
Abstract
In this study, we measured human epidermal growth factor receptor (EGFR) mutations in both tissue and circulating tumor DNA (ctDNA) by using beads, emulsions, amplifications and magnetic polymerase chain reaction (BEAMing PCR). Noninvasive mutation detection by assessing circulating tumor DNA (ctDNA) offers many advantages over tumor biopsy. One hundred non-small cell lung cancer (NSCLC) patients were enrolled, and both preoperative plasma samples and formalin-fixed and paraffin-embedded (FFPE) samples were collected for the study. The EGFR mutation status was determined by BEAMing PCR in ctDNA. Real-time quantitative PCR (qPCR) data were collected from our hospital database (EMR-qPCR, Electronic Medical Records) for comparative analysis. Additionally, qPCR was also performed on FFPE tissues using a Diatech EGFR qPCR kit. The concordance rates were 98.8%, 98.9% and 95.5% for exons 19, 20 and 21, respectively, when the BEAMing data were compared with the EMR-qPCR data. Additionally, when the BEAMing and Diatech qPCR data were compared, 90%, 100%, 96% and 98% of the genes were obtained for exons 19, 20, 21 (L858R) and 21 (L861Q), respectively. For both comparisons, Cohen’s kappa agreement was significant. The advantage of BEAMing is its ability to identify mutated DNA sequences in cancer cells in the background of normal cell DNA contamination. This could be useful for disease monitoring and progression.
Similar content being viewed by others


Introduction
Several oncogenic mechanisms, including epidermal growth factor receptor (EGFR) gene mutation, increased gene copy number, and protein overexpression, contribute to the dysregulation of EGFR-tyrosine kinase (TK) activity1,2. There are two types of mutations reported mostly in the TK domain of EGFR3,4: short deletions in exon 19 and point mutations (G719S, L858R, and L861Q) in exons 19, 20 and 215. Mutations result in destabilization of the domain conformation, constitutive activation of its kinase activity, and activation of downstream signaling pathways6,7, such as the AKT and STAT pathways, which have crucial antiapoptotic functions for cell survival8. Mutations in the EGFR gene have become crucial in predicting the effect of EGFR-TK inhibitors (TKIs) in non-small cell carcinoma (NSCLC) patients.
EGFR mutation detection is usually carried out in surgically resectedtumor tissue or needle biopsy9. Because of the highly heterogeneous nature of the lung, analysis of EGFR mutations could be confusing and misleading. Alternately, detection and monitoring of cancer-specific somatic gene mutations in circulating tumor DNA (ctDNA) is a minimally invasive alternative to tissue biopsy that has shown promise in overcoming some of the challenges associated with sampling from tissue10. Recent data have shown that EGFR mutations can be found in plasma-derived cell-free DNA (cfDNA) from NSCLC patients. Moreover, studies have confirmed that EGFR mutations in plasma can predict the clinical response to targeted therapy11,12. Several studies have suggested that the analysis of cell-free plasma DNA is a clinically useful alternative13,14,15,16.
EGFR mutation testing in advanced NSCLC patients has evolved rapidly over the last decade mainly because of the introduction of the targeted EGFR-TKIs. The clinical gold standard approach for mutational analysis of lung cancers is genotyping of solid tumor biopsies17,18,19. However, a wide range of molecular diagnostic tests are currently available. These tests are suitable for either solid tissues or liquid biopsy samples, like Sanger sequencing20, next generation sequencing21, ARMS, SARMS, ARMS-Plus, Super-ARMS22,23,24, droplet digital PCR25,26.
Here, we describe the comparative results of an observational study using a bead-based PCR assay (Beads, Emulsion, Amplification and Magnetics, BEAMing)27 to detect different activating (exon 19, 21) and resistant EGFR mutations (exon 20) in ctDNA derived from the peripheral blood of NSCLC patients. We evaluated our results to determine the percent agreement between methods (standard qPCR) for the detection of EGFR mutations in tumor tissues using commercially available kits. This is completely a new study using a very sensitive emulsion PCR in our hospital involving large number of patients samples to compare available methodologies designed for non-invasive EGFR mutation testing and to evaluate the concordance between them. This approach promises to be sensitive and reliable for detecting mutations at an early stage of the disease.
