
Abstract
Amphoterin-induced gene and open reading frame 2 (AMIGO2) was previously identified as a driver gene for liver metastasis. AMIGO2 has also been found to be a prognostic factor for cancer patients with a low frequency of spontaneous liver metastasis. Here, we investigated whether AMIGO2 expression affects the acquisition of cancer stem cell-like properties in cancer cells by using human gastric (MKN45) and colon (DLD-1) cancer cell lines. Knockdown of AMIGO2 expression in MKN45 and DLD-1 cells via transfection with short hairpin RNA-AMIGO2 suppressed cell proliferation under several starvation conditions in two-dimensional culture. AMIGO2 knockdown cells showed reduced ability to form multicellular spheres in a three-dimensional culture. Half-maximal inhibitory concentration values of anticancer drugs in sphere-forming cells were decreased by AMIGO2 knockdown. Silencing of AMIGO2 suppressed tumor formation and growth of subcutaneously injected tumors in NOD/SCID mice. Expression levels of AMIGO2 and cancer stem cell markers, such as CD44, CD133, and EpCAM, were almost identical in spherically grown cancer cells. AMIGO2 expression was positively correlated with expression of existing cancer stem cell markers in multiple organ cancer cell lines. These results demonstrate that AMIGO2 accelerates the malignant progression of human cancer cells by inducing a cancer stem cell-like phenotype.
Similar content being viewed by others

Introduction
More than 90% of cancer deaths are not due to the primary tumor growth, but by cancer cells undergo progression that acquires various malignant phenotypes, such as lethal aggressive growth, therapy resistance, survival under low nutrition/hypoxia conditions, recurrence, and metastasis1,2. The process of acquiring malignant phenotype that accelerate cancer cell progression overlaps in many ways with the gain of malignant functions that have been elucidated in so-called cancer stem cells (CSCs). CSCs are a small subpopulation that often accounts for less than 1% of tumor cells, but they have the ability to self-renew, differentiate, form tumors, and respond poorly to therapy, and play a critical role in driving tumor heterogeneity3,4. CSCs were first identified as leukemia stem cells, a subpopulation of tumor cells with unlimited proliferation capacity5. Subsequent studies have revealed the presence of CSCs in a variety of solid tumors, including brain tumors6, head and neck cancer7, breast cancer8, liver cancer9, colorectal cancer10, and melanoma11. A small number of CSCs often grow into large and aggressive tumor masses12,13, demonstrating that CSCs acquire highly malignant tumor properties12,14. Therefore, the presence of CSCs directly leads to poor prognosis for cancer patients. Although CSCs-related biomarkers have been discovered, the search for biomarkers that are common and specific to CSCs and can be used as therapeutic targets has been ongoing for decades but remains a major challenge3,15,16.
Amphoterin-induced gene and open reading frame 2 (AMIGO2), a member of the AMIGO family, is a transmembrane protein that is expressed in the fiber tracts of neuronal tissues and participates in their formation17. In our previous study, we identified AMIGO2 as a driver of liver metastasis by establishing a mouse liver metastasis model18. AMIGO2-mediated liver metastasis has also been extrapolated to human gastric19 and colorectal20,21 cancers. Overexpression of AMIGO2 has been reported in various types of cancer, including gastric cancer19,20,21,22,23, pancreatic cancer24, colorectal cancer20,21,25, breast cancer26, ovarian cancer27, endometrial cancer28, bladder cancer29, melanoma30,31, and pituitary neuroendocrine tumors32. High AMIGO2 expression has been shown to act as a prognostic factor in patients with cancer types, such as ovarian cancer27, endometrial cancer28, bladder cancer29, and pituitary neuroendocrine tumor32, in which spontaneous metastasis to the liver is rarely observed. Therefore, we hypothesized that the essential function of AMIGO2 is to accelerate the progression of cancer cells into more malignant ones, in addition to being involved in liver metastasis.
Knockdown of AMIGO2 expression in AMIGO2-expressing human gastric and colon cancer cell lines suppressed typical malignant properties characterize of CSCs, including two- and three-dimensional culture growth, growth under starvation conditions, anticancer drug resistance, and in vivo growth. AMIGO2 expression was positively correlated with the expression of known CSC markers in cell lines derived from four organ cancers. This study revealed that AMIGO2 functions as a novel CSC-like molecule in cancer cells and may be a therapeutic target for malignant cancer cells.
Results
Silencing AMIGO2 suppresses cancer cell proliferation following reduced serum, glucose, and oxygen concentrations
To elucidate the functional role of AMIGO2 in tumor malignancy, we introduced the pLKO1 expression vector with or without shRNA into the MKN45 gastric cancer and DLD-1 colon cancer cell lines 33. As shown in Supplementary Fig. S1a and S1c, four shAMIGO2 clones (1–1, 1–2, 2–1, and 2–2) with reduced AMIGO2 protein expression, and two empty vector-transfected clones (shControl clone 1 and clone 2) were isolated from MKN45 cells. Four shAMIGO2 clones (1–1, 1–2, 2–1, and 2–2) with reduced AMIGO2 expression and two shControl clones (clone 1 and clone 2) were also isolated from DLD-1 cells (Fig. S1b and S1d).
We examined how AMIGO2 expression affects cell survival and proliferation. In AMIGO2 -silenced MKN45 and DLD-1 cells, cell growth rates were reduced under 10% serum (growth factor-rich) conditions compared with parental and empty vector-transfected cells (Fig. 1a, c). When maintained in 2% serum (growth factor-limited), AMIGO2 -silenced transfectants did not grow or were suppressed, whereas AMIGO2 -expressing parental and vector control cells grew (Fig. 1b,d). To create a hypoxic environment similar to the cancer tissue microenvironment, cells were cultured at 1% O2, following the previous definition of tumor hypoxia (median pO2 < 10 mm Hg, approximately 1.25% O2)34. AMIGO2-silenced cells showed suppressed proliferation under both 10% (Fig. S2a and S2c) and 2% (Fig. S2b and S2d) serum concentrations. Furthermore, when cells were maintained in glucose-free medium under normoxic conditions (21% O2), cell proliferation was suppressed regardless of AMIGO2 expression (Fig. S3a and S3c). On the other hand, when maintained in glucose-free medium under hypoxic conditions (1% O2), cell proliferation of AMIGO2-silenced cell lines was suppressed compared to parental and vector control cells (Fig. S3b and S3d). These results suggest that AMIGO2 expression is involved in the acquisition of cancer cell survival and proliferation under starvation conditions, such as low growth factors, low glucose, and low oxygen.

