
Abstract
The TSC complex formed by TSC1 and TSC2 is the most important upstream negative regulator of mTORC1. Genetic variations in either TSC1 or TSC2 cause tuberous sclerosis complex (TSC) disease which is a rare autosomal dominant disorder resulting in impairment of multiple organ systems. In this study, besides a reported variation, c.2509_2512del (p.Asn837Valfs*11, p.N837fs) in TSC1, we found a de novo TSC2 variation c.1113delG (p.Gln371Hisfs*18, p.Q371fs), which these two mutation influence the formation of TSC complex. We found that the decrease of TSC complex with the appearance of the decreased miR-199b-3p expression. At the same time, the reduction of miR-199b-3p increased the expression of mTOR and the activation of mTORC1 and mTORC2, the additional miR-199b-3p caused the decrease the expression of mTOR and the activation of mTORC1 and mTORC2. In brief, our results may illustrate a novel mechanism of TSC caused by variations in either TSC1 or TSC2, and a new mTOR expression regulator, miR-199b-3p.
Similar content being viewed by others


Introduction
TSC (Tuberous sclerosis complex) (OMIM#191100#613254), with a morbidity of near to 1:6,000–1:10,000 in the world population, is an autosomal dominant disorder that multiple organ systems affected including skin, kidneys, brain, lung and heart1. The prevalence of TSC is independent of nations, ethnicity and sex, however, some studies found that female patients with TSC have a higher incidence of renal disease and lymphangioleiomyomatosis burden than male patients2. Among patients with TSC, more than 80% patients develop multiple and bilateral angiomyolipomas in the kidneys which are the leading cause of adult deaths with this disease3. TSC is usually caused by germline loss-of-function variations in either TSC1 or TSC2, encoding hamartin (TSC1) and tuberin (TSC2), respectively4,5. The complex of TSC1 and TSC2 inactivates the small GTP-binding protein Rheb to negatively regulate mammalian target of rapamycin complex 1 (mTORC1), an important regulator of cellular biosynthesis6, which is now recognized as a classical model of TSC pathogenesis7. Once activated, mTORC1 phosphorylates the downstream effectors, such as p70S6K and eukaryotic initiation factor 4E-binding protein 1 (4EBP1), to promote translation initiation8. mTOR is a component of two functionally and structurally distinct complexes, one is rapamycin-sensetive mTOR complex 1 (mTORC1), the other is rapamycin-insensitive mTOR complex 2 (mTORC2)9. mTORC2 is known to control cell survival and proliferation8.
miRNAs are small, non-coding RNA molecules about 19 to 25 nucleotides which involve in the regulation of gene expression during post-transcriptional stages by pairing to the target mRNA, thereby promoting the degradation of target mRNA or decreasing its translational efficiency10. Many miRNAs are also involved in the pathogenesis of various malignancies that have been evidenced by many studies11. It has been demonstrated that miR-199b is associated with the expression of mTOR12,13, but the molecular mechanism is unclear. Barbara et al.reported that TSC2 might regulate the biogenesis of microRNA in mouse embryonic fibroblasts (MEF), which indicates that the microRNA may play an important role in the pathogenic mechanism of TSC14.
In this study, we reported two variations, include a de novo mutation (c.1113delG) in TSC2 and one reported mutation (c.2509_2512del) in TSC1 that caused TSC in two independent families. Our results showed that TSC2-p.Q371fs influenced the cell proliferation and apoptosis in fibroblast from the patient. Furthermore, the TSC complex may regulate the protein level and activity of mTOR through the manipulation of the level of intracellular miR-199b-3p, which may a novel mechanism of TSC caused by mutation either in TSC1 or TSC2 and expends our understanding of TSC pathogenesis.
Materials and methods
Patients
This study was approved by the Ethics Committees of Shanxi University and the ethics number is SXULL20190049, written informed consent was obtained from all patients and healthy volunteers involved in this study.
Sequencing analysis
Whole-exome sequencing and validation by PCR were performed as described in our previous studies15.
Cell culture
Skin tissue samples were isolated from a TSC patient (22 years old, female) with the variation c.1113delG in TSC2 and two healthy volunteers (female, 23 years old and female, 25 years old). Fibroblasts of skin tissues separated and then cultured in DMEM (Boster) with 10% FBS (Gibco), 100 U/mL penicillin and 0.1 mg/mL streptomycin and grown in 5% CO2at 37 °C as described before16.
HEK293T cells were maintained in DMEM (Boster) supplemented with 10% FBS (BI). Cells were transfected with plasmids in this study using Polythermide (PEI) (PolyScience, Cat.No.23966-2) as described before17.
