centrality scores of the MIF, MK, and SPP1 signaling pathways in different clusters. This reveals that clusters C7 and C10 are the main influencers in the MIF signaling pathway; clusters C2, C5, and C7 are the primary influencers in the MK signaling pathway; and clusters C4 and C7 are the main influencers in the SPP1 signaling pathway (Fig. 4C). By further analyzing the ligand-receptor pairs (L–R pairs) of each signaling pathway, we found that MIF-(CD74 + CXCR4), MK-(MDK + NCL), and SPP1-(SPP1 + CD44) are the most contributive L-R pairs in their respective signaling pathways. These ligand-receptor pairs play a crucial role in facilitating communication among tumor cells and are presented in Fig. 4D and E.

Cellular communication network of the 12 cell clusters. (A) The network centrality scores are displayed via a heatmap, showing the top 19 incoming (left) and outgoing (right) signals across 12 cell clusters. (B) A circle plot displays the complex interactions between different cell populations within the top three signaling pathways. (C) A heatmap displays the top three signaling pathways. (D) The overall contribution of each ligand-receptor pair to the signaling pathways. (E) A circle plot displays the complex interactions between different cell populations for the most important ligand-receptor pairs.
Analysis of major signaling pathways and communication networks between epithelial cells and E-T clusters
Among the 11 key epithelial cell clusters and the E–T cluster, we identified the top three significantly enriched intercellular signaling pathways: MIF, MK, and SPP1 (Fig. 5A and B). These signaling pathways transmit information between cell clusters through specific ligand-receptor pairs, particularly MIF-(CD74 + CXCR4), MK-(MDK + NCL), and SPP1-(SPP1 + CD44) (Fig. 5C). In these pathways, certain cell clusters secrete signaling molecules MIF, MK, and SPP1, while the target cell clusters express their corresponding receptors (CD74, CXCR4, MDK, NCL, and CD44) (Fig. 5D). Through these interactions, a highly complex and dynamic communication network is established between cell clusters11. This process is crucial in the tumor microenvironment, particularly in solid tumors such as ICC, where tumor cells not only rely on intrinsic genomic alterations26 but also continuously interact with surrounding cells to shape the microenvironment and promote tumorigenesis27.

Cellular communication between epithelial cells and E-T clusters. (A)The network centrality scores are displayed in a heatmap, showing the top 3 incoming (left) and outgoing (right) signals across 11 cell clusters. (B) A chord diagram illustrates the complex interactions between cell populations. (C) The overall contribution of each ligand-receptor pair to the signaling pathways. (D) Violin plots show the gene expression of ligand-receptor pairs involved in the three signaling pathways.
Analysis of abnormal signaling pathways and cell communication patterns in ICC tumor clusters
Through the analysis of ten tumor cell subpopulations extracted from ICC samples, we identified 19 abnormally activated intercellular signaling pathways (Fig. 6A). These signaling pathways were grouped based on their functions: GROUP 1: Inflammatory Regulation and Immune Response; GROUP 2: Vascular Function or Angiogenesis Regulation; GROUP 3: Related to Cell Proliferation and Migration; GROUP 4: Related to Growth Factors (Fig. 6B).

Cellular communication between tumor clusters. (A) The network centrality scores are displayed in a heatmap, showing the top 19 incoming (left) and outgoing (right) signals across 10 cell clusters. (B) A scatter plot shows the division of 19 signaling pathways into four groups. (C–E) Figures show the network centrality scores (heatmap), ligand-receptor pairs (bar chart), and gene expression of receptor-ligand pairs (violin plots) for the top three signaling pathways. (F and G) River plots display the communication patterns coordinating secretory cell clusters with outgoing signaling pathways and target cell clusters with incoming signaling pathways.
We focused on analyzing the top three signaling pathways that significantly contribute to the tumor microenvironment—MK, MIF, and SPP1 (Fig. 6C–E). Among them, the main senders of the MIF signaling pathway are the C1-E-T cluster and the C4-Tumor cluster, while the main receiver is the C10-E-T cluster (Fig. 6D). MIF signaling relies on the L-R ligand-receptor pair MIF-(CD74 + CXCR4), where CXCR4 is highly expressed in the C7-E-T, C9-E-T, and C11-E-T clusters, and these clusters widely express receptor and ligand genes throughout the tumor population (Fig. 6D). Using the same analytical approach, we also examined the activities of the MK and SPP1 signaling pathways (Fig. 6C and E). We found that the C7-E-T cluster plays a particularly prominent role in these major signaling pathways, serving as a key influencer across multiple signaling routes.