AMIGO2 silencing suppresses cell proliferation at 10% and 2% serum concentrations MKN45 (a and b) and DLD-1 (c and d) cells and their transfectants were seeded in a 96-well plate at a density of 1 × 103 cells/well and incubated for 7 days. Two types of culture media were prepared, with serum concentrations of 10% (a and c) and 2% (b and d). Cell proliferation was measured every other day from days 1–7 after seeding. The mean ± standard deviation (s.d.) of minimum n = 3 independent experiments is displayed (*p < 0.05 vs parental cancer cells, Dunnett’s test).
AMIGO2 expression is necessary for sphere formation from single cancer cells in three-dimensional cultures
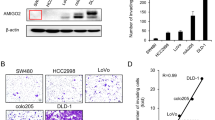
We investigated the role of AMIGO2 in three-dimensional cell growth, from single cancer cells to sphere formation35. Cells were seeded in a low-attachment plate with methylcellulose to prevent them from adhering to each other36. AMIGO2-knockdown MKN45 cells had a reduced ability to form spheres up to 34–62% compared to that of the parental cells (Fig. 2a). AMIGO2-knockdown DLD-1 cells also showed reduced ability to form spheres up to 13–70% compared to that of the parental cells (Fig. 2c). Typical images of spheres formed by MKN45 (Fig. 2b) and DLD-1 (Fig. 2d) cells and their transfectants are shown. Next, we investigated the correlation between the amount of AMIGO2 protein in cancer cells and their sphere-forming ability and found a significant positive correlation in both MKN45 cells (r = 0.8731, p = 0.0102; Fig. S4a) and DLD-1 cells (r = 0.9368, p = 0.0018; Fig. S4b). These results indicate that AMIGO2 is required for the 3-dimensional sphere-forming ability of cancer cells.

AMIGO2 expression is required for self-renewing sphere formation from single cancer cells in three-dimensional cultures MKN45 (a and b) and DLD-1 (c and d) cells and their transfectants were suspended in a medium containing 1.4% methylcellulose and plated in a 96-well plate (1 × 103 cells/well). The number of spheres greater than 50 μm in diameter was counted (a and c). The mean ± s.d. of n = 6 independent experiments is shown. The spheres were photographed (b and d) for day 12 for MKN45 cells and day 10 for DLD-1 cells after seeding. Representative images were presented from three experiments with similar results. Scale bars = 200 μm.
Knockdown of AMIGO2 expression increases drug sensitivity
One of the hallmarks of tumor progression is the generation of subpopulations that are resistant to chemotherapy37. To determine whether AMIGO2 was involved in chemoresistance, we measured the half-maximal inhibitory concentration (IC50) of anticancer drugs using a cancer cell sphere-forming assay. IC50 values of AMIGO2-knockdown clones derived from MKN45 cells for cisplatin (CDDP) ranged from 5.5 to 6.5 nM, an average decrease of 33% compared to 8.9 nM for parental cells (p < 0.05; Fig. 3a). Similarly, the IC50 values of AMIGO2 -knockdown DLD-1 cells for oxaliplatin (L-OHP) ranged from 1.1 to 1.4 µM, an average decrease of 46% compared to 2.3 µM for parental cells (p < 0.05; Fig. 3b). These results suggest that AMIGO2 enhances the survival of cancer cells under antitumor drug treatments and is responsible for their acquisition of the chemoresistance phenotype.

Knockdown of AMIGO2 expression increases drug sensitivity MKN45 (a) and DLD-1 (b) cells and their transfectants were seeded using procedures described in Fig. 2 to induce spherically grown tumors with CDDP (cisplatin, a) or L-OHP (oxaliplatin, b), respectively. IC50 values were determined by the number of spheres and shown in the table. Statistical differences were calculated using Dunnett’s test and compared to parental cells. The mean ± s.d. of minimum n = 3 independent experiments is shown.
AMIGO2 silencing suppresses tumorigenesis
Next, we evaluated the impact of AMIGO2 silencing on the tumorigenicity of cancer cells. MKN45 and DLD-1 cells, and their derived control shRNA and shRNA-AMIGO2 transfectants, were subcutaneously injected into immunodeficient NOD/SCID mice. Tumor size was monitored daily, and the tumors were excised for examination on day 15. Parental MKN45 cells and control shRNA-transfected cells grew to a mean tumor volume of over 34 mm3 by day 15 post-implantation, whereas shRNA-AMIGO2-transfected cells suppressed tumor growth, with a mean tumor volume of less than 18 mm3 (Fig. 4a). Significant suppression of tumor volume by shRNA-AMIGO2 transfected cells was also observed in DLD-1 cells (p < 0.05; Fig. 4d). Tumor growth curves (Fig. 4a, d) and tumor weights (Fig. 4b, e) of AMIGO2-silenced MKN45 cells and DLD-1 cells were also significantly smaller than those of the the parental and control vector-transfected cells.

AMIGO2 silencing suppresses tumorigenesis in vivo in MKN45 and DLD-1 cells Different numbers of MNN45 cells (a–c) and DLD-1 cells (d–f) and their transfectants were injected subcutaneously into non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice and tumor diameters were monitored daily (a and d). Fifteen days after injection, the tumors were excised and weighed (b and e). Macroscopic images of excised tumors were shown in c and f. Scale bars = 10 mm (c and f). Data are represented as the mean ± s.d. of minimum n = 6. Tumor volume and weight were calculated using Dunnett’s test and compared to parental cells.
The resected subcutaneously grown tumors are shown in Fig. 4c, f. The AMIGO2 -silenced subcutaneous tumors were whitish in color compared to the parental and control vector-transfected cells.