The plasmids for overexpression and shRNA
The plasmids pcDNA3-Flag-TSC2-WT and pcDNA3-Myc3-hTSC1-WT were purchased from Addgene. The plasmids pcDNA3-Flag-TSC2-MT containing mutant TSC2 (c.1113delG) and pcDNA3-Myc3-hTSC1-MT containing mutant TSC1 (c.2509_2512del) were constructed using site-specific mutagenesis. Lentiviral plasmids expressing shRNA specific to either TSC1 (NM_000368.5) or TSC2(NM_00548.5) were constructed using the targeting sequences 5’-AAAGAAGAAGCTGCAATATCT-3’18 and 5’-GCTCATCAACAGGCAGTTC TA-3’19 respectively.
Lentiviruses were harvested in HEK293T cells 72 h post co-transfection. The co-transfection of lentiviral plasmid vectors, psPAX and pMD2.G was performed at the ratio of 5:5:1. The medium containing lentiviruses was collected and then purified by centrifugation at 1000×g and filtration through 0.22 μm filter. Cells were transformed with the medium containing the lentiviruses and fresh medium at the ratio of 3:1 and cultured for 72 h, then drug selection with puromycin (1 mg/ml) was performed for 7–10 days to get stable transformed cell lines.
qRT-PCR analysis
The total RNA of fibroblasts from the patient or the healthy volunteers were extracted using RNAiso Plus (TaKaRa, Cat#9109) following the instructions of the supplier. First-stand cDNA for evaluated mRNA was generated using PrimerScriptTMRT Master Mix (TaKaRa, Cat.No.RR0-36A) and cDNA for miRNA was generated using TansScriptmiRNA First-Strand cDNA Synthesis SuperMix (TransGen Biotech, Lot#N10824). Three primer pairs for qRT-PCR of TSC2 were applied for quantification of cDNA located in either 5’ or 3’ terminal and the middle of the mRNA of TSC2. The primer pairs for mTOR and VEGF were obtained from Primer Bank (https://pga.mgh.harvard.edu/primerbank/).
Western blot
The total protein of fibroblasts was extracted by radio-immunoprecipitation buffer (RIPA, Solarbio, R0020) with 1 mM PMSF, 1 mM dithiothreitol and phosphatase inhibitors followed the manufacturers’ instructions. Total protein concentration was measured using a PierceTMBCA protein assay kit (Thermo).
The co-immunoprecipitation was performed as the protocol of IP/Co-IP kit (Absin, Cat. No. abs955). The antibodies involved in this study were as belows: TSC1 monoclonal antibody (Stanta Cruz, Cat.No.sc-377386), TSC2 monoclonal mouse antibody (Stanta Cruz, Cat. No. sc-271314), TSC2 monoclonal rabbit antibody (Cell Signaling. Cat.No.#4308), mouse IgG (Stanta Cruz, Cat.No.sc-2025), mouse anti Myc-tag monoclonal antibody (ABclonal, Cat.No.AE010), DDDDK tag (flag) mouse monoclonal antibody (Proteintech, Cat.No.66008-3-Ig), mTOR (phosphor-S2448) polyclonal antibody (Bioworld, Cat.No.BS4706), Beta actin Polyclonal antibody (Absin, Cat.No.abs132001), 4E-BP1 (53H11) rabbit monoclonal antibody (Cell Signaling, Cat.No.#9644), Phospho-4E-BP1 rabbit monoclonal antibody (Thr37/46) (236B4) (Cell Signaling, Cat.No.#2855), p70S6 Kinase (49D7) rabbit monoclonal antibody (Cell Signaling, Cat. No. #2708), Phospho-p70S6 Kinase rabbit monoclonal antibody (Ser371) (Cell Signaling, Cat. No. #9208), goat anti-rabbit IgG (H + L), AKT mouse monoclonal antibody (Santa cruz, Cat.No.sc-5298), p-AKT mouse monoclonal antibody(Ser 473)( Santa cruz, Cat.No.sc-293125). Secondary antibody, HRP Conjugate (BOSTER, Cat. No. BA1054), HRP-conjugated affinipure goat anti-mouse IgG (Proteintech, Cat.No.SA00001-1).
Running Native gel for the detection of TSC complex was performed using the HEK293T cells. Post-transfection, cells were collected using non-denature lysis buffer on ice. The soluble fraction was then separated by centrifugation at 12,000×g for 10 min at 4 °C. The supernatants were harvested and added with 5×protein non-denaturation loading buffer. All the running buffers and the native gels were without SDS.
Immunofluorescence analysis
Cells grown on coverslips were fixed by 4% paraformaldehyde at 37 °C for 10 min. After permeabilized with 0.5% Triton and X-100 for 15 min, cells were blocked in 5%BSA (Bull Serum Albumin, Solarbio) for 1 h at room temperature. Then the cells were incubated in primary antibody overnight at 4 °C. After washing, the cells were incubated with the secondary antibody for 1 h at room temperature. The cells staining by DAPI (Sigma) after washing, finally mounted on glass slides using gelatin. Images were acquired using a Delta Vision Image Restoration Microscope with a ×63 objective (DeltaVisionElite, GE).
