
Abstract
A series of azo-linked pyrazolophthalazine-5,10-diones (APPDs) were synthesized via multicomponent reaction of phthalic anhydride, synthetized azo-aldehyde, malononitrile, and hydrazine hydrate using the gabapentin (GBP) functionalized silica coated Fe3O4 magnetic nano composites (MT@SP@GBP MNCs) as a catalyst. All derivatives were characterized by FT-IR, 1H NMR, 13C NMR, and mass spectrometry. MT@SP@GBP MNCs was characterized by FT-IR, FE-SEM, TEM, TGA, XRD, EDX and VSM.
Similar content being viewed by others
Introduction
Pyrazolophthalazines (PPs) are one of the important groups of heterocyclic compounds with a pyrazole and a phthalazine ring fused to a hydrazine. PPs play a valuable role in the pharmaceutical and agricultural industries. PPs showed different biological activities such as anti-microbial1, muscle relaxant2, anti-fungal3, anti-convulsant and anti-tumor4, anti-bacterial5 anti-inflammatory and analgesic6, anti-cancer7 and anti-oxidant8.
As a result, the synthesis of nitrogen-containing heterocycles as well as PPs has attracted the attention of researchers because of their wide range of biological activities. Different synthetic procedures have been reported for the synthesis of PPs as ZnFe2O49, Fe3O4@ SiO2-imine/phenoxy-Cu(II)10, PbO11, CuO12, [Bu3NH][HSO4]13, 1-(Cu-ferrite-siloxypropyl)-3-methyl imidazolium polytungstate ionic liquid14, 2,2,2-trifluoroethanol15, diisopropyl ethyl ammonium acetate16, γ-Fe2O3/talc/CuII NPs17, CuI@KSF18, β-cyclodextrin15, 1,1,1,3,3,3-hexafluoro-2-propanol19, hercynite20, uric Acid21, extract of mango peel ash/microwave irradiation22, Fe3O4@GOQDs-N-(β-alanine)23, bovine serum albumin in water24, and [bmim]OH25. Among the various mentioned synthetic methods, nanocatalysts have received more attention compared to other catalysts. High efficiency, high safety, reusable and optimal use of raw materials are the advantages of nanocatalysts. In addition, nanocatalysts do not dissolve in the reaction solution due to their larger dimensions and can be easily separated26.
Azo compounds are the synthetic dyes that were utilized in textile dyeing, textile, leather, tanneries, plastic, and paint industries27,28,29,. In addition, different biological properties were reported for azo heterocycles. For example, M. Alsafy and Alrazzak reported synthesis and antibacterial activities of azo phthalazine30. Malleva et al. Reported azobenzene-nitrazepam as anion-selective Cys-loop receptors31, Mezgebe and Mulugeta reported different heterocycle azo derivatives as anti-viral, anti-convulsant, anti-microbial, anti-diabetic, anti-fungal, anti-inflammatory, and chemosensing agents32.
The application of nanocatalysts in the synthesis of organic compounds has seen a not able rise in recent years, primarily attributed to their straightforward isolation process. Furthermore, their substantial surface area has enhanced their utility in this domain33,34,35,. It is crucial to note that while nanomaterials may display comparable characteristics at the macroscopic level, they possess distinct properties at the nanoscale, and the catalytic efficiency of nanoparticles can be affected by the inherent properties of the nanomaterials36,37,38,39,40,. Nanocatalysts have been documented to exhibit enhanced reaction efficiency, elevated reaction rates, diminished by-product formation, and superior reaction selectivity. Furthermore, these materials have demonstrated comparable outcomes in optimizing the conditions for multicomponent reactions41,42,43,44,45,46,47,48,49,50.
Herein, on continuing of our study on the development of nanocatalysts in azo heterocycle compounds and synthetic procedures, we reported synthesis of APPDs via multicomponent reaction of azo-aldehydes, malononitrle, phthalic anhydride and hydrazine hydrate in MT@SP@GBP MNCs as catalyst (Fig. 1).
Experimental
Materials and instruments
The solvents and chemical materials were obtained from Fluka and Merck. FT-IR spectra were obtained on a Shimadzu (8400 S) spectrometer. Bruker DRX 500 spectrometer at 250 and 62.5 MHz were used for NMR spectrum. The m/e ratio of product ions was obtained by an Agilent instrument (70 eV). FESEM were done by MIRAII, and TEM were obtained on a Zeiss-EM10C (100 KV) scanning microscope. An energy dispersive analysis of X-ray (MIRA II, 20 kV) were applied for the compositional analysis. TGA was carried out at 25–700 °C on a Mettler Toledo apparatus. The BELSORP MINI II were applied for BET analysis and determination of the catalyst’s specific surface area and pore volume. Vibrating sample magnetometer (VSM) (Kavir Kashan company, 500-0.0005 emu, 2 teslas) were used for magnetic properties. Powder X-ray diffraction (XRD) was recorded by Philips Xpert in 0–80° (2θ) and CuKα, radiation, λ = 0.154056.