We further identified different communication patterns between secretory cells and target cells to reveal the complex interactions between cells. For example, output pattern 1 in the C4 tumor cluster coordinates the sending of signals such as SPP1, MIF, VISFATIN, EGF, EDN, and GDF (Fig. 6F); whereas input pattern 4 in the C4 tumor cluster coordinates the reception of signals such as MIF and GALECTIN (Fig. 6G). The analysis of these communication patterns reveals the complex mechanisms of interactions among tumor cell populations, providing insights into the potential roles of different tumor subclusters27.
The critical role of BPTF and CXCR4 in cancer stem cell–driven tumor progression
We used the SCENIC method to study the role of transcription factors (TFs) in tumor progression20,28. We identified 12 transcription factors targeting the MIF-(CD74 + CXCR4) ligand-receptor pair, with BPTF (Bromodomain PHD Finger Transcription Factor) identified as a key regulator of CXCR4 (Fig. 7A). By analyzing the ranking of genes within the module, the height of cumulative curves, and the area under the curve (AUC), we calculated the activity level of BPTF using AUCell (Fig. 7B). The results showed that BPTF expression was significantly increased in tumor tissues compared to normal tissues (Fig. 7C), particularly enriched in the C7-E-T cluster (Fig. 7D). Notably, CXCR4 is an important marker of CSCs29,30, and BPTF plays a critical role in the self-renewal and differentiation of CSCs31,32. The expression of both CXCR4 and BPTF in the C7-E-T cluster further indicates that this cluster possesses CSCs characteristics. Therefore, we designated the C7-E-T cluster as the CXCR4hiBPTFhiE-T subpopulation. Through this analysis, we identified a potential CSCs subpopulation and explored the transcriptional regulatory mechanisms of the MIF signaling molecules secreted by this subpopulation, providing new insights into cell communication and tumor biology during ICC progression.

Transcription factor analysis of tumor clusters. (A) The regulatory network of transcription factors that target genes involved in the MIF-(CD74 + CXCR4) ligand-receptor pair. (B) AUCell quantification of BPTF transcription factor activity. (C) Violin plots showing the expression levels of BPTF across different samples. (D) Box plots displaying BPTF expression across different cell clusters.
Functional validation of BPTF in the malignant progression of ICC
CSCs play a pivotal role in maintaining tumor heterogeneity, promoting disease progression, and conferring therapeutic resistance. To investigate the oncogenic role of the CSC-associated marker BPTF in ICC, we conducted gene knockdown experiments in the ICC cell line HUCCT1 and RBE using three different small interfering RNAs (siRNAs) targeting BPTF. Reverse transcription quantitative PCR (RT-qPCR) confirmed the knockdown efficiency of each siRNA, showing a significant reduction in BPTF mRNA levels (Fig. 8A).Among these, the two siRNAs exhibiting the highest knockdown efficiency, siBPTF-1 and siBPTF-3, were selected and subsequently renamed si-1 and si-2 for further experiments.

BPTF Knockdown Impairs ICC Cell Functions. (A) RT-qPCR analysis demonstrating efficient knockdown of BPTF mRNA in HUCCT1 cells (left) and RBE cells (right) transfected with three distinct BPTF-targeting siRNAs compared to the negative control (NC). *p < 0.05; **p < 0.01; ***p < 0.001. (B) CCK-8 assays showing reduced proliferative capacity of HUCCT1 cells (left) and RBE cells (right) following BPTF knockdown. Cell viability was assessed at 24, 48, 72, and 96 h post-transfection. *p < 0.05; **p < 0.01; ***p < 0.001. (C) Wound healing assays indicating impaired migratory ability of HUCCT1 cells (left) and RBE cells (right) upon BPTF knockdown. Representative images were captured at 0 and 24 h after scratching. (D) Quantification of migration rates in the wound healing assays shown in (C). Bar graphs display data for HUCCT1 cells (left) and RBE cells (right). *p < 0.05; **p < 0.01; ***p < 0.001. (E) Multiplex immunofluorescence (mIF) images illustrating the co-expression of MIF, CD74, CXCR4, and BPTF in intrahepatic cholangiocarcinoma (ICC) tumor tissues and matched adjacent non-tumor tissues. (F) Bar graphs comparing the mean fluorescence intensity of MIF, CD74, CXCR4, and BPTF between normal and tumor tissues. *p < 0.05; **p < 0.01; ***p < 0.001.