Materials and methods
Ethics approval
This is a prospective observational study that investigates the efficiency of BEAMing PCR to detect EGFR mutation from ctDNA of lung cancer patients undergoing treatments at our hospital. Study design and the protocol were approved by the Institutional Review Board (ACTREC-TMC Research Ethics Committee). Each participant was provided with a copy of the original informed consent and were explained about the study. Consent forms were signed by the participants or their legal relatives before the start of the study. All data were maintained confidentially following the Helsinki Declaration. All the research methods were performed in accordance with the relevant guidelines and by following the good clinical practices.
Patient samples and cell lines
Patients with activating EGFR mutations in tumor tissues were selected for the study between 2017 and April 2020 (Table 1). Peripheral blood was collected into 10 ml tubes containing ethylene-diamine-tetraacetic acid (EDTA) as the anticoagulant from 100 patients with lung cancer treated at Tata Memorial Centre-ACTREC, India as per the published criteria28,29,30. All patients provided written informed consent prior to participation. Total genomic DNA isolated from the PC9, H1975 and A549 NSCLC cell lines (a kind gift from Dr. Amit Dutt, ACTRE-TMC and Delhi University South Campus, India) was used as a control. Formalin-fixed paraffin-embedded (FFPE) tumor tissue was obtained from each subject and processed by the Surgical Pathology Laboratory using routine procedures.
Isolation of circulating tumor DNA (ctDNA)
Blood was drawn into EDTA tubes (for plasma). Within one hour, the tubes were subjected to centrifugation at 820 × g for 10 min. One milliliter aliquots of the plasma were transferred to 1.5 ml tubes and centrifuged at 16,000 × g for 10 min to pellet any remaining cellular debris. The supernatants were transferred to fresh tubes and stored at -80°C. Total genomic DNA was extracted from 1 ml of plasma using a DNA Micro Kit (Qiagen) according to the manufacturer’s instructions.
Quantification of total plasma DNA
The isolated ctDNA concentration was estimated using a Nanodrop ND1000 spectrophotometer.
Sensitivity and specificity assay
The sensitivity and specificity of the BEAMing reaction were assessed before the actual assay was started. In this assay, different percentages of mutant DNA from cell lines harboring specific mutations (H1975 and PC9) were mixed in the background of wild-type DNA (A549). Emulsion-based BEAMing PCR was performed, and hybridized samples were run in FACSArea III to detect mutant and wild-type populations.
BEAMing and EGFR mutation detection
We used different primer sets for mutation analysis according to Taniguchi et al.31 (Table 2). The DNA purified from the plasma was used for each BEAMing assay. Initially, amplification with a high-fidelity DNA polymerase was performed in eight separate 25 µl PCRs, each containing template DNA from 250 µl of plasma, 5× Phusion High Fidelity PCR buffer (NEB), 1.5 U of HotStart Phusion polymerase (NEB), 0.2 µM of each primer, 0.25 mM of each dNTP, and 0.5 mM MgCl2. The temperature cycling times were 98°C for 30 s and 35 × (98°C for 10 s, 57°C for 10 s, and 72°C for 10 s). PCR products were pooled and quantified using a Nanodrop spectrophotometer. Emulsion PCR was performed as follows18: Briefly, a 150 µl PCR mixture was prepared containing 18 pg of template DNA, 40 U of Platinum Taq DNA polymerase (Invitrogen), 1× PCR buffer, 0.2 mM dNTPs, 5 mM MgCl2, 0.05 µM Tag1 (5’-tcccgcgaaattaatacgac-3’), 8 µM Tag2 (5’-gctggagctctgcagcta-3’) and ~ 6 × 107 magnetic streptavidin beads (MyOne, Invitrogen) coated with Tag1 oligonucleotide (5’-dual biotin-T-Spacer18- tcccgcgaaattaatacgac-3’). Then, 150 µL of PCR mixture, 600 µL of oil/emulsifier mixture (7% ABIL WE09, 20% mineral oil, 73% TegoSoft DEC (Evonik) and one 5 mm steel bead (Qiagen) were added to a 96-deep-well plate (1.2 ml; Abgene). Microemulsions were prepared by shaking the plate in a TissueLyser (Qiagen) for 10 s at 15 Hz and then for 7 s at 17 Hz. The aqueous compartments were checked under an inverted microscope with 40X magnification for the presence of