Calculations were performed to determine the number of tumor-initiating cells (TIC) of AMIGO2-silenced MKN45 and DLD-1 cells. TIC frequency of parental cell lines was 1:81,279 (MKN45) and 1:108,183 (DLD-1), and the frequency of control shRNA transfectants were comparable to those of the parental cells. In contrast, the TIC frequency of AMIGO2-knockdown cells was significantly increased, ranging from 1:847,909 to 1:1,176,603 (MKN45) and 1:352,837 to 1:1,288,513 (DLD-1) (Table 1). Knockdown of AMIGO2 required 10.4–14.5-fold higher cell numbers in MKN45 cells and 3.3–11.9-fold higher cell numbers in DLD-1 cells to form tumors. This further verifies the crucial role of AMIGO2 in determining the tumorigenic properties in vivo.
Subcellular localization of AMIGO2 and known cancer stem cell markers in spherically growing cancer cells
CD44 and EpCAM have been identified as common cancer stem cell markers in gastric cancer38, and CD44 and CD133 in colorectal cancer39. Therefore, we analyzed the localization of these known cancer stem cell markers and AMIGO2 expression in spherically growing cancer cells by immunocytochemical staining. Figure 5 showed that CD44 and EpCAM in MKN45 spheres were mainly expressed on the cell membrane, whereas AMIGO2 was mostly expressed in the cytoplasm. In DLD-1 spheres, CD44 and CD133 were also detected on the cell surface, and AMIGO2 was predominantly detected in the cytoplasm. Although the localization of AMIGO2 in spherical cancer cells differs from the known cell membrane-localized CSC markers, cancer cells expressing AMIGO2 are consistent with those expressing known CSC markers, suggesting that AMIGO2 serves as an independent malignant progression marker for cancer cells.

Differential subcellular localization of cancer stem cell markers and AMIGO2 expression in spherically proliferating cancer cells Immunocytochemical staining of serial sections of the cancer stem cell markers, CD44, CD133, EpCAM, and AMIGO2, in spheres formed from MKN45 and DLD-1 cells. Nuclei were counter stained with 4′,6-diamidino-2-phenylindole (DAPI). Representative images were shown from three independent immunocytochemical staining with similar results. Scale bar = 50 μm.
Co-expression of AMIGO2 with cancer stem cell markers in multiple organ cancer-derived cell lines
Next, we examined the correlation between AMIGO2 and known CSC markers by mRNA expression using cell lines derived from four organ cancers. AMIGO2 expression was significantly positively correlated with CD44v6 (Fig. 6a) and CD44v9 (Fig. 6b) in gastric cancer cell lines, CD166 (Fig. 6d) in colorectal cancer cell lines, CD44v9 (Fig. 6e) and EpCAM (Fig. 6f) in pancreatic cancer cell lines, and CD44v9 (Fig. 6g) and CD47 (Fig. 6h) in bladder cancer cell lines. CD44v9 expression in colorectal cancer cell lines (Fig. 6c) tended to be positively correlated with AMIGO2 expression, but the difference was not significant.These results suggest that AMIGO2 expression may be a common marker for cancer stem cell-like cells in various organ cancers.

Correlation between AMIGO2 expression and cancer stem cell marker expression in cancer cell lines derived from multiple organ cancers The correlation between AMIGO2 expression and cancer stem cell marker expression in cancer cells was analyzed using Pearson’s correlation coefficient. Expression was analyzed by qRT-PCR (n = 3) for cancer stem cell markers CD44v6 (a) and CD44v9 (b) in gastric cancer cell lines, CD44v9 (c) and CD166 (d) in colorectal cancer cell lines, CD44v9 (e) and EpCAM (f) in pancreatic cancer cell lines, and CD44v9 (g) and CD47 (h) in bladder cancer cell lines.
Preclinical study of AMIGO2 expression inhibition and suppression of cancer stem cell-like malignant phenotypes using clinically approved drug
We previously demonstrated that the clinically approved drug ruxolitinib prevents liver metastasis by pharmacologically suppressing AMIGO2 expression in cancer cells50. We investigated whether ruxolitinib could suppress the cancer stem-like malignant phenotype by inhibiting AMIGO2 expression. Treatment of MKN45 cells or DLD-1 cells with ruxolitinib reduced AMIGO2 expression (Fig. S5a and S5b). Ruxolitinib treatment reduced the number of spheres formed (Fig. S5c and S5d). Furthermore, ruxolitinib suppressed the expression of known cancer stem cell markers, such as CD44v6 (Fig. S5e) and CD44v9 (Fig. S5f.) in MKN45 cells and CD44v9 (Fig. S5g) and CD166 (Fig. S5f.) in DLD-1 cells. In a preclinical study, we then investigated whether ruxolitinib has a therapeutic effect on the subcutaneous growth of AMIGO2-expressing cancer cells. Subcutaneous growth curves of both MKN45 cells (Fig. S6a) and DLD-1 cells (Fig. S6c) showed that daily oral administration of ruxolitinib inhibited tumor growth, but no significant differences were observed. However, tumor weight was significantly suppressed in both cancer cell lines (Fig. S6b and S6d).
As shown in Figure S7, knockdown of AMIGO2 suppressed TGF-β receptor 1 expression in both MKN45 cells (Fig. S7a) and DLD-1 cells (Fig. S7b). Furthermore, a positive correlation between AMIGO2 expression and TGF-β receptor 1 expression in cancer cells was confirmed in MKN45 cells (Fig. S7c) and DLD cells (Fig. S7d). Knockdown of AMIGO2 in cancer cells suppresses cancer stem cell-like malignant traits, suggesting the involvement of TGF-β signaling as one of the molecular mechanisms.
Discussion
In this study, we found that AMIGO2 exerts CSC-like functions, such as inducing tumor cell proliferation under starvation conditions (low growth factor, low glucose, and low oxygen), promoting the formation of multicellular spheres from single tumor cells under three-dimensional culture conditions, acquiring resistance to antitumor drugs, and accelerating tumor development. Furthermore, AMIGO2 expression was positively correlated with the expression of multiple known CSC markers. This study demonstrated using preclinical cell models that AMIGO2 molecule confers CSC-like functions and may represent a novel biomarker as a therapeutic target for malignantly progressed tumor cells.