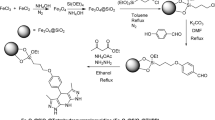
Preparation of MT@SP@GBP MNCs
The synthetized Fe3O4 & Fe3O4@SP-Cl MNPs were synthesized by research group, et al.51,52. In brief, first FeCl2 (2.0 g) and FeCl3 (2.0 g) were stirred in H2O (15 mL) and triethylamine (5 mL) for 24 h. Then, the desired mixture was stirred for half an hour under ultrasonic waves at room temperature. Then CPTES (15 mL) was added to the desired mixture and it was stirred for 24 h. Finally, Fe3O4@SP-Cl was obtained. Then, Fe3O4@SP-Cl MNPs (0.2 g), gabapentin (GBP) (0.2 g), and Et3N (5 mL) were added. It was stirred for 24 h. After stirring with the magnet, separation was performed and incubated in oven at 60 °C for 24 h. The structure of MT@SP@GBP MNCs obtained was confirmed by TEM, FE-SEM, TGAو DLS, VSM, Zeta Potential and FT-IR spectroscopy (Figs. 2, 3, 4, 5, 6, 7, 8 and 9).
Synthesis of APPDs 5a-i
Azo aldehydes (1 mmol), malononitrile (1 mmol, 0.066 g), hydrazine hydrate (1 mmol, 0.050 g), phthalic anhydride (1 mmol, 0.148 g) and MT@SP@GBP MNCs (0.1 g) was mixed and stirred at 25 °C. At the end of the reaction that determined with TLC, MT@SP@GBP MNCs was separated and was washed with hot water (2 × 5.0 mL) and ethanol (2 × 5.0 mL). The solid products were separated by filtration and were recrystallized from ethanol to produce pure products. The spectroscopic data for PPD compounds 5a-i were obtained as below:
(5a): Pale Red solid; Yield: 97%; m.p: 178–180 °C: FT-IR (KBr, cm-1); 3583, 3556 (NH2 stretch), 3413 (O-H stretch), 2952 (C-H stretch), 2259 (CN stretch), 1623 (C = O amide), 1579 (C = C stretch), 1377 (C-N stretch), 1234 (C-O stretch) cm-1. 1H NMR (DMSO-d6, 250 MHz): δH: 6.90 (s, 1H), 7.29 (s, 1H), 7.46 (s, 1H), 7.58 (s, 1H, Ar), 7.70 (s, 1H, Ar), 7.76 (s, br., 3 H, Ar), 8.11 (s, br., 3 H, Ar), 11.49 (s, 1H, OH) ppm; 13C NMR (DMSO-d6, 75 MHz): δC: 63.5, 119.2, 124.6, 125.5, 127.5, 128.9, 130.7, 132.7, 133.0, 135.4, 136.7, 147.0, 148.6, 154.9, 157.4, 160.2, 168.7, 204.0, 208.3; Anal. calcd for C24H14Cl2N6O3: C, 63.16; H, 3.28; N, 17.54. Found: C, 63.15; H, 3.25; N, 17.57.
(5b): Brown solid; Yield: 89%; m.p: 180–182 °C: FT-IR (KBr, cm-1); 3404 (O-H stretch), 2958(C-H stretch), 2255 (CN stretch), 1620 (C = O amide), 1587 (C = C stretch), 1517 and 1346 (NO2 stretch), 1228 (C-N stretch), 1143 (C-O stretch) cm-1. 1H NMR (DMSO-d6, 250 MHz): δH: 2.70 (s, 3 H, CH3), 6.78 (s, 1H), 7.33 (d, J = 6.2 Hz, 1H), 7.59–7.67 (m, 4 H), 7.96 (d, J = 6.2 Hz, 1H), 8.13 (s, 2 H), 8.25 (s, 2 H) ppm; 13C NMR (DMSO-d6, 100 MHz): δC: 22.5, 62.2, 113.2, 113.3, 117.0, 118.2, 119.4, 122.5, 124.1, 124.9, 125.9, 126.7, 128.8, 131.1, 138.6, 139.3, 148.7, 149.3, 152.7, 153.5, 154.0, 163.3, 208.9, 209,8; Anal. calcd for C25H17N7O5: C, 63.16; H, 3.28; N, 17.54. Found: C, 63.14; H, 3.31; N, 17.59.
(5c): Dark orange; Yield: 93%; m.p: 185–187 °C: FT-IR (KBr, cm-1); 3406 (OH stretch), 2129 (CN stretch), 1623 (C = O amide), 1515 and 1344 (NO2 stretch), 1278 (C-O stretch), cm-1. 1H NMR (DMSO-d6, 300 MHz): δH: 6.81 (S, 1H), 7.57–7.60 (m, 4 H), 7.77–7.80 (m, 4 H), 7.88–7.91 (m, 1H), 8.08–8.14 (m, 1H), 13.26 (s, 1H, OH) ppm; 13C NMR (DMSO-d6, 100 MHz): δC: 61.6, 125.2, 125.6, 125.9, 127.6, 128.6, 129.2, 129.5, 130.4, 131.1, 131.3, 131.4, 131.7, 132.5, 133.0, 133.6, 135.8, 136.7, 153.9, 159.1, 201.2, 202.7; Anal. calcd for C24H15N7O5: C, 58.22; H, 3.02; N, 12.93. Found: C, 58.25; H, 2.99; N, 12.90.
(5d): Orange; Yield: 90%; m.p: 186–188 °C: FT-IR (KBr, cm-1); 3407 (O-H stretch), 2241 (CN stretch), 1654 (C = O amide), 1564 (C = C stretch), 1371 (C-N stretch), 1240 (C-O stretch) cm-1. 1H NMR (DMSO-d6, 400 MHz): δH: 6.77 (s, 1H), 7.15 (s, 1H), 7.39–7.60 (m, 6 H, Ar), 7.72 (s, 1H), 7.96–8.08 (m, 4 H, Ar) ppm; 13C NMR (DMSO-d6, 100 MHz): δC: 61.56, 122.5, 122.9, 124.6, 125.1, 125.5, 125.8, 126.3, 128.6, 129.2, 129.9, 130.2, 131.0, 131.3, 131.6, 132.5, 132.9, 133.9, 136.6, 152.2, 200.0, 203.1; Anal. calcd for C24H16N6O3: C, 64.87; H, 3.37; N, 14.41. Found: C, 64.90; H, 3.39; N, 14.38.
