
Abstract
While tumor expression of PD-L1 is useful for predicting NSCLC tumor response to anti-PD-1/PD-L1 therapy (programmed cell death protein 1 (PD-1)/programmed cell death protein ligand 1 (PD-L1)), it is not entirely reliable for therapy response prediction. Here we employ a multiomic phenotypic approach, combining radiomic, clinical and pathologic biomarkers, to develop a radiomic signature for therapy response prediction on conventional pre-therapy imaging. Tumor radiomics were extracted from baseline CT imaging on retrospective cohort (n = 243) of stage 4 NSCLC patients [squamous (n = 33); non-squamous (n = 210)] undergoing first-line anti-PD-1 therapy. Image parameter heterogeneity was mitigated using nested ComBat harmonization. A novel multiomic graph was developed by combining the constituent radiomics and radiological (SUVmax, longest tumor diameter at baseline) and pathological (PD-L1, STK11 and KRAS expression) graphs. The multiomic phenotypes, identified from this graph, were then combined with clinical variables (smoking status, BMI) into a multiomic graph model. The prognostic performance of this model is compared to the combination clinical model, built by concatenation of the various “omics” variables. For the 210 patients’ group, the progression-free survival models yielded the following c-statistics- clinical model: 0.58 (95% CI 0.52–0.61); combination clinical model: 0.68 (95% CI 0.58–0.69); multiomic graph clinical model: 0.71 (95% CI 0.61–0.72). The AIC values for the multiomic graph clinical model, combination clinical model and clinical model are 1278.4, 1284.1 and 1289.6 respectively, showing that the multiomic graph clinical model provides the best fit to the data. This study has constructed a novel “multiomic” signature for prognosis of NSCLC patient response to immunotherapy. The multiomic phenotypes identify patient groups based on various descriptors, thus are well-rounded, and enhance precision in progression-free survival prediction, upon combination with established clinical biomarkers.
Similar content being viewed by others


Introduction
The adaptive upregulation of programmed cell death-ligand 1 (PD-L1) by tumor cells allows them to evade T cell mediated apoptosis promoting neoplastic growth. The PD-1/PD-L1 pathway is therefore a prime target for cancer immunotherapy, leading to the rise of anti-PD-1/PD-L1 immune checkpoint inhibitor therapies, such as pembrolizumab (PEMBRO)1,2,3. However, tumor expression of PD-L1, the currently accepted predictive biomarker for use of PEMBRO, is an imperfect predictor of response to therapy. For example, PD-L1 expression-enriched non-small cell lung cancer (NSCLC) tumors receiving anti-PD-1/PD-L1 first-line monotherapy exhibit a 40–50% rate of response, which increases to 60–70% with the addition of concurrent chemotherapy4. This means that up to 30–50% of NSCLC tumors with high PD-L1 expression fail to respond to PEMBRO 1st line therapy. Furthermore, a subset of tumors with low PD-L1 expression can respond to PEMBRO even without additional chemotherapy5. Use of tumor expression of PD-L1 as a predictive biomarker of therapy response is further complicated by the fact that intra-tumoral expression is heterogenous and that tumor expression levels can fluctuate over time6,7. Hence, tumor expression level of PD-L1 alone is a suboptimal biomarker to predict response to anti-PD-1/PD-L1 therapy. Given the increasing number of therapeutic options available to NSCLC patients, this inaccuracy in the patient selection for PEMBRO based therapy results in a lost opportunity to prolong patient survival and potential cure. Further, the high cost and toxicity of anti-PD-1/PD-L1 agents8and the risk of lowering the patient survival rates makes it critical to identify novel prognostic biomarkers to more accurately select patients that will benefit from PEMBRO.
Radiomic analysis enables the detection of patterns in medical images that are not identifiable by the human eye, providing a comprehensive, non-invasive in-depth analysis of intra-tumoral and peri-tumoral heterogeneity of the entire tumor while in situ9. This enables non-invasive evaluation of the tumor over time which is important since serial tumor biopsy is generally not feasible and carries with it significant patient discomfort and the risk of potential procedure related complications10. Preliminary studies have shown evidence of a correlation between tumoral radiomic signatures and anti-PD-L1/PD-1 therapy response. For example, Valentuzzi et al. developed a NSCLC radiomics signature termed “iRADIOMICS” which was constructed using univariate and multivariate logistic models, using the most promising radiomics features. In a cohort of 30 metastatic NSCLC patients, this multivariate iRADIOMICS signature exhibited promise as a predictive biomarker for PEMBRO response with AUC (95% CI) and accuracy (standard deviation) of 0.9 (0.78-1.00) and 78% (18%). This is compared to the performance of tumor expression of PD-L1 tumor proportion score (TPS) (baseline) of 0.6 (0.37–0.83) and 53% (18%)11. Similarly, Vaidya et al.. developed a random forest classifier, derived from intra-tumoral and peri-tumoral radiomic features, able to distinguish between hyper progressive (HP) (the phenomenon of paradoxical acceleration of disease progression after initiation of immunotherapy12 and other radiographical response patterns with an AUC of 0.85 ± 0.06 in the training (n = 30) and validation (n = 79) sets, in NSCLC patients undergoing monotherapy with PD-1/PD-L1 inhibitors. In addition, there was a clear stratification in overall survival between HPs and non-HPs (HR 2.66, 95% CI 1.27–5.55; p = 0.009)13. Finally, Trebeschi et al.. focused on the development of a non-invasive radiomic biomarker to distinguish between responding and non-responding NSCLC and melanoma on non-first line immunotherapy immune checkpoint inhibitor therapy which exhibited a promising performance in NSCLC (up to 0.83 AUC, p < 0.001) and, to a lesser degree in metastatic melanoma (0.64 AUC, p = 0.05)14.