beads (Fig. 1). Emulsions were dispensed into eight PCR plates and heated at 94°C for 2 min; 3 cycles of 94°C for 10 s and 68°C for 45 s; 70°C for 75 s; 3 cycles of 94°C for 10 s and 65°C for 45 s; 70°C for 75 s; 3 cycles of 94°C for 10 s, 62°C for 45 s, and 70°C for 75 s; and 50 cycles of 94°C for 10 s and 57°C for 45 s and 70°C for 75 s. To disrupt the emulsions, 150 µl of breaking buffer (10 mM Tris-HCl, pH 7.5; 1% Triton-X 100; 1% SDS; 100 mM NaCl; and 1 mM EDTA) was added to each well and mixed with a TissueLyser at 20 Hz for 20 s. The beads were recovered by spinning the suspension at 3,200 × g for 2 min and removing the oil phase. The breaking step was repeated twice. All the beads from the 8 wells were consolidated and washed with 150 µl of wash buffer (20 mM Tris-HCl, pH 8.4, 50 mM KCl). The DNA on the beads was denatured for 5 min with 0.1 M NaOH. Finally, the beads were washed with 150 µl of wash buffer and resuspended in 150 µl of the same buffer. The mutation status of the DNA bound to the beads was determined by allele-specific hybridization. Fluorescently labeled probes complementary to the mutant and wild-type DNA sequences were designed for different mutations (Table 2). The size of the probes ranged from 15 to 18 nt. All the mutant probes were coupled to a Cy5 fluorophore, and all the wild-type probes were coupled to an Alexa488 fluorophore at their 5’ ends (Integrated DNA Technologies). Each allele-specific hybridization reaction contained ~ 1 × 107 beads in 30 µl of wash buffer, 66 µl of 1.5× hybridization buffer (4.5 M tetramethylammonium chloride, 75 mM Tris-HCl pH 7.5, 6 mM EDTA), and 4 µl of a mixture of mutant and wild-type probes, each at 5 µM in TE buffer. The hybridization mixture was heated to 70°C for 10 s and slowly (0.1 °C/s) cooled to 35°C. After incubating at 35°C for 2 min, the mixture was cooled (0.1°C/sec) to room temperature. The beads were collected with a magnet, and the supernatant containing the unbound probes was removed using a pipette. The beads were resuspended in 100 µl of 1× hybridization buffer and heated to 48°C for 5 min to remove unbound probes. After the heating step, the beads were again separated magnetically and washed once with 100 µl of wash buffer. In the final step, the supernatant was removed, and the beads were resuspended in 200 µl of TE buffer for flow cytometric analysis. A FACSAria III flow cytometry system (BD Bioscience) equipped with a high-throughput autosampler was used for the analysis of each bead population. An average of 1 × 106 beads were analyzed for each plasma sample.

Microemulsions were made as described in Materials and Methods. One microliter of the emulsion was added in 1 µl of oil on a microscope slide and photograph was taken. Here in this picture, there were several compartments with a single bead (White arrowheads). Rests contain dual or multiple beads (100X magnification).
Real-time PCR of formalin-fixed paraffin-embedded (FFPE) tissue DNA
Paraffin blocks were collected from the Department of Pathology, Tata Memorial Hospital. The presence of tumor tissues in the blocks was confirmed by pathologists. A total of 50–100 μm sections were used for the extraction of DNA. All specimens underwent histological examination to confirm the presence of tumor tissue. The dissected tumor tissues were digested overnight at 60 °C in 180 µl of ATL buffer (Qiagen) and 20 µl of proteinase K (50 mg/ml; Invitrogen). DNA was isolated using the QIAamp FFPE DNA Kit (Qiagen) following the manufacturer’s protocol. The presence of the most commonly found EGFR mutations was determined by real-time quantitative PCR (qPCR) using an Easy EGFR PCR Kit (Diatech Pharmacogenetics srl, Italy).
Statistical analysis
All the statistical analyses were performed using IBM SPSS Windows version 20.0 software. Diagnostic metrics such as sensitivity, specificity and accuracy were calculated. McNemar’s test was used to test the statistical significance of the difference between the BEAMing and EMR qPCR data as well as between the BEAMing and EasyLine qPCR data. Kappa statistics were used to assess symmetry scores for the two methods. The interpretation of the kappa values was based on data from Altman33.