(5e): Dark red; Yield: 89%; m.p: 181–183 °C: FT-IR (KBr, cm-1); 3429 (O-H stretch), 2956 (C-H stretch), 2252 (CN stretch), 1623 (C = O amide), 1521 (C = C stretch), 1344 (C-N stretch), 1230 (C-O stretch) cm-1. 1H NMR (DMSO-d6, 400 MHz): δH; 2.17 (s, 3 H, CH3), 6.76 (s, 1H), 6.97 (s, 1H), 7.28 (s, 2 H), 7.52 (s, 1H), 7.71–8.03 (m, 4 H, Ar), 8.27 (s, br, 3 H, Ar), 13.10 (s, 1H, OH) ppm; 13C NMR (DMSO-d6, 100 MHz): δC: 14.1, 61.5, 123.7, 125.2, 125.5, 127.6, 128.6, 129.2, 129.7, 130.0, 131.0, 131.4, 131.6, 133.0, 133.9, 135.0, 136.6, 155.1, 163.8, 168.9, 174.2, 201.9, 202.8; Anal. calcd for C25H18N6O3: C, 63.92; H, 3.66; N, 16.94. Found: C, 63.90; H, 3.63; N, 16.96.
(5f): Brown; Yield: 88%; m.p: 182–184 °C: FT-IR (KBr, cm− 1); 3413 (OH stretch), 2246 (CN stretch), 1668 (C = O amide), 1618, 1573 (C = C stretch), 1481 (C-N stretch), 1089 (C-O stretch) cm− 1. 1H NMR (DMSO-d6, 400 MHz): δH: δH; 6.84 (s, 1H), 7.53 (s, br, 3 H, Ar), 7.60 (s, br, 2 H, Ar), 7.74 (s, br, 2 H, Ar), 7.85 (s, br, 2 H, Ar), 8.04 (s, br, 2 H, Ar), 10.41 (s, 1H, OH) ppm; 13C NMR (DMSO-d6, 100 MHz): δC:61.56, 122.1, 124.7, 125.1, 125.5, 127.5, 128.1, 128.6, 129.4, 129.9, 130.2, 131.1, 131.6, 132.1, 133.0, 133.5, 136.6, 137.7, 151.5, 155.1, 202.0, 203.1; Anal. calcd for C24H15IN6O3: C, 64.87; H, 3.37; N, 14.41. Found: C, 64.84; H, 3.34; N, 14.43.
(5 g): Brown; Yield: 93%; m.p: 177–179 °C: FT-IR (KBr, cm− 1); 3475 (OH stretch), 2243 (CN stretch), 1660 (C = O amide), 1625(C = C stretch), 1529, 1348 (NO2 stretch), 1303 (C-N stretch), 1076 (C-O stretch) cm− 1. 1H NMR (DMSO-d6, 400 MHz): δH: 7.02 (s, 1H), 7.56–7.06 (m, 3 H), 7.69–7.92 (m, 4 H), 8.00-8.34 (m, 4 H) ppm; 13C NMR (DMSO-d6, 100 MHz): δC: 61.9, 125.1, 125.5, 125.7, 126.1, 128.6, 129.2, 129.9, 130.1, 131.1, 131.3, 131.6, 132.6, 132.9, 133.9, 136.5, 148.2, 148.7, 151.9, 156.2, 156.6, 163.8, 200.5, 202.1; Anal. calcd for C24H15N7O5: C, 64.87; H, 3.37; N, 14.41. Found: C, 64.90; H, 3.39; N, 14.38.
(5 h): Dark red; Yield: 94%; m.p: 196–198 °C: FT-IR (KBr, cm− 1); 3467 (N-H stretch), 3440 (OH stretch), 2196 (CN stretch), 1643 (C = O amide), 1568 (C = C stretch), 1288 (C-N stretch), 1072 (C-O stretch) cm− 1. 1H NMR (DMSO-d6, 400 MHz): δH: 6.71 (s, 1H), 7.32 (s, br, 1H), 7.55 (s, br, 1H), 7.78 (s, br, 1H), 7.91 (s, br, 1H), 8.07 (s, br, 1H, Ar) ppm; 13C NMR (DMSO-d6, 100 MHz): δC: 61.5, 113.2, 113.3, 118.1, 119.5, 124.8, 125.5, 128.6, 129.2, 131.1, 131.3, 131.6, 133.0, 133.4, 136.6, 148.9, 151.1, 152.2, 163.5, 169.0, 201.7, 202.3; Anal. calcd for C24H15BrN6O3: C, 52.52; H, 2.73; N, 11.67. Found: C, 52.55; H, 2.70; N, 11.70.