While studies such as these are promising, there are several limitations to consider. First, these studies used a heterogenous patient population which included both first line and non-first line immunotherapy, hence introducing variability due to prior therapy-induced alterations in the tumor microenvironment. Further, some studies have focused on identifying individual significant features from the high dimensional radiomic feature sets, rather than constructing radiomic signatures by identifying phenotypic feature patterns. Additionally, the radiomics-based studies also did not address the heterogeneity in radiomic features that arises due to variation in image acquisition (such as scanner type, reconstruction kernel, slice thickness or use of contrast) techniques. Furthermore, to the best of our knowledge prior studies did not fully integrate information from radiomic features with other known prognostic factors into multiomic signatures. The integration of radiomics with radiological and pathological factors into multiomic signatures, for enhancing precision in the prediction of patient response to therapy, is the specific focus of this study.
Multiomic descriptors, built by combining information from various descriptors of the tumor, offer the promise of increased precision in therapy response prediction, by integrating the relative strengths of a variety of biomarkers. They can help provide a deeper, well-rounded overview of the properties of the tumor regions of the patients, as compared to information obtained from individual ‘omics’ descriptors. For example, the clinical variable body mass index (BMI) has been estimated to carry an increased pretherapy hazards ratio for overweight (aHR 0.76; 95% CI 0.68–0.85) and obese patients (aHR 0.68, 95% CI 0.57–0.81)15 a finding supported by other related studies16,17,18. Tumors with KRAS mutations had a 3-month mortality rate of 23.5% compared to 17.7% for tumors with other mutations19. In addition, exploratory predictive biomarkers such as circulating tumor DNA (ctDNA) and tumor mutational burden, have predictive significance to therapy outcome in immunotherapy, but are unlikely to stand alone as predictor of therapy success. Here in our study, we aim to integrate the relative strengths of general predictive biomarkers such as BMI, smoking status and immunotherapy specific markers such as tumor expression of PD-L1, together with novel radiomic biomarker signatures developed in our laboratory.
However, while the integration of multiple predictive variables promises a more precise predictive tool, the increase in number of patient descriptors can introduce problems while selecting the appropriate technique for integration. For instance, a standard concatenation-based combination approach can make the combined signature biased towards information from the largest-sized “omics”. Deep learning methods for multiomic integration and graph networks for analysis of the association between various “omics” are currently being explored as well20,21. However, the performance of these methods often relies on the availability of large-scale input datasets, such as transcriptomic and proteomic datasets. Our study leverages a cohort of 243 patients with stage 4 NSCLC treated at our institution with first line PEMBRO-based therapy, with radiological (maximum standardized uptake volume (SUVmax) and longest tumor diameter at baseline), pathological (PD-L1, STK11 and KRAS expression), radiomics and clinical (smoking status, BMI) information available. Since this sample size is relatively small, compared to the high-dimensional datasets used for current graph network and deep learning multiomic integration approaches, we have developed a novel multiomic graph integration approach for our cohort. The multiomic graph is developed by combining the constituent radiomics, radiological and pathological graphs. The edges in each constituent “omic” graph reflect the connections between the nodes (patients) based on their “omic” information, and the edges in the combined graph reflect all the connections based on each constituent graph. The multiomic phenotypes, identified from the multiomic graph, essentially define patient groups based on information from the various “omics”.
We compare the prognostic accuracy of the multiomic phenotypes to a combination model, built by integrating radiological and pathological variables with the radiomic phenotypes of the patients. The radiomic phenotypes are built using an unsupervised hierarchical clustering approach, that helps reduce the high-dimensional radiomics feature space by identifying statistically significant phenotypic signatures. We demonstrate the efficacy of the Cancer Phenomics Toolkit (CapTk)22an open-source software developed at our institution, that conforms to the Imaging Biomarker Standardization Initiative (IBSI) radiomics standardization protocols for radiomics analysis23. We account for differences in image parameters by using a nested ComBat approach for harmonization by the image acquisition parameters24. This approach enables harmonization of multiple batch effects by implementing sequential harmonization, thus providing a superior approach over standard ComBat techniques, that are unable to adequately harmonize datasets heterogeneous in more than one batch effect, due to their ability to only harmonize by a single batch effect at a time25.
We hypothesize that the addition of the multiomic phenotypes to established clinical biomarkers, such as BMI and smoking status, will improve the accuracy of the prognostic model in the prediction of progression-free survival in patients with NSCLC. We also hypothesize that the multiomic graph clinical model, built by combining the multiomic phenotypes with the clinical biomarkers, would have a higher prognostic performance compared to the combination clinical model, built by combining the radiomic phenotypes, radiological and pathological variables with the clinical biomarkers.
Materials and methods
Study sample and data
This single-center retrospective observational study was conducted at our institution between November 2016 and December 2022. The study was approved by the Institutional Review Board (IRB) committee (IRB protocol number- 848796) under a waiver of informed consent (waiver copy included with submission). All methods in this study were in accordance with the Declaration of Helsinki.
Inclusion criteria
Adult patients of any race or gender with stage 4 NSCLC treated with first line PEMBRO- based therapy at our institution.
Exclusion criteria
Patients without accessible pretherapy imaging within 6 weeks of the start of 1st line therapy with PEMBRO, lack of high-resolution images (CTs from PET/CT, CTs without high resolution 1–3 mm slice thickness images), images degraded by technical artifact, patients without accessible clinical data, children and vulnerable populations (pregnant, prison inmates) were excluded.
Patients with stage 4 NSCLC treated with first line PEMBRO- based therapy at our institution were identified. The details of the monotherapy and combination therapy regimens are included in Supplementary Table S1. A subset of 107 patients from this cohort had been analyzed in two of our previous studies, that studied the improvement in performance of prognostic models of progression-free survival upon addition of radiomic phenotypes to standard clinical prognostic factors26, and the effect of image parameter heterogeneity-mitigation techniques on the reproducibility of radiomic signatures27. Our current study focuses on the development of prognostic multiomic phenotypes for the patients using conventional CT. The shooting ranges of the CT scans were the thoracic regions of the patients. The 3D tumor CT volumes were extracted from baseline scans obtained immediately prior to the start of immunotherapy through manual segmentation by board-certified, fellowship-trained thoracic radiologists S.I.K (with 18 years of clinical experience) and L.R. (with 10 years of clinical experience) using the semi-automated ITK-SNAP software (version 3.6.0)28. Segmentation was performed on the lung CT reconstruction windows, with exclusion of perilesional ground glass, if such were present. Only the primary lesion was segmented in each scan. Large bronchi and cavitary components were excluded from the segmentation, as were large vessels, when possible.