To assess the impact of BPTF deficiency on cellular behavior, we first performed a CCK-8 assay to evaluate cell proliferation. The results demonstrated that BPTF silencing significantly suppressed the proliferative capacity of HUCCT1 cells (Fig. 8B). Furthermore, wound-healing assays revealed that BPTF knockdown also impaired the migratory ability of this cell line (Fig. 8C and D). We speculate that the downregulation of BPTF may compromise the tumorigenic potential driven by CSCs, thereby attenuating the proliferative and migratory tendencies of tumor cells. These findings suggest that BPTF may serve as a key driver of ICC progression by maintaining CSC-like properties.
To further validate the conclusions drawn from our single-cell transcriptomic analyses, we collected pathological tissue sections from ten ICC patients treated at our center, including tumor samples and their matched adjacent non-tumor tissues. Multiplex immunofluorescence (mIF) staining was performed to examine the spatial distribution and protein expression levels of MIF, CD74, CXCR4, and BPTF—key signaling axis components identified in the single-cell analysis. The results demonstrated that the co-expression levels of these markers were markedly higher in tumor tissues compared to adjacent normal tissues, providing additional evidence supporting the activation of these critical genes in ICC (Fig. 8E and F and Supplemental Fig. S1A).
The role of the MIF signaling pathway in ICC tumor cells and its regulatory mechanisms of key genes
The MIF cytokine forms a complex with the type II transmembrane receptor CD74 and CXCR4, thereby activating multiple signaling pathways33. To investigate the role of the MIF signaling pathway in tumor cells, we analyzed 1,072 epithelial/tumor cells. The results showed that MYC, CDK4 and ODC1 were significantly overexpressed in tumor cells, and these genes are key members of the MYC pathway (Supplemental Fig. S2A–C). To investigate the potential function of BPTF in tumor cells, we performed data analysis using the Gene Transcription Regulation Database (https://gtrd20-06.biouml.org/). The results indicated that BPTF is a co-regulatory factor essential for MYC function and can modulate MYC transcriptional activity.
We further identified marker genes enriched in the tumor cell clusters (Supplemental Fig. S2D–F), discovering a total of 8 genes that participate in cancer occurrence, development, and metastasis through various pathways and mechanisms. These genes include: FOSL1: promoting tumor cell proliferation; HGF: promoting tumor cell invasion and metastasis; CD55: helping tumor cells evade immune surveillance; CDKN2A: associated with cell cycle regulation and apoptosis; FDCSP: related to immune regulation. The roles of these genes in different cancers vary depending on the signaling pathways and cellular processes involved. Some genes (such as FOSL1) can simultaneously affect multiple malignant phenotypes, indicating their significant roles in cancer progression and potential interactions and cross-effects.
The above research indicates that the MIF signaling pathway may promote malignant phenotypes by activating the corresponding receptors in target cell clusters, such as MYC, CDK4, and ODC1. This finding provides important clues for further understanding the role of MIF in ICC progression and suggests its potential therapeutic target value in regulating tumor cell behavior34.
Discussion
Although we have some understanding of the tumor microenvironment in ICC27,35,36, elucidating the key molecular pathways in tumorigenesis requires complex models to analyze the details of cellular interactions in the tumor microenvironment. The specific mechanisms of signaling pathways regulating ICC remain an unresolved issue. Abnormal intercellular communication may lead to uncontrolled cell proliferation, driving tumorigenesis and progression11. Intercellular interactions are crucial for maintaining the state of tumor stem cell subpopulations and regulating their differentiation fate37. Increasing evidence confirms that epithelial-mesenchymal transition (EMT) plays a critical role in the progression of epithelial tumors from initiation to various stages, and it is expected to become an important direction for novel targeted therapies38. During the transition of epithelial tumors from normal epithelium to tumor cells, some cells exhibit both epithelial and tumor characteristics, which constitute the E-T cell cluster, providing unique insights into the study of tumorigenesis and progression39.