Using repeated in vivo selection procedures, we previously established a tumor cell line with high tropism for liver metastasis from a murine fibrosarcoma cell line that rarely causes liver metastasis, facilitating the identification of AMIGO2 as the driver gene for liver metastasis18. We revealed that AMIGO2-induced liver metastasis is due to adhesion between tumor cells and hepatic sinusoidal endothelial cells, which is the first step in distant hematogenous metastasis, i.e., specific binding by homophilic or heterophilic binding between tumor cells expressing AMIGO2 and hepatic sinusoidal endothelial cells expressing AMIGO family proteins (AMIGO1, AMIGO2, and AMIGO3)18. Extrapolation to human cancer was validated by showing a significant correlation between the high expression of AMIGO2 in primary tissues of gastric cancer19 and colorectal cancer20,21 with liver metastases. We also found that the prognosis of these AMIGO2-high-expressing patients with cancer was significantly worse than that of AMIGO2-low-expressing patients19,20,21. Furthermore, high expression levels of AMIGO2 in the primary tumor tissues of ovarian cancer27, endometrial cancer28, bladder cancer29, and pituitary neuroendocrine tumor32, which rarely cause liver metastasis, did not correlate with liver metastasis but was correlated with worse patient prognosis. Moreover, AMIGO2 expression is involved in the acquisition of peritoneal dissemination in ovarian cancer27, growth and survival of melanoma31, and epithelial–mesenchymal transition (EMT) in prostate cancer40 and colorectal cancer41. Our results combined with those of others suggest that AMIGO2 not only functions as a driver of liver metastasis but also exerts CSC-like functions that directly promote the malignant progression of cancer cells. To the best of our knowledge, this is the first report that AMIGO2 is involved in the malignant progression of cancer cells by possessing CSC-like function.
Although a definitive marker molecule for identifying CSCs has not yet been established, several surface markers such as CD44 and EpCAM, and transcription factors such as SOX2 and OCT4 have been confirmed as markers of CSCs42. Six attempts have been made to target and eradicate CSCs as new cancer treatment strategies42. i) Targeting by antibody therapy against cell surface markers expressing CSCs43. ii) Drug-induced inhibition of CSC-related signaling pathways44. iii) Drug therapy targeting metabolism characteristic quiescent CSCs45. iv) Epigenetic therapy with demethylase or histone deacetylase inhibitors to deplete CSCs46,47. v) Antagonist therapy against secreted proteins or receptors that form the CSC protective niche48. vi) Chimeric antigen receptor (CAR)-T cell immunotherapy targeting CSC surface markers49. In addition to these six promising attempts, CSC therapy for cancer cells with high AMIGO2 expression requires the development of drugs that can specifically and persistently suppress AMIGO2 expression by gene transfer, such as the short hairpin RNA-AMIGO2 shown in this study. In this regard, through screening of small molecule compounds and drug repositioning, we identified ruxolitinib, a drug that suppresses AMIGO2 mRNA/protein expression in cancer cells and inhibits adhesion to hepatic endothelial cells to prevent liver metastasis50. Ruxolitinib, a clinically available JAK-STAT pathway inhibitor, stably suppresses AMIGO2 for a long period of time50 and may therefore be able to eliminate various malignant cancer cells with high AMIGO2 expression. It is noteworthy that conventional cancer stem cell markers derived from single organ cancers required specific therapeutic drugs for this marker. Therefore, specific therapeutic drugs are required for cancer stem cells in each organ. Because AMIGO2 is expressed in many types of organ cancers, administration of drugs that inhibit AMIGO2 expression, such as ruxolitinib may offer a therapeutic option for cancer cells that have acquired cancer stem cell-like malignant traits, as shown in Fig. S5 and S6.
Among cancer cells derived from the same organ, some express AMIGO2 strongly, while others express it weakly33. Molecular mechanisms underlying the malignant characteristics of cancer cells can be divided into AMIGO2-dependent and AMIGO2-independent mechanisms. In AMIGO2-dependent cancer cells, the maintenance of the CSC-like phenotype is tightly controlled by AMIGO2 expression, whereas in AMIGO2-independent cancer cells, alterations in other carcinogenesis-related gene are probably involved. Our results showed that suppression of AMIGO2 expression in AMIGO2-high-expressing cancer cells resulted in the loss of the malignant phenotype. Because AMIGO2-dependent malignant cancer cells are the therapeutic target, it is necessary to distinguish between AMIGO2-dependent and AMIGO2 -independent cancers when administering molecular targeted therapy.
This study has four limitations that limit its ability to evaluate AMIGO2 as a cancer stem cell marker.First, it lacks the functional analysis necessary to identify AMIGO2 as a novel CSC marker. Further validation should address the following five points: i) whether a subpopulation of stem-like cancer cells that have acquired various malignant characteristics due to AMIGO2 expression can be detected or enriched in the side population fractionated by flow cytometry. ii) whether AMIGO2-expressing cells display representative CSC metabolic features, such as high aldehyde dehydrogenase 1 activity 51. iii) whether the spheres generated in this study have ability to self-renewal by serial subculture and further by anti-aggregation treatment. iv) whether tumorigenicity is maintained through serial transplantation, in which transplanted tumors are recovered and repeatedly transplanted into new mice. v) whether lineage tracing and transcriptional profiling of cancer cells with different AMIGO2 expression levels are consistent with known CSC profiles using scRNA-seq and other methods. Second, AMIGO2 as a common driver molecule conferring CSC-like properties, including metastasis, needs to be verified in mouse autologous carcinogenesis models, orthotopic tumor models, patient-derived xenograft models, and other human cancer types. Third, AMIGO2 expressing cancer cells induces EMT41 and promotes adhesion to liver endothelial cells18,33, the extent to which these functions contribute to CSC-like functions remains to be elucidated. Fourth, it remains to be determined whether forced expression of AMIGO2 in cancer cells that do not express AMIGO2 confers the cancer stem cell-like malignant phenotypes shown in this study.