The list of demographic, clinical, pathological and radiological covariates for our patient cohort is included in supplementary table S2. The list of demographic variables also contains “race”- this information was collected from the patients’ electronic health records, as an important variable in the study of patients with advanced stage NSCLC.
Clinical and pathologic sub-cohorts
Recent studies have shown the merit of paying attention to KRAS and STK11 mutations while studying response to therapy in patients with NSCLC treated with anti-PD-1/PD-L1 checkpoint inhibitor therapy29,30. Reduced efficacy (worse progression-free survival) of immunotherapy has been observed in patients with KRAS and STK11 mutations. Thus, in our study, we have also included the gene mutational status (solid tumor genomic sequencing) of KRAS and STK11 for the subset of 184 patients for which this information was available. The mutation categories and number of patients in each category for this patient subset has been included in supplementary table S3. The list of mutations in both genes is included in supplementary table S4.
The patients were divided into two subgroups based on histology- squamous carcinoma (n = 33) and non-squamous carcinoma (n = 210). In the case of 210 patients with non-squamous carcinoma, the longest tumor diameter at baseline and SUVmax values are taken as the radiological variables and the PD-L1 expression categories are taken as the pathological variable. The SUVmax values were obtained from the clinical reports of the 18 F-FDG PET-CT images obtained immediately prior to the initiation of therapy. For a subset of 166 patients in the non-squamous carcinoma subgroup, the list of pathological variables also contains KRAS and STK11 mutation categories. Similarly, for the squamous carcinoma subgroup, in the case of 33 patients, the longest tumor diameter at baseline and SUVmax values are taken as the radiological variables and the PD-L1 expression categories are taken as the pathological variable. For a subset of 18 patients in the squamous carcinoma subgroup, the list of pathological variables also contains KRAS and STK11 mutation categories.
Radiomic feature extraction
The CaPTk software was used to extract 102 radiomic features from each tumor’s entire segmented volume17. The radiomic features represent the following descriptors: (1) Intensity features (2) Histogram-based features (3) Volumetric features (4) Morphologic features (5) Gray level run length matrix features (6) Neighboring gray tone difference matrix features (7) Gray level size zone matrix features (8) Local binary pattern features. A description of the radiomic features types is in the supplement (“radiomic feature description”); the list of features belonging to each type and their formulae are included in supplementary tables S5 and S6.
Radiomic feature harmonization
The impact of heterogeneity introduced by the image acquisition parameters was mitigated using a nested ComBat approach, to correct for multiple batch effects24,25. The nested approach was initialized with the original radiomic features as the input data and a list of batch effects- pixel spacing, slice thickness, contrast enhancement and kernel resolution (Supplementary Table S7-non-squamous carcinoma group, Supplementary Table S8-squamous carcinoma group). Features significantly affected by batch effects after harmonization were deemed non-robust31 and discarded from further analysis. The resulting feature set was then subjected to z-score normalization. Further details on harmonization are in the supplement (“details on radiomic feature harmonization”).
Radiomic phenotype identification
To identify intrinsic radiomic phenotypes, unsupervised hierarchical clustering was performed on the harmonized and reduced feature set described above32. An agglomerative approach was used to create a hierarchical clustering of the patients using Euclidean distance as distance between feature vectors and Ward’s minimum variance method as the clustering criterion33. The optimal number of stable phenotypes k was determined using consensus clustering34. Statistical significance of the stable phenotypes was evaluated using the SigClust method35,36. The significance of the cluster index was tested against a null distribution, and the test was performed at each phenotype split to determine statistical significance (p < 0.05). Further details on radiomic phenotype identification are in the supplement (“details on radiomic phenotype identification”).
Construction of multiomic graph phenotypes
The multiomic graph was constructed to define the relationship between patients based on the information from the various “omics” descriptors. The phenotypes obtained from this multiomic graph can hence be thought of as communities the patients were divided into, derived from the associations between the patients, based on the various available sources of information. To reduce the high dimensionality of the radiomics feature set, Principal Components Analysis was performed on the heterogeneity mitigated radiomics feature set, and the first principal component (PC) was taken as the representative radiomics feature. A radiomics graph was computed, such that an edge exists between the graph nodes (representative of patients), if the patients are k nearest neighbors of each other (here, we have chosen the value of k to be 2), i.e., are similar to each other, based on their representative radiomics feature. This step was repeated for the radiological and pathological variables as well, i.e., a radiological graph was constructed, based on patient similarities derived from their radiological information, and a pathological graph was constructed, based on patient similarities derived from their pathological information. A combined graph was constructed, preserving the connections between patients based on the constituent graphs. In other words, if a connection existed between nodes A and B based on their radiomics information (value ‘1’ in cell AB of the radiomics adjacency matrix, else 0), and between nodes A and C based on their radiological information (value ‘1’ in cell AC of the radiological adjacency matrix, else 0), then edges AB and AC would exist in the combined graph, with edge weights ‘1’ (value ‘1’ in cells AB and AC of the combined adjacency matrix). Further, if an edge exists between A and B, based both on their radiomics and radiological information, then the edge weights would be added in the combined graph, and their edge weight of AB would be ‘2’ (value ‘2’ in the combined adjacency matrix). These combined edge weights were then normalized by dividing each weight by the number of constituent graphs (i.e., 3). The Kernighan Lin bisection method, that divides a graph into two communities by maximizing the modularity (i.e., dense connections between the nodes within the community and sparse connections between nodes of the two communities) was applied on this weighted multiomic graph, to detect two multiomic graph phenotypes37,38.