(5i): Brown; Yield: 93%; m.p: 178–180 °C: FT-IR (KBr, cm− 1); 3404 (OH stretch), 3082 (C-H stretch), 2966 (C-H stretch), 2189 (CN stretch), 1662, 1569 (C = O amide), 1404 (C-N stretch), 1278 (C-O stretch) cm− 1. 1H NMR (DMSO-d6, 400 MHz): δH: 1H NMR (DMSO-d6, 300 MHz): δH; 7.00 (s, 1H), 7.34 (d, J = 1.5 Hz, 1H), 7.56–7.63 (m, 4 H), 7.73–7.77 (m, 3 H), 7.85–7.89 (m, 2 H), 7.92–7.97 (m, 1H) ppm; 13C NMR (DMSO-d6, 100 MHz): δC: 61.6, 119.9, 123.9, 124.6, 125.1, 125.6, 128.8, 129.2, 129.3, 129.4, 130.0, 131.1, 131.8, 133.0, 133.5, 148.9, 150.7, 152.2, 163.8, 201.1, 202.6; Anal. calcd for C24H15ClN6O3: C, 71.73; H, 4.38; N, 15.21. Found: C, 71.72; H, 4.41; N, 15.20.
Results and discussion
Characterization of MT@SP@GBP MNCs
In our previous research, we reported synthesis of azo-linked heterocyclic compounds via eco-friendly protocols51,52,53,54,55,56,. So, in this study, after preparation of MT@SP@GBP MNCs, different technical analyses including FT-IR, FE-SEM, TEM, TGA, XRD, EDX, VSM and BET were used for nanocatalyst characterization.
The MT@SP@GBP MNCs was prepared and fully characterized as detailed in the Supplementary Materials. In brief, the MT@SP core-shell structures were subjected to sequential treatment with 3-chloropropyltriethoxysilane (CPTES) and GBP, resulting in the production of the desired MNC (Fig. 2).
FT-IR spectroscopy of MT@SP@GBP MNCs was performed to identify the functional groups of the synthesized nanoparticles. The vibrations of O-H band from -COOH group were observed in 2980–3490 cm− 1 and the strong intense band at 3524 cm− 1 is related to the N-H stretching vibrations of nano-catalyst, The band at 1638 cm− 1 is related to the C = O stretching vibrations of imine group, The bands at 1512 and 1463 cm− 1 are related to the C = C vibrations of aromatic part, The strong intense band at 1163 and 1029 cm− 1 is related to C-O and Si-O stretching vibrations respectively and vibrations of Fe-O band were observed in 639 cm− 1 (Fig. 3).

Synthesis of novel APPD 5a-i with MT@SP@GBP MNCs.

The synthesis of MT@SP@GBP MNCs.

The FT-IR spectra of (a) MT, (b) MT@SP, and (c) synthesized MT@SP@GBP MNCs.
The size and morphology of the MT@SP@GBP MNCs was studied by TEM and FE-SEM. The following figure shows the TEM images of the sample at different scales of 47 and 72 nm. TEM and FE-SEM images of the MT@SP@GBP MNCs reveal that MNCs are formed with nearly spherical morphology having a particle size of 47–72 nm. Furthermore, TEM images show some aggregation, which was illustrated the successful grafting of the GBP (Figs. 4).
Figure 5 displays the VSM plot of the MT@SP@GBP MNCs. Based on the VSM spectrum analysis of the specimen; results indicate that an elevation in external magnetic field intensity along the x-axis translates to a concurrent increase in the magnetic susceptibility of the material. At approximately 5300 Tesla, the magnetic property becomes saturated and attains a value of 39.83 as the Hysteresis saturated (Hs) value (Fig. 5).
Figure 6 displays TGA spectrum of MT@SP@GBP MNCs. Based on Fig. 6, in temperatures between 25 and 100 °C, MT@SP@GBP MNCs lost 11% of their weight after removing moisture absorbed on the surface. Then, the sample lost 32% of weight from organic carbon compounds (GBP) at 290–350 °C and in the temperature range of 430–450 °C, the weight loss (10%) is due to the melting of parts of the sample due to the breaking of oxygen-metal bonds. So, the sample has about 53% weight loss until its pure form at 450 °C. Therefore, the stability of the catalyst is 280 °C (Fig. 6).

(A) The TEM image and (B) FE-SEM images of MT@SP@GBP MNCs.

The VSM image of synthesized MT@SP@GBP MNCs.

The TGA image of synthesized MT@SP@GBP MNCs.
To check the structure of MT@SP@GBP MNCs more closely, DLS analysis and Zeta Potential were also performed. According to the data of DLS analysis, the nano particle size is 34.63 nm and according to Zeta Potential analysis, the surface tension of the composition is equal to 34.6 mV (Fig. 7).
The present study employs XRD analysis to examine the MT@SP@GBP MNCs in comparison to pure Fe3O4. The presented pattern showed that the Fe3O4 iron oxide phase attains the foremost ranking amidst other phases, thereby denoting its singular phase composition (Fig. 8).
The energy-dispersive X-ray spectrum (EDS) dedicated the C (13.16 w/w %), O (33.78 w/w %), N (3.89 w/w %), Si (6.33 w/w %) and Fe (42.83 w/w %) atoms in the structure of MT@SP@GBP MNCs (Fig. 9).

The DLS and Zeta potential of synthesized MT@SP@GBP MNCs.

The XRD of synthesized MT@SP@GBP MNCs.

The EDX of synthesized MT@SP@GBP MNCs.
In the continuing on our previous research and studying the efficiency of the MT@SP@GBP MNCs, the preparation of azo-linked PPDs via the one-pot reaction of hydrazine hydrate, phthalic anhydride, azo aldehydes and malononitrile by MT@SP@GBP MNCs were studied. At first, the reaction parameters were optimized and we used the reaction of 2-hydroxy-5-((2,4-dichlorophenyl)diazenyl)benzaldehyde 1a, malononitrile 2, phthalic anhydride 3 and hydrazine hydrate 4 as a model reaction.