Results
Concentration of ctDNA
We could effectively isolate ctDNA from the plasma of all the samples. The lowest and highest concentrations were 5.2 and 31.23 ng/mL, respectively, with a mean of 15.74 (Fig. 2). Moreover, the mean A260/280 ratio of the samples was 1.81 (Fig. 3).

Concentration of ctDNA in 100 samples (ng/mL).

Purity of the ctDNA (A260/A280).
Analysis of the accuracy and sensitivity of BEAMing using cell line DNA
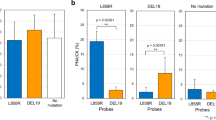
Wild-type EGFR DNA from A549 cells was mixed with a mutated EGFR DNA fragment (PC9 for Exon 19, H1975 for Exon 20 and 21 in separate tubes) at 1% and 0.1% ratios. Each PCR mixture contained a total of 100 ng of DNA, which was used as a template for the genomic and subsequent BEAMing emulsion PCR assays. The mutated DNA fractions were estimated by the ratio of the number of beads labeled with Alexa Fluor 647 (mutant) to that labeled with Alexa Fluor 488 (wild-type). Due to the limit of detection, our assay was able to detect mutant EGFR alleles in samples containing as little as 0.1 ng of mutant DNA per 100 ng of DNA. Below this concentration, there was no amplification of the mutant allele (Fig. 4).

Sensitivity and specificity analysis of the BEAMing PCR. A decreasing percentage of EGFR mutant DNA was added to the background of the wild-type DNA. Genomic and subsequent BEAMing PCR was carried out. The samples were hybridized and run in a FACSArea III. The mutant (Q1) and wild-type (Q3) populations were separated and are indicated. If the aqueous compartments were all the same size, the expected Poisson distribution was obtained. However, because they are not uniform, some compartments are larger and contain more than one template. This raises the number of double template beads (Q2).
Mutation assay by BEAMing and its comparison with hospital qPCR data
BEAMing PCR was carried out on 100 ctDNA samples for the detection of different mutations in exons 19, 20 and 21 of EGFR. Real-time PCR (Therascreen) data were collected from our hospital database (EMR-qPCR, Electronic Medical Records). Additionally, a qPCR assay was performed with the available FFPE tissues using an EasyLine qPCR Kit (Diatech-qPCR). The percentages of true positives and true negatives were calculated with respect to the EMR-qPCR and Diatech-qPCR data (Tables 3 and 4). When the BEAMing data was compared with the EMR-qPCR data, 98.8%, 98.9% and 95.4% positive concordance was detected for exons 19, 20 and 21, respectively (Table 3). Moreover, when the BEAMing and Diatech-qPCR data were compared, 90%, 100%, 96% and 98% positive concordance for exons 19, 20, 21-L858R and 21-L861Q, respectively (Table 4) was observed. We aim to compare our EMR-qPCR data (FFPE tissue) with those of the BEAMing assay using ctDNA to determine whether ctDNA is an early and additional or supplementary diagnostic approach for EMR-qPCR as well as Diatech qPCR, thereby saving time and repeating biopsies. When BEAMing and EMR-qPCR were compared, the Cohen’s kappa coefficient was 0.969, 1.000 and 0.774 (Table 3) for exons 19, 20 and 21, respectively. This represents the proportion of agreement between the two methodologies beyond the agreement occurring due to chance. In this case, a P value of p < 0.001 suggested that the kappa value was significantly different from zero. This finding does not imply statistically significant agreement, and we interpreted the magnitude of agreement from the reported kappa value. To assess the level of agreement between the two methodologies, we interpreted the kappa value based on definitions outlined by Altman33. Our results showed good agreement (0.969 and 0.774 for exons 19 and 21, respectively) and very good agreement (1.000 for exon 19) between the two methodologies (BEAMing vs. EMR-qPCR). Moreover, the kappa score was also calculated for different exons when the BEAMing and Diatech-qPCR methods were compared. For exon 19, the kappa score was 0.648, and for exon 21-L858R, it was 0.778. Both values showed significant symmetry. No statistical data were computed because Diatech Kit-Exon 20, BEAMing-Exon 20 and Diatech Kit-L861Q Exon 21 were constants (Table 4). These represent the proportion of agreement between the two methodologies beyond the agreement occurring due to chance. Here, the P-value of p < 0.001 suggests that the kappa value is statistically significant from zero. This does not imply statistically significant agreement and we can interpret the magnitude of agreement from the reported kappa value. Hence, from the above results, we conclude that there is concordance between BEAMing and EMR-qPCR and Diatech-qPCR. However, we also detected few false positives and false negatives in our assay (Tables 3 and 4). This could be because of the deamination of nucleotides in FFPE samples, which causes a C: G > T:A change that, in turn, yields a false-positive mutation result19. To prevent this, uracil DNA glycosylase (UDG), which cleaves deaminated cytosine (uracil) was used. The results of the comparison between the exon 20-T790M qPCR and the BEAMing and Diatech kit data were not estimable since all the cases were negative according to one of the comparative methodologies.