This study aims to provide a panoramic view of single-cell transcriptomics in ICC and investigate a series of signaling pathways that mutually regulate through intracellular signaling events. We identified 19 aberrantly activated signaling pathways in the E-T and tumor cell clusters, with the C7-E-T cluster playing a significant role in intercellular signaling through pathways like MIF, MK, and SPP1. The multifunctional pro-inflammatory protein MIF-related pathways play a key role, as MIF is closely associated with tumorigenesis, angiogenesis, and metastasis in various cancer phenotypes40,41,42. MIF binds to the cell surface CD74, and in certain circumstances, CD74 forms complexes with CXCR2 or CXCR4, enhancing the biological effects of MIF, activating downstream signaling pathways, and regulating the survival, invasion, and metastasis of tumor cells, thereby providing mechanistic support for tumor progression43.
TFs are intracellular proteins that play a key role in regulating gene expression. They control the transcription process by specifically recognizing and binding to particular sequences on DNA44. Through transcription factor analysis, we identified BPTF as a key transcription factor that plays an important role in gene regulation. Some studies indicate that BPTF helps maintain the unique epigenetic state of CSCs31,32.
ICC is a highly aggressive malignancy characterized by pronounced cellular heterogeneity and a complex TME. While previous studies have primarily focused on genetic and epigenetic alterations as the principal drivers of tumorigenesis, these mechanisms alone fail to fully explain the dynamic progression of ICC. Increasing evidence suggests that intercellular communication plays a pivotal role in regulating tumor behavior and therapeutic responses.
CSCs are a small subset of tumor cells with self-renewal and persistent proliferation capabilities, driving tumorigenesis, metastasis, and maintaining tumor heterogeneity45. In-depth exploration of the regulatory mechanisms of CSCs, especially the key signaling pathways involved, can reveal the role of CSCs in maintaining tumor heterogeneity and driving tumor progression46. Through intercellular communication analysis, we found that signaling in the MIF pathway primarily operates through the ligand-receptor pair MIF-(CD74 + CXCR4)43. Studies show that CXCR4 is highly expressed in certain populations of tumor stem cells, which possess strong self-renewal and tumor-forming capabilities29,30. Although components of the MIF pathway have been reported in various malignancies, this study provides the first integrative characterization of its role in ICC at single-cell resolution. Notably, we further linked this extracellular signaling axis to downstream oncogenic programs such as MYC activation, suggesting a potential role for this pathway in bridging external TME cues with intracellular transcriptional reprogramming.
To functionally validate this mechanism, we focused on BPTF, a chromatin remodeling factor highly expressed in the CXCR4hi subpopulation and implicated in the regulation of CSCs. In vitro experiments demonstrated that BPTF knockdown significantly inhibited the proliferation and migration of ICC cells, supporting its role in maintaining stem-like properties and tumor aggressiveness. These results indicate that BPTF may function not only as a downstream effector of MIF signaling but also as a promising therapeutic target.
To enhance the translational relevance of our findings, we conducted multiplex immunofluorescence staining in an independent clinical cohort comprising tumor and paired adjacent non-tumor tissues from ICC patients. This analysis revealed co-expression of MIF, CD74, CXCR4, and BPTF at the protein level within tumor tissues, further supporting the activation of the MIF signaling axis in ICC.
However, we are also fully aware that the current validation has clear limitations: the present experiments only indirectly suggest that the CXCR4hiBPTFhi E-T subpopulation may possess cancer stem cell characteristics such as self-renewal, multipotent differentiation, and high tumorigenicity. We fully acknowledge the necessity of additional experiments to rigorously confirm the stemness properties of this subpopulation. In future studies, we plan to perform CXCR4hi cell sorting and BPTF overexpression or knockdown rescue experiments to further elucidate the mechanistic role of this subpopulation in ICC progression. In parallel, we will focus on investigating its interactions with tumor microenvironment components, such as immune cells and fibroblasts. Future studies should incorporate animal models and larger, multi-center clinical cohorts to evaluate the robustness and clinical feasibility of these findings. These planned efforts will help address the current limitations and provide a more comprehensive understanding of the biological significance and therapeutic potential of this cancer stem cell subpopulation in ICC.