The AMIGO family genes (AMIGO1, AMIGO2, and AMIGO3) encoding type I transmembrane proteins containing transmembrane domains that have been reported to localize to the cell membrane17. These AMIGOs were first shown to bind to each other and function as intercellular adhesion molecules for axon tract formation during development17. AMIGO2 localizes to the cell membrane surface and functions as a scaffolding protein to bind to several cell membrane proteins, such as the β subunit of the voltage-gated potassium channel (Kv) superfamily52, kinase-dead receptor pseudo-tyrosine kinase (PTK7)31, and 3-phosphoinositide-dependent kinase 1 (PDK1). For example, in endothelial cells, AMIGO2 has been reported to physically interact with PDK1, localizing it to the cell membrane and acting as an activator of Akt53. However, immunocytochemical staining of AMIGO2 in spherically grown cancer cells revealed that AMIGO2 was partially expressed on the cell membrane, with its expression mainly in the cytoplasm and nucleus (Fig. 5). In gastric19 and colorectal20,21 cancers, we observed that the intercellular localization of AMIGO2 changed from the cell membrane to the cytoplasm, which was also demonstrated in another study in melanoma31. Moreover, analysis of the subcellular localization of AMIGOs based on UniProtKB/Swiss-Prot information using the human gene database, GeneCards (https://www.genecards.org/), showed that AMIGO1 and AMIGO3 were localized to the cell membrane, whereas AMIGO2 was detected in the cell membrane, nucleus, cytosol, and Golgi apparatus (https://www.genecards.org/cgi-bin/carddisp.pl?gene=AMIGO2#localization). Based on our current data, the cytoplasmic/nuclear localization of AMIGO2 appears to be necessary for maintaining malignant potential of cancer cells. In particular, AMIGO2 expression has been shown to be localized to the nucleus in cancer cells invading toward the invasive front in tumor tissues29,41. These findings, combined with partial evidence that AMIGO2 expression correlates with TGF-β receptor 1 expression (Fig. S7), suggest that nuclear localization of AMIGO2 may contribute to cancer cell invasion via EMT mediated by the TGF-β signaling pathway41. However, the detailed molecular mechanisms remain to be elucidated. Increased nuclear expression of AMIGO2 in cancer cells may be a novel indicator of malignant potential in the future.
In conclusion, our data revealed that AMIGO2 expression conferred malignant potential in two human cancer cell lines. Since these malignant properties are consistent with those of CSCs2, AMIGO2 appears to be a novel regulator of multiple cancer-associated CSC-like cells that controls the malignant progression of tumor cells, including liver metastasis. AMIGO2 may be a promising therapeutic target to prevent the progression of malignancy limited to cancer cells with high AMIGO2 expression in various organ cancers.
Methods
Cell culture and transfection
Gastric cancer cell lines MKN7 were obtained from Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University (Sendai, Japan), MKN45 and MKN74 from Japanese Collection of Research Bioresources (JCRB; Osaka, Japan) cell bank, and TMK-1 from the founder54. Pancreatic cancer cell lines PANC-1, RT4 and T24 were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA), and SUIT-2 was obtained from JCRB. The colorectal cancer cell lines DLD-1 and LoVo were obtained from JCRB cell bank, CACO-2 and KP4 from the Riken Bioresource Center (Tsukuba, Japan), and COLO 205 was obtained from ATCC. All bladder cancer cell lines (5637, SCaBER, RT4, and T24) were obtained from ATCC. CACO-2, COLO 205, DLD-1, KP4, LoVo, MKN7, MKN45, MKN74, PANC-1, RT4, SUIT-2, T24, and TMK-1 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, 05919; Shimadzu Diagnostics Corp., Kyoto, Japan). SCaBER and 5637 cells were maintained in Eagle’s minimum essential medium (MEM, 05900; Nissui Pharmaceuticals, Tokyo, Japan) and Roswell Park Memorial Institute medium (RPMI1640, 05918; Nissui Pharmaceuticals), respectively. DMEM medium without glucose (09891–25; Nacalai Tesque, Kyoto, Japan) was also used. All cell lines were cultured in their respective culture media supplemented with 10% fetal bovine serum (1370978; Gibco, Gaithersburg, MD, USA) and L-glutamine (0.3 mg/mL; 16919–84; Nacalai Tesque) at 37 °C and 5% CO2 in a humidified incubator until use in analysis. For the hypoxic environment, according to the previous definition of tumor hypoxia (median pO2 < 10 mm Hg, approximately 1.25% O234, cells in the hypoxic group were incubated at 1% O2 in a hypoxic chamber (Wakenyaku Co. Ltd., Tokyo) gassed with 1% O2, 94% N2, and 5% CO2.
Human AMIGO2-specific short hairpin RNA (shRNA) plasmid, pLKO.1-AMIGO2-shRNA 1 and 2, and empty vector pLKO.1 were purchased from Sigma-Aldrich (St. Louis, MO, USA). For AMIGO2 shRNA 1 sequence, the sense strand was 5′-CCGGGCCTAAAGCAAAGTTAGATTTCTCGAGAAATCTAACTTTGCTTTAGGCTTTTTTG-3′ and for AMIGO2 shRNA 2 sequence, the sense strand was 5′-CCGGGTCTTGACTTATCGTCCAATACTCGAGTATTGGACGATAAGTCAAGACTTTTTTG-3′. DLD-1 and MKN45 cells were transfected with pLKO.1-AMIGO2-shRNA 1 and 2 and pLKO.1 empty vector using Lipofectamine 2000 reagent (11668019; Thermo Fisher Scientific Japan, Tokyo, Japan). Cells were selected using puromycin at a concentration of 3 μg/mL (DLD-1) or 1 μg/mL (MKN45).
RNA extraction, cDNA preparation, and qRT-PCR analysis
Total RNA was extracted from tumor cells using the TRI reagent (TR118; Molecular Research Center, Cincinnati, OH, USA) according to the manufacturer’s protocol. For qRT-PCR, 0.4 µg of total RNA was used for cDNA synthesis in a 10 μL reaction mixture containing PrimeScript™ RT Master Mix (RR036A; Takara Bio, Shiga, Japan). PCR amplification of cDNA was performed using TB Green Premix Ex Taq II (RR820; Takara Bio). The PCR cycles consisted of 30 s initial denaturation at 95 °C, followed by 40 cycles at 95 °C for 15 s each, and 60 °C for 30 s in a thermal cycler (Viia7; Thermo Fisher Scientific). The changes in mRNA levels were calculated using the delta-delta CT method with β-actin as an endogenous control.