Details of prognostic models and their association with survival outcome
The radiomic phenotypes, radiological variables and pathological variables constitute the combination model, and when added to the clinical variables, they constitute the combination clinical model. The prognostic performance of the following five models was evaluated in the prediction of progression-free survival: clinical model, combination model, combination clinical model, multiomic graph phenotypes and multiomic graph clinical model. The five models were evaluated for the group of 210 patients and the subset of 166 patients for whom the mutational status of KRAS and STK11 was available. The prognostic performance of the model built using tumor volumes, for patients with non-squamous carcinoma, and comparison with the prognostic performance of the model with the multiomic graph phenotypes added to the tumor volumes, for (n = 210) and (n = 166) non-squamous carcinoma patient groups was also evaluated.
Kaplan–Meier (K–M) curves and log-rank test using the entire cohort assessed the performance of the following models in separating participants above versus below the median score during the prognosis of progression-free survival (PFS)39: clinical model, combination model, combination clinical model, multiomic graph phenotypes and multiomic graph clinical model. The p value from the log-rank test indicates if the model can provide a statistically significant (p < 0.05) separation between patients with prognostic scores above and below the median prognostic score. Further, fivefold cross-validated Cox proportional-hazards regression analysis with 200 iterations was performed on all the models described above. The discrimination capacity of the models was assessed using the concordance statistic (c-statistic), taking the average of the c-statistic obtained over the 200 iterations. The 95% confidence interval of the average c-statistic was calculated using 100,000 bootstrap replicates40. The Akaike Information Criterion (AIC)41used as a measure of model fit, was used to compare the following models for progression-free survival: multiomic graph clinical model, clinical model, and combination clinical model.
Assessment of the biological significance of the multiomic graph phenotypes
The distribution of the longest tumor diameter at baseline and the tumor volume across patients belonging to the two multiomic graph phenotypes was compared using boxplots and the Mann–Whitney U (MWU) test was performed to test for significant difference in the mean of the distributions42.
We performed all data manipulation and statistical analysis using Python (Ver. 3.7, Anaconda) and the R programming language (Ver. 3.5.1)43,44.
Results
Patient characteristics
The median age of the patients was 66 years (range [30, 91] years), 47.7% of the cohort was male and 38.3% of the cohort consisted of current smokers (Supplementary Table S2). The entire cohort received first-line therapy with PEMBRO. For the group of 210 patients with non-squamous carcinoma, there were 143 survival events (progression). For the subset of 166 patients, there were 111 survival events (progression).
Radiomic feature harmonization
The percentage of features with significantly different distributions arising from the batch effects was reduced after applying nested ComBat to the original features. A total of 78.4% of the original radiomic features were robust to differences introduced by the batch effects.
Radiomic phenotype identification
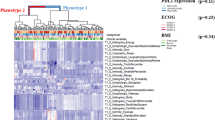
For the group of 210 patients, two distinct radiomic phenotypes were identified, with 67 patients in phenotype 1 and 143 patients in phenotype 2 (p = 0.032 for SigClust test of two clusters versus one). For the subset of 166 patients, two distinct radiomic phenotypes were identified, with 55 patients in phenotype 1 and 111 patients in phenotype 2 (p = 0.041 for SigClust test of two clusters versus one). Figure 1 shows radiomic phenotypes identified by unsupervised hierarchical clustering, in non-squamous carcinoma patients (n = 210). The radiomic phenotypes identified in the squamous carcinoma patient group are illustrated in Supplementary Fig. S1.

Heatmap of radiomic derived features (created using R programming language (ver. 3.5.1). Unsupervised hierarchical clustering identifies two distinct, and statistically significant (p value for phenotype split for two phenotypes, p = 0.032) tumor radiomic phenotypes in the group of patients with non-squamous carcinoma (n = 210). Chi square test p values quantifying the association of these phenotypes with BMI, smoking status and PDL1 expression are also included in the figure.
Construction of multiomic graph phenotypes
For the group of 210 patients, two multiomic graph phenotypes were identified, with 105 patients in each phenotype. For the subset of 166 patients, two multiomic graph phenotypes were identified, with 82 patients in phenotype 1 and 84 patients in phenotype 2. Figure 2 illustrates the multiomic graph construction process and Fig. 3 summarizes the multiomic graph phenotypes construction process. Figure 4 shows the multiomic graph clinical model and the combination clinical model, for prognosis of progression-free survival.

Construction of multiomic graph.

Flowchart summarizing the steps for the construction of multiomic graph phenotypes.

(a) Multiomic graph clinical model and (b) combination clinical model for prognosis of progression-free survival.
Progression-free survival analysis results
For the group of 210 patients, the median PFS was 166 days for patients belonging to multiomic graph phenotype 1 and 130 days for those belonging to multiomic graph phenotype 2. The various models for PFS resulted in the following statistically significant log-rank p values for patients above versus below the median prognostic score-clinical model: p = 0.031; combination model: p = 0.0062; combination clinical model: p = 0.0017; multiomic graph phenotypes p = 0.021 and multiomic graph clinical model: p = 0.00046 (K–M curves included in Fig. 5). The various PFS models yielded the following c-statistics- clinical model: 0.58 (95% CI 0.52–0.61); combination model: 0.63 (95% CI 0.53–0.64); combination clinical model: 0.68 (95% CI 0.58–0.69); multiomic graph phenotypes: 0.65 (95% CI 0.56–0.67) and multiomic graph clinical model: 0.71 (95% CI 0.61–0.72). The AIC values for the multiomic graph clinical model, combination clinical model and clinical model are 1278.4, 1284.1 and 1289.6 respectively, showing that the multiomic graph clinical model provides the best fit to the data when compared to the clinical and combination clinical models. However, since the AIC values are comparatively similar, this is just a suggestive observation (results in Tables 1 and 2). The prognostic performance of the model built using tumor volumes, for patients with non-squamous carcinoma (n = 210), and comparison with the prognostic performance of the model with the multiomic graph phenotypes added to the tumor volumes, is included in the Supplementary Tables S9a,b. The K–M curves for the survival analysis on the group of 33 squamous carcinoma patients has been included in Supplementary Figure S2. The progression-free survival analysis c-scores and AIC values for the group of 33 squamous carcinoma patients have been included in Supplementary Tables S10 and S11 respectively.