Despite these limitations, the high concordance observed between transcriptomic and proteomic data in independent samples strongly supports the reliability of our conclusions and lays a solid foundation for subsequent functional investigations. Collectively, our findings provide valuable insights into the development of targeted therapeutic strategies against TME- and CSC-related pathways in ICC.
In conclusion, the single-cell transcriptomic atlas reveals the characteristics of the TME in ICC and its adjacent tissues. By studying the key mediators in the MIF signaling pathway, we identified an aberrantly activated CSCs subcluster, namely the CXCR4hiBPTFhiE-T subcluster. The abnormally secreted signaling molecules from this subcluster may provide important clues for further investigation into their biological functions and molecular mechanisms, aiding in the formulation or improvement of treatment strategies for ICC.
Data availability
The data is presented by the public database. The scRNA-seq data used in this study are openly available from the GEO databas (https://www.ncbi.nlm.nih.gov/geo/). Transcriptional regulation analysis from the Gene Transcription Regulation Database (https://gtrd20-06.biouml.org/). In addition, gene expression data and survival analyses based on TCGA-CHOL (The Cancer Genome Atlas—Cholangiocarcinoma) cohort and GEPIA (http://gepia.cancer-pku.cn/) were used to further validate our findings.
References
Wang, J., Liu, S., Cao, Y. & Chen, Y. Overcoming treatment resistance in cholangiocarcinoma: current strategies, challenges, and prospects. Front Cell Dev. Biol. 12, 1408852. https://doi.org/10.3389/fcell.2024.1408852 (2024).
Moeini, A., Sia, D., Bardeesy, N., Mazzaferro, V. & Llovet, J. M. Molecular pathogenesis and targeted therapies for intrahepatic cholangiocarcinoma. Clin. Cancer Res. 22, 291–300. https://doi.org/10.1158/1078-0432.Ccr-14-3296 (2016).
Wei, M. et al. Multiple cellular origins and molecular evolution of intrahepatic cholangiocarcinoma. Cancer Lett. 379, 253–261. https://doi.org/10.1016/j.canlet.2016.02.038 (2016).
Varghese, T. P., John, A. & Mathew, J. Revolutionizing cancer treatment: The role of radiopharmaceuticals in modern cancer therapy. Precis. Radiat. Oncol. 8, 145–152. https://doi.org/10.1002/pro6.1239 (2024).
Shen, G. et al. Bridging chronic inflammation and digestive cancer: The critical role of innate lymphoid cells in tumor microenvironments. Int. J. Biol. Sci. 20, 4799–4818. https://doi.org/10.7150/ijbs.96338 (2024).
Huang, D. et al. Advances in single-cell RNA sequencing and its applications in cancer research. J. Hematol. Oncol. 16, 98. https://doi.org/10.1186/s13045-023-01494-6 (2023).
Ye, B. et al. Single-cell RNA sequencing identifies a novel proliferation cell type affecting clinical outcome of pancreatic ductal adenocarcinoma. Front Oncol. 13, 1236435. https://doi.org/10.3389/fonc.2023.1236435 (2023).
Chen, Y., Anwar, M., Wang, X., Zhang, B. & Ma, B. Integrative transcriptomic and single-cell analysis reveals IL27RA as a key immune regulator and therapeutic indicator in breast cancer. Discov. Oncol. 16, 977. https://doi.org/10.1007/s12672-025-02811-w (2025).
Zhang, J. et al. Single-cell transcriptome sequencing reveals aberrantly activated inter-tumor cell signaling pathways in the development of clear cell renal cell carcinoma. J. Transl. Med. 22, 37. https://doi.org/10.1186/s12967-023-04818-9 (2024).
Wu, Z. et al. Mitochondrial-related drug resistance lncRNAs as prognostic biomarkers in laryngeal squamous cell carcinoma. Discov. Oncol. 15, 785. https://doi.org/10.1007/s12672-024-01690-x (2024).
Su, J. et al. Cell-cell communication: new insights and clinical implications. Signal Transduct. Target Ther. 9, 196. https://doi.org/10.1038/s41392-024-01888-z (2024).
An, L., Yu, R., Han, Y. & Zhou, Z. Decoding the intercellular communication network during tumorigenesis. Cancer Biol. Med. 19, 265–272. https://doi.org/10.20892/j.issn.2095-3941.2021.0558 (2021).