Primer design
Human gene-specific primers were designed to span the non-coding intron region: AMIGO2 upstream: 5′-CCCCTGCAAGTGTAAAACCA-3′; AMIGO2 downstream: 5′-AGGGGTTCCAAAAACACCAC-3′; CD44v6 upstream: 5′-CAACGGAAGAAACAGCTACC-3′; CD44v6 downstream: 5′-CTGTTGTCGAATGGGAGTCT-3′; CD44v9 upstream: 5′-AGCAGAGTAATTCTCAGAGCTT-3′; CD44v9 downstream: 5′-TGCTTGATGTCAGAGTAGAAGT-3′; CD47 upstream: 5′-CATGGCCCTCTTCTGATTTC-3′; CD47 downstream: 5′-GGAGGTTGTATAGTCTTCTGATTGG-3′; CD166 upstream: 5′-CATACCTTGCCGACTTGACG-3′; CD166 downstream: 5′-GAAGGCAATAAATACTGGGGAGC-3′; EpCAM upstream: 5′-CGCAGCTCAGGAAGAATGTG-3′; EpCAM downstream: 5′-TGAAGTACACTGGCATTGACG-3′; ß-actin upstream: 5′-CGTGGGCCGCCCTAGGCACC-3′; and ß-actin downstream: 5′-TTGGCTTAGGGTTCAGGGGG-3′.
Immunoblotting analysis
Cells were lysed in the RIPA buffer (1% NP-40, 50 mM Tris [pH7.5], 165 mM NaCl, 10 mM EGTA, 1 mM Na3VO4, 10 mM NaF, and 1 mM PMSF) with protease inhibitors, aprotinin and leupeptin. Protein samples were separated on 10% sodium dodecyl sulfate–polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (USEQ00010; Merck Millipore, Darmstadt, Germany). The membranes were treated with a 5% skim milk solution for 1 h at 25 °C. The following antibodies were used for immunoblotting: rat monoclonal anti-AMIGO2 (1:8000, rTNK1b012a)20 and mouse monoclonal anti-β-actin (1:4000, A5441; Sigma-Aldrich). The relative intensities of the bands were scanned and quantified using the ImageJ software. The specificity of the rTNK1b012a antibody was verified by establishing expressing cell lines by transfecting HepG2 cells with a plasmid containing the AMIGO1, AMIGO2, or AMIGO3 gene. The results confirmed that the rTNK1b012a antibody is specific for the human AMIGO2 protein and does not cross-react with AMIGO1 or AMIGO320.
Cell proliferation assay
For the cell proliferation assay, cells were seeded in a 96-well plate at a density of 1 × 103 cells/well, cultured in different concentrations of serum (10% and 2%), and incubated for 1–7 days. The medium was changed every two days. The cell proliferation assay was performed as previously reported 55, with some modifications as described below. On days 1, 3, 5 and 7 after seeding, cells were fixed with 5% glutaraldehyde, stained with 0.05% crystal violet in CAPS buffer, rinsed with tap water, and dried, followed by the addition of 10% acetic acid to each well. Absorbance was measured at 495 nm using a plate reader (Infinite M200 PRO; Tecan Japan, Kawasaki, Japan).
Sphere formation assay
All cell lines and transfected cells were plated in an ultra-low-attachment 96-well plate (3474; Corning, Kennebunk, ME, USA) at a density of 1 × 103 cells/well in the culture medium with 1.75% methylcellulose (138–05052; Fujifilm Wako Pure Chemical, Osaka, Japan) to prevent cells from adhering to each other to form colonies. The anticancer drug was added at the same time as cells were seeded, and the cells were treated with the same drug until the end of the experiment, when the number of spheroid cells was counted. The number of spherical cells with a diameter of 50 μm or greater was counted for MKN45 cells on day 12 of CDDP treatment, and for DLD-1 cells on day 10 of L-OHP treatment, and IC50 values were calculated according to a previous report56. CDDP and L-OHP were obtained from Nichiiko (G00400; Fuji, Japan) and Fujifilm Wako Pure Chemical (156–02691), respectively.
Immunocytochemistry and immunofluorescence assay for spheres
Ten-to-twelve days after sphere formation by MKN45 and DLD-1 cells and their transfectants, the spheres were jellified with iPGell (Genostaff, Tokyo, Japan), according to the manufacturer’s protocol, and fixed in 4% paraformaldehyde for 24 h. The jellified spheres were embedded in paraffin and tissue blocks were prepared. Serial sections (3 μm) were cut and placed on slides for immunofluorescence analysis. After treatment with 10% normal goat serum (426042; Nichirei, Tokyo, Japan) for 10 min at 25 °C, sections were incubated with the following primary antibodies at 4 °C for 16 h: rat monoclonal anti-AMIGO2 (1:1000, rTNK1b012a)20, rabbit polyclonal anti-CD44 (1:3000, 15675–1-AP; Proteintech), rabbit polyclonal anti-CD133 (1:1000, 18470–1-AP; Proteintech), and rabbit polyclonal anti-EpCAM (1:3000, 21050–1-AP; Proteintech) antibodies. Multiple primary antibodies were added simultaneously for co-staining. Anti-rat IgG Alexa Fluor488 (1:1000, ab150157; Abcam, Cambridge, UK) and anti-rabbit IgG Alexa Fluor555 (1:1000, ab150057; Abcam) antibodies were used as secondary antibodies. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (340–07971; Dojindo, Kumamoto, Japan).