Progression-free survival analysis for 210 patients with non-squamous carcinoma built using (a) clinical model (BMI, smoking status) (b) combination model (radiomic phenotypes, radiological variables (SUVmax, longest tumor diameter at baseline), pathological variable (PD-L1 expression) (c) combination clinical model (d) multiomic graph phenotypes and (e) multiomic graph clinical model.
For the subset of 166 patients, the various models for PFS resulted in the following log-rank p values for patients above versus below the median prognostic score- clinical model: p = 0.073; combination model: p = 0.063; combination clinical model: p = 0.024 (statistically significant); multiomic graph phenotypes p = 0.01 and multiomic graph clinical model: p = 0.0063 (statistically significant) (K–M curves included in Fig. 6). The K–M curves for the survival analysis on the group of 18 squamous carcinoma patients has been included in Supplementary Fig. S3. The various PFS models yielded the following c-statistics-clinical model: 0.56 (95% CI 0.51–0.59); combination model: 0.6 (95% CI 0.52–0.61); combination clinical model: 0.64 (95% CI 0.57–0.68); multiomic graph phenotypes: 0.62 (95% CI 0.58–0.67) and multiomic graph clinical model: 0.68 (95% CI 0.58–0.69). The AIC values for the multiomic graph clinical model, combination clinical model and clinical model are 947.5, 952.3 and 960.4 respectively, showing that the multiomic graph clinical model provides the best fit to the data when compared to the clinical and combination clinical models. However, since the AIC values are comparatively similar, this is just a suggestive observation (results in Tables 3 and 4). The prognostic performance of the model built using tumor volumes, for patients with non-squamous carcinoma (n = 166), and comparison with the prognostic performance of the model with the multiomic graph phenotypes added to the tumor volumes, is included in the Supplementary Tables S12a,b. The progression-free survival analysis c-scores and AIC values for the group of 18 squamous carcinoma patients have been included in Supplementary Tables S13 and S14 respectively.

Progression-free survival analysis for subset of 166 non-squamous carcinoma patients with available mutation status of KRAS and STK11 genes, using (a) clinical model (BMI, smoking status) (b) combination model (radiomic phenotypes, radiological variables (SUVmax, longest tumor diameter at baseline), pathological variables (PD-L1 expression, KRAS expression, STK11 expression) (c) combination clinical model (d) multiomic graph phenotypes and (e) multiomic graph clinical model.
Assessment of the biological significance of the multiomic graph phenotypes
For the group of 210 patients, the average value of longest tumor diameter at baseline for patients belonging to multiomic graph phenotype 1 was 62.6 mm and those belonging to phenotype 2 was 31.6 mm. The difference in the mean value of the longest tumor diameter at baseline for patients belonging to phenotype 1 and 2 is statistically significant (MWU test p value-1.3 × 10e−20). The average value of tumor volume for patients belonging to multiomic graph phenotype 1 was 138.9 cm3 and those belonging to phenotype 2 was 19.8 cm3. The difference in the mean value of the tumor volumes for patients belonging to phenotype 1 and 2 is statistically significant (MWU test p value- 2.1 × 10e−19) (boxplots included in Fig. 7).

Distribution of longest tumor diameter at baseline and tumor volume for multiomic graph phenotypes 1 and 2 (n = 210 patients). The average value of longest tumor diameter at baseline for patients belonging to multiomic graph phenotype 1 was 62.6 mm and those belonging to phenotype 2 was 31.6 mm. The average value of tumor volume for patients belonging to multiomic graph phenotype 1 was 138.9 cm3 and those belonging to phenotype 2 was 19.8 cm3. The difference in the mean value of longest tumor diameter at baseline and the tumor volumes for patients belonging to phenotype 1 and 2 is statistically significant.
Discussion
In this study, we have developed a multiomic approach to therapy response prediction for anti-PD-1/PD-L1 therapy in NSCLC, integrating known clinical and pathologic predictive variables with our own novel radiomic signature predictive of therapy response. If successfully translated, this new multiomic tool could improve the precision in patient selection for anti-PD-1/PD-L1 based therapy potentially resulting in improved patient survival and reduced exposure to unwanted therapeutic toxicity on an ineffective therapy. Traditional multiomic integration processes involve concatenation of the various “omics” descriptors into a single large matrix. This often introduces “the curse of dimensionality”—as the number of descriptors increases, while the number of observations stays the same. The size imbalance between different “omics” can also bias the training algorithm to learn from the “omics” with the largest size, while overlooking the other “omics”. Deep learning techniques have also been adopted by recent studies for the purpose of multiomic integration. For instance, Lin et al. extracted deep learning-based features using subnetworks on each omics dataset and concatenated the deep features. A final neural network was used on these features for breast cancer subtypes classification20. Similarly, Islam et al. predicted breast cancer subtypes learning features through convolutional neural networks applied on gene expression and copy number variation datasets45. In the study by Yang et al., the team developed a multimodal autoencoder capable of taking all omics datasets as input, compacting them into the central layer and reconstructing them at the end. The resulting deep features were used to discover cancer subtypes46. For studies involving genomic and genetic data, graph network analysis-based approaches have been developed for understanding complex associations between various genes. For instance, Li et al. developed a weighted graphical lasso approach for gene association network reconstruction using gene expression data21.
However, the performance of graph network and deep learning-based approaches relies heavily on the availability of high-dimensional input data, including genomic and epigenomic data, obtained from high-throughput technologies, such as microarrays and next-generation sequencing. Since our study had a relatively small cohort size (243 patients), compared to the traditionally large datasets used for current graph network and deep learning multiomic integration approaches, we developed a novel multiomic graph integration approach for our cohort. The multiomic graph was developed by integration of the constituent “omics” graphs- essentially the radiomics, radiological and pathological graphs. The edges in the combined graph combine the connections based on all the constituent graphs- thus encompassing the patient relationships from all “omics” sources. The multiomic phenotypes identified from the multiomic graph identify patient groups based on the combination of various patient descriptors and can thus be considered well-rounded.