Catalytic studies
To synthesize derivatives of PPDs via the reaction of 1a (1 mmol), malononitrile 2 (1 mmol), phthalic anhydride 3 (1 mmol) and hydrazine hydrate 4 (1 mmol) and 0.1 g of various catalysts were studied at a temperature of 60 °C (Table 1).
Influence of temperatures on reaction time
The synthesis of PPD 5a involved the reaction of 1a (1 mmol), malononitrile 2 (1 mmol), phthalic anhydride 3 (1 mmol) and hydrazine hydrate 4 (1 mmol) with MT@SP@GBP (0.1 g). This study was investigated via changing the temperatures and the results can be ascribed that the best temperature in this synthesis, is room temperature (Table 2).
Effect of MNCs catalyst value
The results of the investigation of the quantities of nanocatalyst were shown in Table 3. The findings confirmed that 0.1 g of the nanocatalyst per 1 mmol of azo-aldehyde resulted in higher yield and shorter reaction time (Table 3).
The recyclability and reusability of catalyst was studied in the model one-pot reaction among 1a (1 mmol), malononitrile 2 (1 mmol), phthalic anhydride 3 (1 mmol) and hydrazine hydrate 4 (1 mmol), and 0.1 g of MT@SP@GBP. The separated catalyst is reusable after the reaction by washing with warm EtOH and drying at 80 °C. MT@SP@GBP was used again for subsequent experiments under similar reaction conditions. The catalyst is reusable for the next cycle without noticeable loss of its activity. After six cycles of reusing the catalyst, yields of the product decreased only slightly. Experience has shown that reusing the catalyst for the seventh time greatly reduced the efficiency of the reaction (Table 4).
To evaluate the efficacy of the reported procedure, different azo-linked aldehydes, malononitrile, phthalic anhydride, hydrazine hydrate and MT@SP@GBP MNCs were applied to reaction in H2O at 25 °C (Fig. 1; Table 5).
Conclusions
In summary, we have reported a new protocol for the synthesis of novel azo-linked PPDs by reaction of azo-linked aldehydes, malononitrile phthalic anhydride, hydrazine hydrate with MT@SP@GBP MNCs. The reaction proceeds the in room-temperature to produce the PPDS in good yields (88–97%). Room temperature reaction conditions, easy separation, and recyclable MT@SP@GBP nanocatalyst are the notable aspects of this study.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
References
Said, M. S. et al. Total synthesis of (-)-2-methoxy-2-butenolide-3-cinnamate and its antimicrobial potentials. Nat. Prod. Res. 35, 5177–5182. https://doi.org/10.1080/ 14786419.2020.1789979 (2021).
Awadallah, F. M., Saleh, D. O. & El-Eraky, W. Synthesis, vasorelaxant activity, and molecular modeling study of some new phthalazine derivatives. Eu J. Med. Chem. 52 (1), 14–21. https://doi.org/10.1016/j.ejmech.2012.02.051 (2012).
Khalaf, M. New phthalazine based nickel (II), Cobalt (II), and copper (II) mixed-ligand complexes; characterization, physicochemical properties, anti-inflammatory, antifungal, antibacterial, DFT and molecular Docking exploration. J. Ind. Chem. Soc. 101 (8), 101191. https://doi.org/10.1016/j.jics.2024.101191 (2024).
Abu El-Azm, S. M., Mahmoud, F. R., Hekal, H. & M. & Recent developments in chemistry of phthalazines. Org. Chem. Curr. Res. 4 (1), 1000132. https://doi.org/10.4172/2161-0401.1000132 (2015).
Bachhar, V., Joshi, V., Singh, A., Mir, M. A. & Bhardwaj, A. Antibacterial, antioxidant, and antidiabetic activities of TiO2 nanoparticles synthesized through ultrasonication assisted cold maceration from stem extract of euphorbia Hirta. Lett. Appl. Nano Bio Sci. 14 (1). https://doi.org/10.33263/LIANBS141.001 (2025).
Da-Chuan, L., Gong, G. H., Wei, C. X., Jin, X. J. & Quan Zh. Sh. Synthesis and anti-inflammatory activity evaluation of a novel series of 6-phenoxy- [1,2,4]triazolo[3,4-a]phthalazine-3-carboxamide derivatives. Bio Med. Chem. Lett. 26 (6), 1576–1579. https://doi.org/10.1016/j.bmcl.2016.02.008 (2016).
Li, J., Zhao, Y. F., Yuan, X. Y., Xu, J. X. & Gong, P. Synthesis and anticancer activities of novel 1,4-disubstituted phthalazines. Molecules 11 (7), 574–582. https://doi.org/10.3390/11070574 (2006).
Badiger, K. B., Sannegowda, L. K. & Kamanna, K. Microwave-assisted one-pot synthesis of tetrahydrobenzo [b] Pyrans in the presence of WEWFPA and their electrochemical studies. Org. Commun. 15 (2), 148–166. https://doi.org/10.25135/acg.oc.124.2111.2263 (2022).
Mhaske, A. K., Gadhave, A. G., Dholi, A. G. & Uphade, B. K. One-pot, green synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-diones by ZnFe2O4 nanoparticles an efficient nanocatalyst. J. Inorg. Organomet. Polym. Mater. 34 (3), 999–1014. https://doi.org/10.1007/s10904-023-02875-7 (2024).