Discussion
Circulating tumor DNA testing to genotype patients has facilitated improved care of EGFR mutation-positive NSCLC patients. We have reported here that ctDNA is a promising biomarker for the early detection of EGFR mutations as well as for following the course of therapy in NSCLC patients. We could effectively isolate and measure the ctDNA concentration from as many as 100 patient samples. However, whether ctDNA levels are exactly proportional to tumor burden cannot be ascertained conclusively because of the varying levels of ctDNA and because there is no independent way to measure total systemic burden at this time (Figs. 2 and 3). BEAMing PCR can detect a very small amount of mutant DNA sequences in a larger pool of fragments containing wild-type DNA, in order of a single mutant allele in a background of 10,000 wild-type alleles, and is able to enable copy-number quantification31,34. The technique is based on a combination of emulsion digital PCR and flow cytometry, with beads, emulsification, amplification and magnetics to achieve the necessary level of sensitivity.
In the present study, we compared the concordance between tissue EGFR qPCR (both via the Therascreen and Diatech kits) and ctDNA-BEAMing PCR. As the fraction of mutant alleles in the isolated DNA was minimal, we first designed a sensitivity assay by mixing different percentages of mutant DNA in the background of wild-type DNA to determine whether BEAMing PCR could detect the mutant alleles. Our results showed that this assay could detect mutant alleles as low as 0.1% in the background of 99.9% of the wild-type allele. The percentage of concordance was high when the BEAMing data were compared with both sets of qPCR data (Tables 3 and 4). Since the P value is greater than the significance level α = 0.001, the null hypothesis cannot be rejected, and we can assume that there is no significant difference between BEAMing and EMR-qPCR or Diatech-qPCR methodology.
The most frequently encountered chemical alteration of FFPE-DNA is due to spontaneous deamination of cytosine. A base substitution of C to T (C > T) or G to A (G > A, from C > T on the antisense strand), collectively C: G > T:A, is one of the most common artifacts, and it is caused by a chemical reaction called cytosine deamination35,36. This change spontaneously occurs in vivo and is automatically corrected by the intracellular enzymes uracil DNA glycosylase (UDG) and 5-methylcytosine DNA glycosylase37,38. To reduce deamination-induced errors in FFPE tissue blocks, some researchers have tried pretreating FFPE blocks with UDG in their NGS studies with an expectation of eliminating the error39,40.
Based on the positive and meaningful results in this study, we can conclude that performing BEAMing PCR on ctDNA is an effective and sensitive assay for determining the mutation status of the EGFR gene in NSCLC patients. However, large sample studies involving thousands of patients in a multicentric setup are required to effectively determine the cause of the mutation. This will further validate the conclusion of the present study. This technique provides an alternative to invasive biopsies that retrieve tumor tissue for analysis. Moreover, future treatment decisions for lung cancer are likely to be based on molecular subtypes reflecting tumor biology rather than clinical features or histologic subtypes. Discoveries linking two areas of seemingly unconnected research tend to reframe existing knowledge in ways that permit new interpretations and new understanding of the field of study addressed by this research.
Data availability
Raw data in pdf and excel file have been uploaded as Supplementary files. This work is observational and diagnostic in nature. No new mutations were discovered. No data was submitted anywhere. In the raw data patients’ identity or case numbers were not disclosed.