The multiomic graph phenotypes were added to established clinical prognostic variables to build a multiomic graph clinical model. For the group of 210 patients, the multiomic graph clinical model yielded a c-statistic of 0.71 (95% CI 0.61–0.72) in the prediction of progression-free survival, while the clinical model and the combination clinical model yielded c-statistics of 0.58 (95% CI 0.52–0.61), and 0.68 (95% CI (0.58–0.69) respectively. The AIC values for the multiomic graph clinical model, combination clinical model and clinical model are 1278.4, 1284.1 and 1289.6 respectively, showing that the multiomic graph clinical model provides the best fit to the data when compared to the clinical and combination clinical models. However, since the AIC values are comparatively similar, this is just a suggestive observation. For the subset of 166 patients, the multiomic graph clinical model yielded a c-statistic of 0.68 (95% CI 0.58–0.69) in the prediction of progression-free survival, while the clinical model and the combination clinical model yielded c-statistics of 0.56 (95% CI 0.51–0.59) and 0.64 (95% CI 0.57–0.68) respectively. The AIC values for the multiomic graph clinical model, combination clinical model and clinical model are 947.5, 952.3 and 960.4 respectively, showing that the multiomic graph clinical model provides the best fit to the data when compared to the clinical and combination clinical models. However, since the AIC values are comparatively similar, this is just a suggestive observation. For the group of 33 patients with squamous carcinoma, the multiomic graph clinical model yielded a c-statistic of 0.6 (95% CI 0.53–0.61) in the prediction of progression-free survival, while the clinical and combination clinical model yielded c-statistics of 0.52 (95% CI 0.5–0.56) and 0.57 (95% CI 0.51–0.59) respectively (supplementary information table S10). The AIC values for the multiomic graph clinical model, combination clinical model and clinical model are 131.6, 137.8 and 143.5 respectively, showing that the multiomic graph clinical model provides the best fit to the data (since AIC values are similar, this is a suggestive observation) (Supplementary Table S11).Thus, we show that the multiomic graph clinical model enhances therapy response prediction when compared to the clinical model and the combination clinical model. The multiomic graph phenotypes also improve the prognostic performance when added to the model built using tumor volume (n = 210 patients, c-statistics (tumor volume): 0.56 (95% CI 0.51–0.6); c-statistics (multiomic graph phenotypes + tumor volume): 0.66 (95% CI 0.53–0.67), another important prognostic marker explored in previous studies47,48.

(A1, A2,A3) Representative tumors (soft tissue window) belonging to phenotype 1. (B1,B2,B3) Representative tumors (soft tissue window) belonging to phenotype 2.
The tumors between the two multiomic graph phenotypes demonstrate statistically significant differences in terms of size (longest tumor diameter at baseline, tumor volume) as shown by MWU test p values < 0.05), with tumors belonging to phenotype 1 apparently bigger than tumors belonging to phenotype 2. The average value of tumor volume for patients belonging to multiomic graph phenotype 1 was 138.9 cm3 and those belonging to phenotype 2 was 19.8 cm3. The tumor volumes of the representative tumors shown in Fig. 8A belonging to phenotype 1 are 412.5 cm3 (A1), 89.1 cm3 (A2) and 251.1 cm3 (A3). The tumor volumes of the representative tumors shown in Fig. 8B belonging to phenotype 2 are 15.3 cm3 (B1), 22.7 cm3 (B2) and 8.8 cm3 (B3). The above observation suggests that although tumor size might be the only difference between the tumors appreciable visually, the multiomic signature may be able to capture additional patterns in the tumor ROIs that are relevant to prognosis and patient response to therapy. Further, larger tumors correspond to a higher t-stage in the NSCLC staging system and have been known to have a worse prognosis with increase in size. In our future work, we can derive delta multiomic graph phenotypes from the radiological, pathological and delta radiomics information obtained using the pre- and post-treatment scans of the patients. These delta multiomic graph phenotypes can help capture the changes in the descriptors of the tumor region over time, in response to therapy. We can assess the correlation between the delta multiomic graph phenotypes and the change in the volume of the tumor ROIs over time, which will further be an important prognostic indicator of the patient response to therapy.
To our knowledge, our study is the first to construct a “multiomic” signature for prognosis of NSCLC patient response to immunotherapy, in contrast to prior radiogenomic approaches leveraging a radiomics signature to identify patient categories based on a genomic biomarker-based classification. For instance, Chen et al. evaluated patient sustainability for PD-1 or PD-L1 checkpoint inhibitor immunotherapy49 using a CT radiomics-based signature for a cohort of 194 NSCLC patients (discovery cohort-85 patients, two testing cohorts-66 and 43 patients). Taking the standardized CD274 count as the response vector, a linear regression step was applied to select the radiomics features most relevant for CD274 prediction, and the weighted sum of these features was used to establish a radiogenomics signature- the lung cancer immunotherapy-radiomics prediction vector (LCI-RPV). In the NSCLC testing cohorts, LCI-RPV predicted PD-L1 positivity (AUC-0.7, 95% CI (0.57–0.84) and AUC-0.7, 95% CI (0.46–0.94)).
Previous radiomics studies have shown the effect of differences in image acquisition protocols on the reproducibility of radiomics features. For instance, in the study by Zhao et al., on a cohort of same day repeat CT lung cancer cohort, with each scan reconstructed at one of the six imaging settings (1.25 mm, 2.5 mm, 5 mm slice thickness with smooth and sharp reconstruction algorithms), found that interchanging smooth and sharp reconstruction algorithms can reduce feature reproducibility50. Similarly, the study by Meyer et al., involving CT scans obtained at different dose levels, section thickness, kernels and reconstruction algorithms settings, showed that the reproducibility of most radiomic features was highly affected by CT acquisition and reconstruction settings51. However, construction of radiomic signatures that are robust to the variation in the image acquisition protocol remains a largely unmet need. We believe that our study has addressed a relatively less explored need in the NSCLC immunotherapy literature by addressing the impact of heterogeneity in the image acquisition and reconstruction parameters on the information extracted from the radiomic descriptors of the tumor ROIs.