Tumorigenicity in non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice
Female NOD/SCID mice (4-weeks-old) were obtained from Nippon SLC (Hamamatsu, Japan) and maintained under specific pathogen-free conditions with light–dark cycles (12 h each) at 23 ± 3 °C and 50 ± 10% humidity at the Institute for Animal Experimentation of Tottori University. Animals were used after one week of acclimation. Food and water were supplied ad libitum throughout the experiment. All experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee of Tottori University (Permission No. 22-Y-21) and were performed in accordance with approved institutional and ARRIVE guidelines. MKN45 cells (1 × 106, 5 × 105, and 2.5 × 105 cells), DLD-1 cells (5 × 106, 5 × 105, and 5 × 104 cells) and their transfectants were suspended in 100 μL of media and injected subcutaneously into NOD/SCID mice. Tumor development was checked by palpation and measured daily using Vernier calipers. The tumor volume was calculated as length × (width)2/2. Mice were sacrificed on the 15th day after tumor implantation by placing the mouse cage in a chamber without anesthesia and injecting compressed carbon dioxide (CO2) gas in accordance with the AVMA Guidelines for the Euthanasia of Animals (2020 edition). In order to shorten the time from CO2 exposure to loss of consciousness, the CO2 concentration was gradually increased at a replacement rate of 30–70% of the volume in the chamber per minute in accordance with the AVMA guidelines. After confirming that the mouse had stopped breathing, CO2 was infused for more than 1 min. Based on the incidence of tumorigenicity, the number of tumor-initiating cells (TIC) was calculated using the extreme limiting dilution analysis software (https://bioinf.wehi.edu.au/software/elda/).
Statistical analysis
Differences in the number of 3D spheres, AMIGO2 mRNA and protein expression, in vitro cell growth, and in vivo tumor growth and weight were analyzed using Dunnett’s test. Correlation between AMIGO2 expression in tumor cells and cancer stem cell marker expression was analyzed using Pearson’s correlation coefficients. Tumor-inducing potential (TIC) was calculated based on the incidence of tumor formation in parental cells, shControl, and shAMIGO2-transfected cells using the Extreme Limiting Dilution Analysis (ELDA) software (https://bioinf.wehi.edu.au/software/elda/). For statistical comparisons, significance was set at p < 0.05.
Data availability
Data is provided within the manuscript or supplementary information files. The data that support the findings of this study are available from the corresponding author (E-mail: fuokada@tottori-u.ac.jp) upon reasonable request.
Abbreviations
- AMIGO2:
-
Amphoterin-induced gene and open reading frame 2
- CSCs:
-
Cancer stem cells
- EpCAM:
-
Epithelial cell adhesion molecule
- IC50 :
-
Half-maximal inhibitory concentration
- NOD/SCID:
-
Non-obese diabetic/severe combined immunodeficient
- TIC:
-
Tumor-initiating cells
References
Fares, J. et al. Molecular principles of metastasis: a hallmark of cancer revisited. Sig. Transduct. Target Ther. 5, 28 (2020).
Jeevisha, B., Emily, D. & Tannishtha, R. Stem cells in cancer initiation and progression. J. Cell Biol. 219, e201911053 (2020).
Liu, L. & Wang, H. Biomarkers and targeted therapy for cancer stem cells. Trends Pharmacol. Sci. 45, 56–66 (2024).
Walcher, L. et al. Cancer stem cells –origins and biomarkers: perspectives for targeted personalized therapies. Front. Immunol. 11, 1280 (2020).
Vetrie, D., Helgason, G. V. & Copland, M. The leukaemia stem cell: similarities, differences and clinical prospects in CML and AML. Nat. Rev. Cancer. 20, 158–173 (2020).
Singh, S. K. et al. Identification of human brain tumour initiating cells. Nature 432, 396–401 (2004).
Krishnamurthy, S. & Nör, J. E. Head and neck cancer stem cells. J. Dent. Res. 91, 334–340 (2012).
Zeng, X. et al. Breast cancer stem cells, heterogeneity, targeting therapies and therapeutic implications. Pharmacol. Res. 163, 105320 (2021).
Lee, T. K., Guan, X. Y. & Ma, S. Cancer stem cells in hepatocellular carcinoma – from origin to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 19, 26–44 (2022).
Dalerba, P. et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. U. S. A. 104, 10158–10163 (2007).
Schatton, T. & Frank, M. H. Cancer stem cells and human malignant melanoma. Pigment Cell Melanoma Res. 21, 39–55 (2008).
Batlle, E. & Clevers, H. Cancer stem cells revisited. Nat. Med. 23, 1124–1134 (2017).
Paul, R., Dorsey, J. F. & Fan, Y. Cell plasticity, senescence, and quiescence in cancer stem cells: biological and therapeutic implications. Pharmacol. Ther. 231, 107985 (2022).
Clevers, H. The cancer stem cell: premises, promises and challenges. Nat. Med. 17, 313–319 (2011).
Desai, A., Yan, Y. & Gerson, S. L. Concise reviews: cancer stem cell targeted therapies: toward clinical success. Stem Cells Transl. Med. 8, 75–81 (2019).
Walcher, L. et al. Cancer stem cells-origins and biomarkers: perspectives for targeted personalized therapies. Front Immunol. 11, 1280 (2020).
Kuja-Panula, J. et al. AMIGO, A Transmembrane protein implicated in axon tract development, defines a novel protein family with leucine-rich repeats. J. Cell Biol. 160, 963–973 (2003).
Kanda, Y. et al. Amigo2-upregulation in tumour cells facilitates their attachment to liver endothelial cells resulting in liver metastases. Sci. Rep. 7, 43567 (2017).
Goto, K. et al. The impact of AMIGO2 on prognosis and hepatic metastasis in gastric cancer patients. BMC Cancer 22, 280 (2022).
Goto, K. et al. Establishment of an antibody specific for AMIGO2 improves immunohistochemical evaluation of liver metastases and clinical outcomes in patients with colorectal cancer. Diagn. Pathol. 17, 16 (2022).
Tanio, A. et al. AMIGO2 as a novel indicator of liver metastasis in patients with colorectal cancer. Oncol. Lett. 21, 278 (2021).
Nakamura, S. et al. AMIGO2 expression as a potential prognostic biomarker for gastric cancer. Anticancer Res. 40, 6713–6721 (2022).
Rabenau, K. E. et al. DEGA/AMIGO-2, a leucine-rich repeat family member, differentially expressed in human gastric adenocarcinoma: effects on ploidy, chromosomal stability, cell adhesion/migration and tumorigenicity. Oncogene 23, 5056–5067 (2004).
Shen, S., Gui, T. & Ma, C. Identification of molecular biomarkers for pancreatic cancer with mRMR shortest path method. Oncotarget 8, 41432–41439 (2017).
Huo, T. et al. Colorectal cancer stages transcriptome analysis. PLoS ONE 12, e0188697 (2017).