Shimokawa, M. et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 545, 187–192 (2017).
Shibata, M. & Hoque, M. O. Targeting cancer stem cells: a strategy for effective eradication of cancer. Cancers 11, 732 (2019).
Razi, S. et al. The role of tumor microenvironment on cancer stem cell fate in solid tumors. Cell Commun. Signal 21, 143. https://doi.org/10.1186/s12964-023-01129-w (2023).
Zhang, M. et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 73, 1118–1130. https://doi.org/10.1016/j.jhep.2020.05.039 (2020).
Jerby-Arnon, L. et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984-997.e924. https://doi.org/10.1016/j.cell.2018.09.006 (2018).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. https://doi.org/10.1038/nmeth.4402 (2017).
Jin, S. et al. Inference and analysis of cell-cell communication using cell chat. Nat. Commun. 12, 1088. https://doi.org/10.1038/s41467-021-21246-9 (2021).
Aibar, S. et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086. https://doi.org/10.1038/nmeth.4463 (2017).
Song, G. et al. Single-cell transcriptomic analysis suggests two molecularly subtypes of intrahepatic cholangiocarcinoma. Nat. Commun. 13, 1642. https://doi.org/10.1038/s41467-022-29164-0 (2022).
Cecchi, F., Rabe, D. C. & Bottaro, D. P. Targeting the HGF/Met signalling pathway in cancer. Eur. J. Cancer 46, 1260–1270. https://doi.org/10.1016/j.ejca.2010.02.028 (2010).
Johnson, C., Burkhart, D. L. & Haigis, K. M. Classification of KRAS-activating mutations and the implications for therapeutic intervention. Cancer Discov. 12, 913–923. https://doi.org/10.1158/2159-8290.Cd-22-0035 (2022).
Choi, J. H. & Thung, S. N. Recent advances in pathology of intrahepatic cholangiocarcinoma. Cancers 16, 1537 (2024).
Shao, X., Lu, X., Liao, J., Chen, H. & Fan, X. New avenues for systematically inferring cell-cell communication: Through single-cell transcriptomics data. Protein Cell 11, 866–880. https://doi.org/10.1007/s13238-020-00727-5 (2020).
Warren, E. A. K. & Maithel, S. K. Molecular pathology for cholangiocarcinoma: A review of actionable genetic targets and their relevance to adjuvant & neoadjuvant therapy, staging, follow-up, and determination of minimal residual disease. Hepatobiliary Surg. Nutr. 13, 29–38. https://doi.org/10.21037/hbsn-22-563 (2024).
Fabris, L., Sato, K., Alpini, G. & Strazzabosco, M. The tumor microenvironment in cholangiocarcinoma progression. Hepatology 73(Suppl 1), 75–85. https://doi.org/10.1002/hep.31410 (2021).
Van de Sande, B. et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 15, 2247–2276. https://doi.org/10.1038/s41596-020-0336-2 (2020).
Hermann, P. C. et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1, 313–323. https://doi.org/10.1016/j.stem.2007.06.002 (2007).
Gelmini, S., Mangoni, M., Serio, M., Romagnani, P. & Lazzeri, E. The critical role of SDF-1/CXCR4 axis in cancer and cancer stem cells metastasis. J. Endocrinol. Invest. 31, 809–819. https://doi.org/10.1007/BF03349262 (2008).
Zhao, X. et al. BPTF promotes hepatocellular carcinoma growth by modulating hTERT signaling and cancer stem cell traits. Redox Biol 20, 427–441. https://doi.org/10.1016/j.redox.2018.10.018 (2019).
Frey, W. D. et al. BPTF maintains chromatin accessibility and the self-renewal capacity of mammary gland stem cells. Stem Cell Rep. 9, 23–31. https://doi.org/10.1016/j.stemcr.2017.04.031 (2017).
Klasen, C. et al. MIF promotes B cell chemotaxis through the receptors CXCR4 and CD74 and ZAP-70 signaling. J. Immunol. 192, 5273–5284 (2014).
Wang, Q. et al. IL1RN and PRRX1 as a prognostic biomarker correlated with immune infiltrates in colorectal cancer: Evidence from bioinformatic analysis. Int. J. Genom. 2022, 2723264. https://doi.org/10.1155/2022/2723264 (2022).