There are important limitations to our study. We were not able to explore multiomic integration approaches based on deep learning due to the relatively small sample size of the study. The retrospective, observational nature of the study did not allow the standardization of image acquisition protocols. Thus, we had to apply a nested ComBat approach to harmonize the differences in radiomic feature distribution that arose due to the variation in image acquisition parameters. Further, since this study has a clinical cohort composed of patients from a single institution, there is a potential bias stemming from therapy management strategies and from patient demographics, given that this patient population is largely derived from a large urban setting. This also poses a limitation on the assessment of the robustness of our prediction models due to the lack of independent external validation data. In addition, we have performed manual segmentation of the tumor regions, which can introduce a bias in tumor boundary selection. Previous radiomic studies have indicated the effect of inter-reader variability on tumor segmentation and subsequently, on the radiomic signatures developed from the tumor ROIs52. However, a recent preliminary study showed that radiomic features extracted from segmentations by different observers were highly correlated and had similar predictive value53. Our future work can involve exploration of fully automated tumor segmentation algorithms and evaluation of the effect of inter-reader segmentation variability on the radiomic features as well as application of our algorithms on larger cohorts derived from multicenter data.
Conclusion
In this study, we develop novel multiomic phenotypes by integration of radiomics, radiological and pathological information of the patients, to enhance precision in progression-free survival prediction, when combined with prognostic clinical variables, in patients with stage 4 NSCLC treated with first line PEMBRO (pembrolizumab) monotherapy or combination therapy. We have developed a multiomic graph integration approach for our cohort. The multiomic graph is developed by combining constituent radiomics, radiological and pathological graphs. The multiomic phenotypes, identified from the multiomic graph, essentially define patient groups based on information from the various “omics”. The addition of the multiomic phenotypes to established clinical biomarkers including BMI and smoking status, improved the accuracy of the prognostic model in the prediction of progression-free survival in patients with NSCLC. Our future work will explore the prognostic capability of delta radiomic signatures, capturing changes in tumor properties over time, in a longitudinal dataset obtained for this cohort.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed death-ligand 1
- PEMBRO:
-
Pembrolizumab
- NSCLC:
-
Non-small cell lung cancer
- PFS:
-
Progression-free survival
- SUVmax :
-
Maximum standardized uptake volume
- BMI:
-
Body mass index
References
Blank, C. & Mackensen, A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol. Immunother. 56, 739–745 (2007).
Hui, R. et al. Pembrolizumab as first-line therapy for patients with PD-L1-positive advanced non-small cell lung cancer: a phase 1 trial. Ann. Oncol. 28 (4), 874–881 (2017).
Kyi, C. & Postow, M. A. Immune checkpoint inhibitor combinations in solid tumors: opportunities and challenges. Immunotherapy 8 (7), 821–837 (2016).
Reck, M. et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N. Engl. J. Med. 375 (19), 1823–1833 (2016).
Rizvi, N. A. et al. Nivolumab in combination with platinum-based doublet chemotherapy for first-line treatment of advanced non–small-cell lung cancer. J. Clin. Oncol. 34 (25), 2969 (2016).
McLaughlin, J. et al. Quantitative assessment of the heterogeneity of PD-L1 expression in non–small-cell lung cancer. JAMA Oncol. 2 (1), 46–54 (2016).
Wang, Y. et al. PD-L1 expression in circulating tumor cells increases during radio (chemo) therapy and indicates poor prognosis in non-small cell lung cancer. Sci. Rep. 9 (1), 566 (2019).
Baraibar, I. et al. Safety and tolerability of immune checkpoint inhibitors (PD-1 and PD-L1) in cancer. Drug Saf. 42, 281–294 (2019).
Aerts, H. J. W. L. The potential of radiomic-based phenotyping in precision medicine: a review. JAMA Oncol. 2(12), 1636–1642 (2016).
Rolfo, C. et al. Liquid biopsy for advanced NSCLC: a consensus statement from the international association for the study of lung cancer. J. Thorac. Oncol. 16 (10), 1647–1662 (2021).
Valentinuzzi, D. et al. [F] FDG PET immunotherapy radiomics signature (iRADIOMICS) predicts response of non-small-cell lung cancer patients treated with pembrolizumab. Radiol. Oncol. 54 (3), 285–294 (2020).
Champiat, S. et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin. Cancer Res. 23 (8), 1920–1928 (2017).
Vaidya, P. et al. Novel, non-invasive imaging approach to identify patients with advanced non-small cell lung cancer at risk of hyperprogressive disease with immune checkpoint Blockade. J. Immunother. Cancer 8(2) (2020).
Trebeschi, S. et al. Predicting response to cancer immunotherapy using noninvasive radiomic biomarkers. Ann. Oncol. 30 (6), 998–1004 (2019).
Gupta, A. et al. Premorbid body mass index and mortality in patients with lung cancer: a systematic review and meta-analysis. Lung Cancer. 102, 49–59 (2016).
Petrelli, F. et al. Association of obesity with survival outcomes in patients with cancer: a systematic review and meta-analysis. JAMA Netw. Open. 4 (3), e213520–e213520 (2021).
Shukla, S. et al. Impact of body mass index on survival and serious adverse events in advanced non-small cell lung cancer treated with bevacizumab: a meta-analysis of randomized clinical trials. Curr. Med. Res. Opin. 37 (5), 811–817 (2021).
Jiang, M. et al. The relationship between body-mass index and overall survival in non-small cell lung cancer by sex, smoking status, and race: A pooled analysis of 20,937 international lung cancer consortium (ILCCO) patients. Lung Cancer. 152, 58–65 (2021).
Hoang, T. C. T. et al. Risk factors for early mortality from lung cancer: evolution over the last 20 years in the French nationwide KBP cohorts. ESMO Open. 9 (6), 103594 (2024).
Lin, Y. et al. Classifying breast cancer subtypes using deep neural networks based on multi-omics data. Genes 11(8), 888 (2020).
Li, Y., Scott, A. & Jackson Gene network reconstruction by integration of prior biological knowledge. G3 Genes Genomes Genet. 5(6), 1075–1079 (2015).