Abbreviations
- EGFR:
-
Epidermal growth factor receptor
- TK:
-
Tyrosine kinase
- TKI:
-
Tyrosine kinase inhibitor
- NSCLC:
-
Non-small cell lung carcinoma
- ctDNA:
-
Circulating tumor DNA
- cfDNA:
-
Cell free DNA
- BEAMing:
-
Bead emulsion amplification and magnetics
- FFPE:
-
Formalin-fixed paraffin-embedded
- qPCR:
-
Quantitative polymerase chain reaction
References
Ciardiello, F. & Tortora, G. EGFR antagonists in cancer treatment. N Engl. J. Med. 358, 1160–1174 (2008).
Hynes, N. E. & MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell. Biol. 21, 177–184 (2009).
Lynch, T. J. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small cell lung cancer to gefitinib. N Engl. J. Med. 50, 2129–2139 (2004).
Paez, J. G. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 (2004).
Stewart, E. L., Tan, S. Z., Liu, G. & Tsao, M. S. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations—A review. Transl Lung Cancer Res. 4, 67–81 (2015).
Yun, C. H. et al. Structures of lung cancer–derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 11, 217–227 (2007).
Kumar, A., Petri, E. T., Halmos, B. & Boggon, T. J. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J. Clin. Oncol. 26, 1742–1751 (2008).
Sordella, R., Bell, D. W., Haber, D. A. & Settleman, J. Gefitinib-sensitizing EGFR mutations in lung cancer activate antiapoptotic pathways. Science 305, 1163–1167 (2004).
Tiseo, M. et al. Predictors of gefitinib outcomes in advanced non-small cell lung cancer (NSCLC): study of a comprehensive panel of molecular markers. Lung Cancer. 67, 355–360 (2010).
Diaz, L. A. Jr & Bardelli, A. Liquid biopsies: genotyping circulating tumor DNA. J. Clin. Oncol. 32, 579–586 (2014).
Seki, Y. et al. Circulating cell-free plasma tumour DNA shows a higher incidence of EGFR mutations in patients with extrathoracic disease progression. ESMO Open. 3, e000292 (2018).
Vallee, A. et al. Plasma is a better source of tumor-derived circulating cell-free DNA than serum for the detection of EGFR alterations in lung tumor patients. Lung Cancer. 82, 373–374 (2013).
Buder, A., Tomuta, C. & Filipits, M. The potential of liquid biopsies. Curr. Opin. Oncol. 28, 130–134 (2016).
Oxnard, G. R. et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin. Cancer Res. 20, 1698–1705 (2014).
Sacher, A. G. et al. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol. 2(8), 1014–1022 (2016).
Buder, A. et al. Cell-free plasma DNA-guided treatment with osimertinib in patients with advanced EGFR-mutated NSCLC. J. Thorac. Oncol. 13(6), 821–830 (2018).
Lindeman, N. I. et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the College of American Pathologists, the International Association for the study of Lung Cancer, and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 142(3), 321–346 (2018).
Singh, A. P., Li, S. & Cheng, H. Circulating DNA in EGFR-mutated lung cancer. Ann. Transl Med. 5(18), 379–379 (2017).
Normanno, N., Denis, M. G., Thress, K. S., Ratcliffe, M. & Reck, M. Guide to detecting epidermal growth factor receptor (EGFR) mutations in ctDNA of patients with advanced non-small-cell lung cancer. Oncotarget 8(7), 12501–12516 (2017).
Jing, C. et al. Next-generation sequencing-based detection of EGFR, KRAS, BRAF, NRAS, PIK3CA, Her2 and TP53 mutations in patients with non-small cell lung cancer. Mol. Med. Rep. 18(2), 2191–2197 (2018).
Deeb, K. K., Hohman, C. M., Risch, N. F., Metzger, D. J. & Starostik, P. Routine clinical mutation profiling of non–small cell Lung Cancer using next-generation sequencing. Arch. Pathol. Lab. Med. 139(7), 913–921 (2015).
Ellison, G. et al. A comparison of ARMS and DNA sequencing for mutation analysis in clinical biopsy samples. J. Exp. Clin. Can. Res. 29, 132–139 (2010).
Reck, M. et al. ctDNA determination of EGFR mutation status in European and Japanese patients with advanced NSCLC: the ASSESS study. J. Thorac. Oncol. 11(10), 1682–1689 (2016).