Wang, J. & Ilyas, S. Targeting the tumor microenvironment in cholangiocarcinoma: Implications for therapy. Exp. Opin. Investig. Drugs 30, 429–438. https://doi.org/10.1080/13543784.2021.1865308 (2021).
Li, Z., Li, J., Bai, X., Huang, X. & Wang, Q. Tumor microenvironment as a complex milieu driving cancer progression: A mini review. Clin. Transl. Oncol. 27, 1943–1952. https://doi.org/10.1007/s12094-024-03697-w (2025).
Ebrahim, T., Ebrahim, A. S. & Kandouz, M. Diversity of intercellular communication modes: A cancer biology perspective. Cells https://doi.org/10.3390/cells13060495 (2024).
Meng, K. & Lu, H. Clinical application of high-LET radiotherapy combined with immunotherapy in malignant tumors. Precis. Radiat. Oncol. 8, 42–46. https://doi.org/10.1002/pro6.1225 (2024).
Li, K. H. et al. Clinical outcomes of multisite moderate to high dose radiotherapy for patients with metastatic melanoma. Precis. Radiat. Oncol. 8, 62–69. https://doi.org/10.1002/pro6.1224 (2024).
Chen, Y. et al. Prognostic value of preoperative modified Glasgow prognostic score in predicting overall survival in breast cancer patients: A retrospective cohort study. Oncol. Lett. 29, 180. https://doi.org/10.3892/ol.2025.14926 (2025).
Lue, H. et al. Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene 26, 5046–5059. https://doi.org/10.1038/sj.onc.1210318 (2007).
Wang, Q., Wang, H., Ding, Y., Wan, M. & Xu, M. The role of adipokines in pancreatic cancer. Front Oncol. 12, 926230. https://doi.org/10.3389/fonc.2022.926230 (2022).
Penticuff, J. C., Woolbright, B. L., Sielecki, T. M., Weir, S. J. & Taylor, J. A. MIF family proteins in genitourinary cancer: Tumorigenic roles and therapeutic potential. Nat. Rev. Urol. 16, 318–328. https://doi.org/10.1038/s41585-019-0171-9 (2019).
Zeng, X. et al. Effects of micronutrients and macronutrients on risk of allergic disease in the European population: A Mendelian randomization study. Food Agric. Immunol. 35, 2442369 (2024).
Yadav, A. K. & Desai, N. S. Cancer stem cells: Acquisition, characteristics, therapeutic implications, targeting strategies and future prospects. Stem Cell Rev. Rep. 15, 331–355. https://doi.org/10.1007/s12015-019-09887-2 (2019).
Li, Y. R., Fang, Y., Lyu, Z., Zhu, Y. & Yang, L. Exploring the dynamic interplay between cancer stem cells and the tumor microenvironment: Implications for novel therapeutic strategies. J. Transl. Med. 21, 686. https://doi.org/10.1186/s12967-023-04575-9 (2023).
Funding
This study was financially supported by the open project of Xinjiang Uygur Autonomous Region Key Laboratory (2022D04021), the Xinjiang Uygur Autonomous Region Natural Science Foundation (2022D01C290) and the Tianchi Young Talent Doctoral Program (2022TCYCDXG).
Author information
Authors and Affiliations
Contributions
Xiaogang Dong contributed to study concept and design. Xiaowei Gu and Fuzhong Liu collected data. Junfeng Zhang, Nigeerayi Nuermaimaiti, and Wenjia Guo contributed to analyze the data. Xiaowei Gu and Fuzhong Liu contributed to prepare the manuscript. Xiaogang Dong provided critical feedback on methods, supervised the study. All authors approved the final version to be published.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
This study was approved by the Ethics Committee of Affiliated Cancer Hospital of Xinjiang Medical University (Approval No. K-2024217) in accordance with the Declaration of Helsinki, and written informed consent was obtained from all participants or their legal guardians.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gu, X., Liu, F., Zhang, J. et al. Single-cell transcriptome sequencing reveals tumor stem cells and their molecular characteristics in intrahepatic cholangiocarcinoma. Sci Rep 15, 31170 (2025). https://doi.org/10.1038/s41598-025-17102-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-17102-1