Rathore, S. et al. Brain cancer imaging phenomics toolkit (brain-CaPTk): an interactive platform for quantitative analysis of glioblastoma. Brainlesion: Glioma, Multiple Sclerosis, Stroke and Traumatic Brain Injuries: Third International Workshop, BrainLes 2017, Held in Conjunction with MICCAI 2017, Quebec City, QC, Canada, September 14, 2017, Revised Selected Papers 3. (Springer International Publishing, 2018).
Zwanenburg, A. et al. Image biomarker standardisation initiative. arXiv preprint arXiv:1612.07003 (2016).
Horng, H. et al. Improved generalized combat methods for harmonization of radiomic features. Sci. Rep. 12 (1), 19009 (2022).
Fortin, J. P. et al. Harmonization of cortical thickness measurements across scanners and sites. Neuroimage 167, 104–120 (2018).
Singh, A. et al. Development of a robust radiomic biomarker of progression-free survival in advanced non-small cell lung cancer patients treated with first-line immunotherapy. Sci. Rep. 12 (1), 9993 (2022).
Singh, A. et al. Resampling and harmonization for mitigation of heterogeneity in image parameters of baseline scans. Sci. Rep. 12 (1), 21505 (2022).
Yushkevich, P. A., Gao, Y. & Gerig, G. ITK-SNAP: An interactive tool for semi-automatic segmentation of multi-modality biomedical images. In 2016 38th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC) (IEEE, 2016).
Davis, A. P. et al. Efficacy of immunotherapy in KRAS-mutant non-small-cell lung cancer with comutations. Immunotherapy 13 (11), 941–952 (2021).
Ricciuti, B. et al. Diminished efficacy of programmed death-(ligand) 1 Inhibition in STK11-and KEAP1-mutant lung adenocarcinoma is affected by KRAS mutation status. J. Thorac. Oncol. 17 (3), 399–410 (2022).
Anderson, T. W. & Donald, A. Darling. A test of goodness of fit. J. Am. Stat. Assoc. 49 (268), 765–769 (1954).
Kassambara, A. Practical Guide To Cluster Analysis in R: Unsupervised Machine Learning, vol. 1 (Sthda, 2017).
Ward, H. Jr Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 58 (301), 236–244 (1963).
Monti, S. et al. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Mach. Learn. 52, 91–118 (2003).
Liu, Y. et al. Statistical significance of clustering for high-dimension, low–sample size data. J. Am. Stat. Assoc. 103 (483), 1281–1293 (2008).
Chitalia, R. D. et al. Imaging phenotypes of breast cancer heterogeneity in preoperative breast dynamic contrast enhanced magnetic resonance imaging (DCE-MRI) scans predict 10-year recurrence. Clin. Cancer Res. 26 (4), 862–869 (2020).
Kernighan, B. W. & Shen, L. An efficient heuristic procedure for partitioning graphs. Bell Syst. Tech. J. 49(2), 291–307 (1970).
Newman, M. E. J. & Girvan, M. Finding and evaluating community structure in networks. Phys. Rev. E. 69 (2), 026113 (2004).
Bland, J. M. & Altman, D. G. Survival probabilities (the Kaplan–Meier method). BMJ. 317 (7172), 1572–1580 (1998).
Uno, H. et al. On the C-statistics for evaluating overall adequacy of risk prediction procedures with censored survival data. Stat. Med. 30 (10), 1105–1117 (2011).
Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 19(6), 716–723 (1974).
Mann, H. B. & Donald, R. Whitney. On a test of whether one of two random variables is stochastically larger than the other. Ann. Math. Stat. 50–60 (1947).
Van der Walt, S. et al. scikit-image: image processing in Python. PeerJ. 2, e453 (2014).
Core Team, R. R. R: A language and environment for statistical computing (2013).
Islam, M. et al. An integrative deep learning framework for classifying molecular subtypes of breast cancer. Comput. Struct. Biotechnol. J. 18, 2185–2199v (2020).
Yang, H. et al. Subtype-GAN: a deep learning approach for integrative cancer subtyping of multi-omics data. Bioinformatics. 37 (16), 2231–2237 (2021).
Cangır, A. et al. Prognostic value of tumor size in non-small cell lung cancer larger than five centimeters in diameter. Lung Cancer. 46 (3), 325–331 (2004).
Zhang, J. et al. Relationship between tumor size and survival in non-small-cell lung cancer (NSCLC): an analysis of the surveillance, epidemiology, and end results (SEER) registry. J. Thorac. Oncol. 10 (4), 682–690 (2015).
Chen, M. et al. A novel radiogenomics biomarker for predicting treatment response and pneumotoxicity from programmed cell death protein or ligand-1 Inhibition immunotherapy in NSCLC. J. Thorac. Oncol. 18 (6), 718–730 (2023).
Zhao, B. et al. Reproducibility of radiomics for deciphering tumor phenotype with imaging. Sci. Rep. 6 (1), 23428 (2016).
Meyer, M. et al. Reproducibility of CT radiomic features within the same patient: influence of radiation dose and CT reconstruction settings. Radiology 293 (3), 583–591 (2019).
Pavic, M. et al. Influence of inter-observer delineation variability on radiomics stability in different tumor sites. Acta Oncol. 57 (8), 1070–1074 (2018).
Hershman, M. et al. Impact of interobserver variability in manual segmentation of non-small cell lung cancer (NSCLC) applying low-rank radiomic representation on computed tomography. Cancers. 13(23), 5985 (2021).
Author information
Authors and Affiliations
Contributions
Overseeing the work, D.K.; clinical studies, L.R., M.H., C.A., E.C., S.I.K.; all experiments (except nested ComBat analysis), writing the manuscript, A.S.; nested ComBat analysis, H.H.; contribution to method development, A.S., H.H., E.A.C.; manuscript revision, all authors; approval of final version of submitted manuscript, all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Singh, A., Horng, H., Roshkovan, L. et al. Development of a prognostic multiomic biomarker of progression-free survival in advanced non-small cell lung cancer patients treated with first line immunotherapy. Sci Rep 15, 34591 (2025). https://doi.org/10.1038/s41598-025-18101-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-18101-y