Nesarvand, M., Azarifar, D. & Ebrahimiasl, H. One-pot and green synthesis 1H-pyrazolo[1,2-b]phthalazine-5,10-dione and dihydropyrano[3,2-c]chromene derivatives by Fe3O4@SiO2-imine/phenoxy-Cu(II) as an efficient and reusable catalyst. Res. Chem. Intermed. 47, 3629–3644. https://doi.org/10.1007/s11164-021-04498-4 (2021).
Tayebee, R., Maleki, B. & Sabeti, M. A new simple method for the Preparation of PbO nanoparticles and implementation of an efficient and reusable catalytic system for the synthesis of 1H-pyrazolo[1,2- b]phthalazine-5,10-diones. J. Iran. Chem. Soc. 14, 1179–1188. https://doi.org/10.1007/s13738-017-1068-2 (2017).
Patil, S., Mane, A. & Dhongade-Desai, S. CuO nanoparticles as a reusable catalyst for the synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-dione derivatives under solvent-free conditions. J. Iran. Chem. Soc. 16, 1665–1675. https://doi.org/10.1007/s13738-019-01640-3 (2019).
Shaikh, M. A., Farooqui, M. & Abed, S. [Bu3NH][HSO4] catalyzed: an eco-efficient synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-diones and 2H-indazolo[2,1-b]phthalazine-triones under solvent-free conditions. Res. Chem. Intermed. 44, 5483–5500. https://doi.org/10.1007/s11164-018-3435-8 (2018).
Saadati-Moshtaghin, H. R. & Zonoz, F. M. Synthesis and characterization of magnetically recoverable 1-(copperferritesiloxypropyl)-3-methylimidazolium heteropolytungstate ionic liquid as a new nanocatalyst for the Preparation of 1H-pyrazolo[1,2-b]phthalazine-5,10-diones. J. Nanostruct. Chem. 7, 317–325. https://doi.org/10.1007/s40097-017-0241-6 (2017).
Mohamadpour, F. Polyfluorinated alcohol as a reusable media promoted catalyst-free green synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-dione derivatives. Results Chem. 7, 101546. https://doi.org/10.1016/j.rechem.2024.101546 (2024).
Jadhav, C. K., Nipate, A. S., Chate, A. V., Kulkarni, M. V. & Gill, C. H. Microwave-Assisted chemistry: new synthetic application for the rapid construction of 1H-Pyrazolo[1,2-b]Phthalazine-5,10-Dione derivatives in diisopropyl Ethyl ammonium acetate. Polycycl. Aromat. Compd. 43, 895–914. https://doi.org/10.1080/10406638.2021.2021252 (2023).
Chalaki, B., Akhlaghinia, B. & S. & CuII anchored onto the magnetic talc: a new magnetic nanostructured catalyst for the one-pot gram-scale synthesis of 1H-Pyrazolo[1,2-b]phthalazine-5,10-dione derivatives. Chem. Select. 5 (35), 11010–11019. https://doi.org/10.1002/slct.202002099 (2020).
Mahmoodi, N. O., Shahabbasi, A., Yazdani Nyaki, H. & Taherpour, H. Flexible green synthesis of 2-benzyl-3hydroxy-1H-pyrazolo([1,2-b]phthalazine and [1,2-a] pyridazine)dione derivatives using CuI@KSF nanocatalyst under solvent-free conditions. J. Mol. Struct. 1321, 139813. https://doi.org/10.1016/j.molstruc.2024.139813 (2025).
Mohamadpour, F. Synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-dione derivatives in a reusable catalyst/solvent; 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP). Curr. Res. Green. Sustain. 9, 100427. https://doi.org/10.1016/j.crgsc.2024.100427 (2024).
Paknejadi, Z. & Kefayati, H. Appl. Chem. Today 19, 183–200. https://doi.org/10.22075/chem.2024.30688.2177 (2024).
Mohamadpour, F., Maghsoodlou, M. T., Heydari, R. & Lashkari, M. Uric acid as a natural and reusable catalyst for synthesis of biologically significant 3,4-dihydropyrimidinones/thiones, 1H-pyrazolo[1,2-b]phthalazine-5,10-diones and poly substituted dihydropyrrol-2-ones. Org. Prep. Proc. Int. 56(2), 118-129 (2024). https://doi.org/10.1080/00304948.2023.2235948
Hiremath, B., Kantharauj, K. & P. & Microwave-Accelerated facile synthesis of 1H-Pyrazolo[1,2-b]Phthalazine-5,10-Dione derivatives catalyzed by WEMPA. Polycycl. Aromat. Compd. 42, 2162–2178. https://doi.org/10.1080/10406638.2020.1830129 (2020).
Khaleghiabbasabadi, M. & Azarifar, D. β-Alanine-functionalized magnetic graphene oxide quantum dots: an efficient and recyclable heterogeneous basic catalyst for the synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-dione and 2,3-dihydroquinazolin-4(1H)-one derivatives. Appl. Organomet. Chem. 34, e5872. https://doi.org/10.1002/aoc.5872 (2020).
Farahanipour, A., Bavandi, H., Shahedi, M. & Habib, Z. Synthesis of 1H-Pyrazolo[1,2-b]phthalazine-5,10-dione and 1H-pyrazolo[1,2-a]pyridazine-5,8-dione derivatives by bovine serum albumin in water. Polycycl. Aromat. Compd. 43, 7042–7051. https://doi.org/10.1080/10406638.2022.2128829 (2023).