Sonzogni, O. et al. Reporters to mark and eliminate basal or luminal epithelial cells in culture and in vivo. PLoS Biol. 16, e2004049 (2018).
Liu, Y. et al. In vivo selection of highly metastatic human ovarian cancer sublines reveals role for AMIGO2 in intra-peritoneal metastatic regulation. Cancer Lett. 503, 163–173 (2021).
Simmen, F. A. et al. The Krüppel-like factor 9 (KLF9) network in HEC-1-A endometrial carcinoma cells suggests the carcinogenic potential of dys-regulated KLF9 expression. Reprod. Biol. Endocrinol. 6, 41 (2008).
Yamamoto, A. et al. AMIGO2 expression at the invasive front of bladder cancer predicts recurrence-free and overall survival after radical cystectomy. Oncol. Lett. 30, 339 (2025).
Bi, O. et al. SFPQ promotes an oncogenic transcriptomic state in melanoma. Oncogene 40, 5192–5203 (2021).
Fontanals-Cirera, B. et al. Harnessing BET inhibitor sensitivity reveals AMIGO2 as a melanoma survival gene. Mol. Cell. 68, 731–744 (2017).
Cui, Y. et al. Single-cell transcriptome and genome analyses of pituitary neuroendocrine tumors. Neuro. Oncol. 23, 1859–1871 (2021).
Izutsu, R. et al. Liver metastasis formation is defined by AMIGO2 expression via adhesion to hepatic endothelial cells in human gastric and colorectal cancer cells. Path. Res. Pract. 237, 154015 (2022).
Natsuizaka, M. et al. Synergistic up-regulation of hexokinase-2, glucose transporters and angiogenic factors in pancreatic cancer cells by glucose deprivation and hypoxia. Exp. Cell Res. 313, 3337–3348 (2007).
Chen, S. F. et al. Non adhesive culture system as a model of rapid sphere formation with cancer stem cell properties. PLoS ONE 7, e31864 (2012).
Coles-Takabe, B. L. et al. Don’t look: growing clonal versus nonclonal neural stem cell colonies. Stem Cells. 26, 2938–2944 (2008).
Makena, M. R. et al. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165339 (2020).
Han, M. E. et al. Cancer spheres from gastric cancer patients provide an ideal model system for cancer stem cell research. Cell Mol. Life Sci. 68, 3589 (2011).
Horst, D. et al. Prognostic significance of the cancer stem cell markers CD133, CD44, and CD166 in colorectal cancer. Cancer Invest. 27, 844–850 (2009).
Han, Z. et al. Integrated analysis reveals prognostic value and progression-related role of AMIGO2 in prostate cancer. Transl. 11, 914–928 (2022).
Izutsu, R. et al. AMIGO2 enhances the invasive potential of colorectal cancer by inducing EMT. Cancer Gene Ther. 31, 1786–1795 (2024).
Zhou, H. et al. Cancer stem cells: recent insights and therapies. Biochem. Pharmacol. 209, 115441 (2023).
Salles, G. et al. Rituximab in B-cell hematologic malignancies: a review of 20 years of clinical experience. Adv. Ther. 34, 2232–2273 (2017).
Lagadinou, E. D. et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12, 329–341 (2013).
Farge, T. et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 7, 716–735 (2017).
Maes, T. et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell 33, 495-511.e12 (2018).
Dombret, H. et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 126, 291–299 (2015).
Wang, J. et al. CXCR4 antagonist AMD3100 (plerixafor): from an impurity to a therapeutic agent. Pharmacol. Res. 159, 105010 (2020).
Spiegel, J. Y. et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat. Med. 27, 1419–1431 (2021).
Seong, H. K. et al. Prevention of liver metastasis via the pharmacological suppression of AMIGO2 expression in tumor cells. Sci. Rep. 14, 28183 (2024).
Wei, Y. et al. ALDH1: A potential therapeutic target for cancer stem cells in solid tumors. Front. Oncol. 12, 1026278 (2022).
Maverick, E. E., Leek, A. N. & Tamkun, M. M. Kv2 channel-AMIGO β-subunit assembly modulates both channel function and cell adhesion molecule surface trafficking. J. Cell Sci. 134, jcs256339 (2021).
Park, H. et al. AMIGO2, a novel membrane anchor of PDK1, controls cell survival and angiogenesis via Akt activation. J. Cell Biol. 211, 619–637 (2015).
Ito, H. et al. Cytogenetical analysis of the human gastric carcinoma cell line TMK-1. Hiroshima J. Med. Sci. 38, 121–124 (1989).
Taniguchi, N. et al. Bisphosphonate-induced reactive oxygen species inhibit proliferation and migration of oral fibroblasts: A pathogenesis of bisphosphonate-related osteonecrosis of the jaw. J Periodontol. 91, 947–955 (2020).
Rodbard, D. Statistical estimation of the minimal detectable concentration (“sensitivity”) for radioligand assay. Anal. Biochem. 90, 1–12 (1978).
Acknowledgements
The authors would like to thank Drs. Hiroyuki Satofuka, Yasuhiro Kazuki, Hiroyuki Kugoh, and Mitsuo Oshimura for the development and supply of the specific monoclonal antibody, rTNK1A0012, against human AMIGO2.
Funding
This work was partially supported by a Grant-in-Aid for Scientific Research to FO (20K07447 & 23K06482), and a Grant-in-Aid for Research Activity Start-up (23K19538) to RI from the Japanese Ministry of Education, Culture, Sports, Science, and Technology.
Author information
Authors and Affiliations
Contributions
HKS, RI, JPJ, RSat, RSas, and AY performed in vitro experiments; HKS, MO, RSat, and FO performed in vivo experiments; HKS, MO, and FO contributed to the formulation of the experimental design; HKS, MO, RI, JPJ, RSat, RSas, JH, AY, AT, and FO contributed to the interpretation and discussion of the results; HKS conceived the concept of this study and wrote the original draft; FO designed and arranged all experiments and wrote the manuscript, and edited. The authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Seong, H.K., Osaki, M., Izutsu, R. et al. AMIGO2 accelerates tumor progression by inducing a cancer stem cell-like phenotype. Sci Rep 15, 32861 (2025). https://doi.org/10.1038/s41598-025-18095-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-18095-7