Zhu, Y. et al. A novel ARMS-based assay for the quantification of EGFR mutations in patients with lung adenocarcinoma. Oncol. Lett. 15(3), 2905–2912 (2018).
Li, Y. et al. Comprehensive analysis of EGFR T790M detection by ddPCR and ARMS-PCR and the effect of mutant abundance on the efficacy of osimertinib in NSCLC patients. J. Thorac. Dis. 11(7), 3004–3014 (2019).
Feng, Q. et al. A comparison of QuantStudio 3D digital PCR and ARMS-PCR for measuring plasma EGFR T790M mutations of NSCLC patients. Cancer Manag Res. 10, 115–121 (2018).
Dressman, D., Yan, H., Traverso, G., Kinzler, K. W. & Vogelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Nat. Acad. Sci. 100, 8817–8822 (2003).
Szpechcinski, A. et al. Detection of EGFR mutations in liquid biopsy samples using allele-specific quantitative PCR: a comparative real-world evaluation of two popular diagnostic systems. Adv. Med. Sci. 66(2), 336–342 (2021).
Guo, K. et al. Detection of epidermal growth factor receptor (EGFR) mutations from preoperative circulating tumor DNA (ctDNA) as a prognostic predictor for stage I–III non-small cell lung cancer (NSCLC) patients with baseline tissue EGFR mutations. Transl Lung Can. Res. 10(7), 3213–3225 (2021).
Yamaguchi, O. et al. Predictive significance of circulating tumor DNA against patients with T790M-positive EGFR-mutant NSCLC receiving osimertinib. Sci. Rep. 13, 20848 (2023).
Taniguchi, K. et al. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin. Can. Res. 17, 7808–7815 (2011).
Diehl, F. et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 14, 985–990 (2008).
Altman, D. G. Practical Statistics for Medical Research (Chapman & Hall/CRC, 1999).
Diehl, F. et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA. 102, 16368–16373 (2005).
Fryxell, K. J. & Zuckerkandl, E. Cytosine deamination plays a primary role in the evolution of mammalian isochores. Mol. Biol. Evol. 17, 1371–1383 (2000).
Zhu, J. K. Active DNA demethylation mediated by DNA glycosylases. Ann. Rev. Genet. 43, 143–166 (2009).
Krokan, H. E., Drablos, F. & Slupphaug, G. Uracil in DNA: occurrence, consequences and repair. Oncogene 21, 8935–8948 (2002).
Kavli, B., Otterlei, M., Slupphaug, G. & Krokan, H. E. Uracil in DNA: general mutagen, but normal intermediate in acquired immunity. DNA Repair. (Amst). 6, 505–516 (2007).
Wong, S. Q. et al. CANCER 2015 Cohort sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med. Genomics. 7, 23 (2014).
Bourgon, R. et al. High-throughput detection of clinically relevant mutations in archived tumor samples by multiplexed PCR and next-generation sequencing. Clin. Can. Res. 20, 2080–2091 (2014).
Acknowledgements
We would like to thank Evonik Corporation, Germany, for providing the TegoSoft and Abil WE09 reagents as gifts. Special thanks to Mr. Satish Munnolli for doing the plagiarism check of the manuscript with the iThenticate software and Ms. Pallavi Rane, TMC Central Biostatistical Cell for the statistical analysis of the data.
Funding
This study was supported by the Department of Biotechnology, Government of India (Grant No: BT/PR10867/MED/30/1419/2014) and TMC-TRAC (IEC No: 900117).
Author information
Authors and Affiliations
Contributions
RB conceived and designed the experiments, analyzed the data and wrote and approved the final manuscript. DM and NB performed the experiments and analyzed the data. KP, VN, AJ, CSP and JK were responsible for the treatment and selection of patients. RKK and SR provided the tissue blocks and contributed to identifying the tumor samples by histopathology. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declarations
This study was approved by the ACTREC-TMC institutional review board, and all participants provided informed consent.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mirikar, D., Banerjee, N., Prabhash, K. et al. Comparative analysis of EGFR mutations in circulating tumor DNA and primary tumor tissues from lung cancer patients using BEAMing PCR. Sci Rep 15, 1252 (2025). https://doi.org/10.1038/s41598-025-85160-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-85160-6