Loka Maheshwari, D. P. & Satyanarayana, B. One pot, Four component synthesis of phthalazine-1,4-diones derivatives in [BMIM][OH] medium. Asian. J. Chem. 32 (1), 45-48 (2020). https://doi.org/10.14233/ajchem.2020.22180
Somwanshi, B., Somvanshi, B. & Kharat, P. Nanocatalyst: A brief review on synthesis to applications. J. Phys. Conf. Ser. 1644, 012046. https://doi.org/10.1088/1742-6596/ 1644/1/012046 (2020).
Lipskikh, O. I., Korotkova, E. I., Khristunova, Y. P., Barek, J. & Kratochvil, B. Sensors for voltammetric determination of food Azo dyes - A critical review. Electrochim. Acta. 260, 974–985. https://doi.org/10.1016/j.electacta.2017.12.027 (2018).
Alsantali, I. Miscellaneous Azo dyes: a comprehensive review on recent advancements in biological and industrial applications. Dyes Pigm. 199, 110050. https://doi.org/10.1016/j.dyepig.2021.110050 (2022).
Benkhaya, S., M’rabet, S. & El Harf, A. Classifications, properties, recent synthesis and applications of Azo dyes. Heliyon 6, e03271. https://doi.org/10.1016/j.heliyon.2020.e03271 (2020).
Alsafy, S. M. & Alrazzak, N. A. Synthesis characterization, and biological activity study of new heterocyclic compounds. Eng. Proc. 59 (1), 178 (2023). https://doi.org/10.3390/engproc2023059178
Maleeva, G. et al. A photoswitchable GABA receptor channel blocker. Britisch J. Pharm. 176, 2661–2677. https://doi.org/10.1111/bph.14689 (2019).
Mezgebe, K. & Mulugeta, E. Synthesis and Pharmacological activities of Azo dye derivatives incorporating heterocyclic scaffolds: a review. RSC Adv. 12, 25932–25946. https://doi.org/10.1039/D2RA04934A (2022).
Baruah, M. J., Dutta, R., Zaki, M. E. A. & Bania, K. K. Heterogeneous Iron-Based catalysts for organic transformation reactions: A brief overview. Molecules 29 (13), 3177. https://doi.org/10.3390/molecules29133177 (2024).
Izquierdo, S. et al. Green heterogeneous catalysts for cleaner solvent-free production of acetates. J. Porous Mater. 30, 847–858. https://doi.org/10.1007/s10934-022-01376-1 (2023).
Aneksomboonpol, P. et al. Surface structure characteristics of dental implants and their potential changes following installation: a literature review. J. Korean Assoc. Oral Maxillofac. Surg. 49 (3), 114–124. https://doi.org/10.5125/jkaoms.2023.49.3.114 (2023).
Hashmi, A. W., Mali, H. S., Meena, A., Hashmi, M. F. & Bokde, N. D. Surface characteristics mmeasurement using computer vision: a review. Comput. Model. Eng. Sci. 135 (2), 917–1005. https://doi.org/10.32604/cmes.2023.021223 (2023).
Osman, A. I. et al. Synthesis of green nanoparticles for energy, biomedical, environmental, agricultural, and food applications: a review. Environ. Chem. Lett. 22, 841. https://doi.org/10.1007/s10311-023-01682-3 (2024).
Goudarziafshar, H., Zafari, M. & Moosavi-Zare, A. R. Porous carbon/Fe3O4 nanocomposite as a new magnetically recoverable catalyst for the Preparation of polyhydroquinolines. RSC Adv. 14, 27565–27574. https://doi.org/10.1039/D4RA05432F (2024).
Moosavi-Zare, A. R., Najafi, R. & Goudarziafshar, H. The Preparation of [1,2,4]triazolo[1,5-a]pyrimidines catalyzed by schiff base zinc(II) complex supported on magnetite nanoparticles under mild conditions. RSC Adv. 14, 19167–19173. https://doi.org/10.1039/D4RA02339K (2024).
Esmaili, S., Moosavi-Zare, A. R. & Khazaei, A. Nano-[Fe3O4@SiO2/N-propyl-1-(thiophen-2-yl)ethanimine][ZnCl2] as a nano magnetite schiff base complex and heterogeneous catalyst for the synthesis of pyrimido[4,5-b]quinolones. RSC Adv. 12, 5386–5394. https://doi.org/10.1039/D2RA00213B (2022).
Ojaghi Aghbash, K., Noroozi Pesyan, N. & Batmani, H. Fe3O4@silica-MCM-41@DABCO: A novel magnetically reusable nanostructured catalyst for clean in situ synthesis of substituted 2-aminodihydropyrano[3,2-b]pyran-3-cyano. Appl. Orgmetal Chem. 33, e5227. https://doi.org/10.1002/aoc.5227 (2019).
Aalinejad, M., Noroozi Pesyan, N., Heidari, N. & Batmani, H. Danandeh Asl, A. Supramolecular Fe3O4@PEG/–diaza crown ether@Ni: a novel magnetically reusable nano catalyst for the clean synthesis of 2-aryl-2,3-dihydroquinazolin-4(1H)-ones. Appl. Orgmetal Chem. 33, e4878. https://doi.org/10.1002/aoc.4878 (2019).
Noroozi, N., Batmani, H., Havasi, F. & P. & Copper supported on functionalized MCM-41 as a novel and a powerful heterogeneous nanocatalyst for the synthesis of benzothiazoles. Polyhedron 158, 248–254. https://doi.org/10.1016/j.poly.2018.11.005 (2019).
Mohaddesi, M., Pesyan, N. & Fe, N. 3O4@SiO2 nanoparticle-immobilized Cu(II)-benzoylthiourea complex as a magnetically reusable catalyst for the synthesis of benzo[d]imidazo[2,1-b]thiazole-1-ium hydroxide derivatives in water. J. Iran. Chem. Soc. 17, 2105–2117. https://doi.org/10.1007/s13738-020-01915-0 (2020).
Ojaghi Aghbash, K., Noroozi Pesyan, N. & Batmani, H. Cu-Kojic acid complex anchored to functionalized Silica-MCM-41: A promising regioselective and reusable nanocatalyst for click reaction. ACS Omega. 5 (35), 22099–22108. https://doi.org/10.1021/acsomega.0c02115 (2020).
Hassanloie, N. et al. Anchoring of Cu–dimethylglyoxime complex in MCM-41 matrix: a new, recyclable, and highly efficient nanocatalyst for the green Preparation of 2,3-dihydroquinazolin-4(1H)-ones. Monatsh Chem. 152, 833–844. https://doi.org/10.1007/s00706-021-02793-9 (2021).
Taghizadeh, A., Noroozi Pesyan, N., Alamgholiloo, H. & Sheykhaghaei, G. Immobilization of nickel on Kryptofix 222 modified Fe3O4@PEG core-shell nanosphere for the clean synthesis of 2-Aryl-2,3-dihydroquinazolin-4(1H)-ones. Appl. Orgmetal Chem. 36 (9), e6787. https://doi.org/10.1002/aoc.6787 (2022).
Taghizadeh, A. & Noroozi Pesyan, N. Nickel supported Fe3O4@PEG/methyl o-phenylenediamine nanosphere: an eco-friendly strategy for improved synthesis of benzothiazoles under sonication. Inorg. Chem. Commun. 175, 114127. https://doi.org/10.1016/j.inoche.2025.114127 (2025).
Ojaghi Aghbash, K. & Noroozi Pesyan, N. Copper-kryptofix-21-Fe3O4/MCM-41: A powerfully recoverable nanocatalyst for clean regioselective 1,2,3-triazole synthesis-click reaction. Appl. Orgmetal. Chem. 38 (7), e7515 (2024). https://doi.org/10.1002/aoc.7515.
Hassanloie, N. et al. Preparation of Fe3O4@SiO2@Cu-MoO3 core-shell nanostructures: Synergistics effects of copper and molybdenum for catalytic enhancement. J. Porous Mater. 30, 859-869. https://doi.org/10.1007/s10934-022-01379-y (2023).
Nikpassand, M., Rafat, F. & Zare Fekri, L. Greener synthesis of novel azo-linked 1,2,4-triazolidine-3-ones (thiones) using recyclable Fe3O4@SP@vanillin@thioglycolic acid magnetic nanocomposite. Org. Prep Proced. Int. 56, 234–242. https://doi.org/10.1080/00304948.2023.2259779 (2024).
Nikpassand, M., Zare Fekri, L. & Varma, R. S. Sustainable conversion of carbon dioxide into novel 5-aryldiazenyl-1,2,4-triazol-3-ones using Fe3O4@SP-vanillin-TGA nanocomposite. Heterocycl. Commun. 29 (1), 20220164. https://doi.org/10.1515/hc-2022-0164 (2023).
Shafaati, T., Nikpassand, M. & Mokhtary, M. Zare Fekri, L. Synthesis of novel 4-arylidene-benzodiazepines using Cellulose@SiPr@Catechin@Fe3O4. Results Chem. 7, 101525. https://doi.org/10.1016/j.rechem.2024.101525 (2024).
Jafarpour, N., Nikpassand, M. & Faramarzi, M. Conjugation of folic acid onto Poly (acrylic acid-co-allylamine)-grafted mesoporous silica nanoparticles for controlled methotrexate delivery. J. Drug Delivery Sci. Tech. 96, 105667. https://doi.org/10.1016/j.jddst.2024.105667 (2024).
Kandelous, Y. M., Nikpassand, M. & Fekri, L. Z. Recent focuses in the syntheses and applications of magnetic Metal–Organic frameworks. Top. Curr. Chem. (Z). 382, 30. https://doi.org/10.1007/s41061-024-00475-8 (2024).
Gharib, M., Nikpassand, M., Mokhtary, M. & Zare Fekri, L. Glucono-delta-lactone as a substrate for preparation of new magnetically recoverable nanocatalyst for the environmentally benign synthesis of novel azo linked 3-methyl-isoxazol-5-ones. Synth. Commun. 53, 1935-1953. https://doi.org/10.1080/00397911.2023.2258529 (2023).
Acknowledgements
Financial support from the Research Council of Rasht Branch, Islamic Azad University, Rasht, Iran is sincerely acknowledged.
Author information
Authors and Affiliations
Contributions
Z.A. and M.N. wrote the main manuscript text and Z.A. prepared figures. L.Z. and M.M. reviewed. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Alizadeh, Z., Nikpassand, M., Mokhtary, M. et al. Preparation, characterization and application of MT@SP@GBP and their use for synthesis of novel pyrazolophthalazine-5,10-diones. Sci Rep 15, 35351 (2025). https://doi.org/10.1038/s41598-025-19325-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-19325-8