Measurement of apoptosis
Cell apoptosis assays were performed by incubation with Fas-ligand (Peprotech, Cat: #310–03 H-10VG) at 1 µg/ml for 24 h, then extra incubation with 5 µg/ml CHX (Cycloheximide) (MCE, Cat.No. HY-12320) and 50 µg/ml TNFα (MACS, Lot. No. 51809100455) for 24 h. Finally, cells were treated with trypsin (without EDTA) and collected for apoptosis analysis by flow cytometry using AnnexinVPE/7-AAD Apoptosis detection Kit (Biotech, Cat.No.20180426) according to the manufacturer’s instructions.
Cell proliferation assay
The fibroblast proliferation activity was measured by Cell Counting Kit-8 (CCK-8, BOSTER, Cat.No. AR1160). Fibroblasts were seeded in 96-well plate at the density of 1,000 cells per well and cultured for 1–7 days depending assay schedules, at every detection time point of cell proliferation, cells were fed with fresh medium with the addition of 10 µl CCK-8 solutions for 1 h at 37 °C, optical densities were measured on a micro plate reader at 450 nm.
Luciferase reporter assay
HEK293T cells were seeded into 24-well plates at a density of 1.5 × 105 per well. The cells were co-transfected psiCheck-2 with insertion of the 3′ UTR of mTOR and miR-199-3p inhibitors or negative control. The analysis of luciferase activities was using the TransDetect Double-Luciferase Reporter Assay Kit (Transgen, Beijing, China).
Statistical analysis
All the data are showed as mean ± SD from independent experiments at least three times. The two-tailed Student’s t-test was applied for the determination of the p-values. p < 0.05 was considered as significant difference.
Results
Family Pedigree/Patient information
All patients included in this study were recruited from two unrelated Chinese families. The blood samples were obtained for whole-exome sequencing for the detection of the variations in the probands. The information of all the patients was listed in Table 1. The proband in family I was diagnosed as TSC when she was 16 with facial angiofibromas and renal angiomyolipoma. Her daugher was suspected as TSC because of cardiac rhabdomyomas during prenatal ultrasound. The patients in family II were not diagnosed as TSC but the patients in this family were all with epilepsy.
The proband (II-1) of family I (Fig. 1A) showed the typical symptom of TSC-facial angiofibromas (Fig. 1B), however, epilepsy was not present in the proband but in her daughter (III-1). A de novo mutation in TSC2 (c.1113delG, p.Gln371Hisfs*18) was detected in this patient as well as her daughter (III-1) (Fig. 1C). The deletion of one base pair caused amino acid change at Gln371, and resulting in frameshift and premature truncation of the protein at the highly conserved region imply that this mutation is very likely to be the major pathogenic cause of TSC (Fig. 1D).

Family pedigree and sequencing results. (A, E) Family pedigree, the families involved in this study. Squares indicate males, circles indicate females, the patients are shown as filled symbols. The proband is indicated by arrows(↗), the individual showed no genotype under the squares or circles indicate the genotype was not tested during this study. (B) The facial angiofibromas of the proband in family I. (C, F) Sanger sequencing analysis, the gene variation is shown by red arrows. (D, G) Evolutionary conservation of amino acid residues altered by the variations across different species.
The proband of family II (Fig. 1E) was detected a reported variation (c.2509-2512del, p.Asn837Valfs*11) in TSC1, his mother and his son also detected the same variation in TSC1 by sanger sequencing (Fig. 1F). This variation caused a translational frame shift, resulting in premature truncation at the highly conserved domains of TSC1 (Fig. 1G).
TSC2-Q371fs influence the TSC2 expression and the phenotype of fibroblasts
The heterozygous variation in TSC2 (c.1113delG) resulted in frameshift and a truncated protein (Fig. 2A). The mRNA and protein level of endogenous wild type TSC2 in TSC2-Q371fs fibroblasts were much lower than TSC2-WT fibroblasts (Fig. 2A, B and C). The proliferation assays showed that the proliferation of the fibroblasts with the mutation TSC2-Q371fs from patient was significantly increased compared to TSC2-WT fibroblasts from healthy volunteers (Fig. 2D). The apoptosis analyses revealed that the apoptosis was declined in TSC2-Q371fs fibroblasts (Fig. 2E).

Expression of TSC2 and phenotypes of the fibroblasts with or without TSC2-Q371fs, the WT was present as the average of all healthy volunteers. (A) Linear schematic of the protein structure of TSC2-WT, TSC2-Q371fs and the location of the primes and the qRT-PCR results, relative expression levels were normalized with GAPDH. (B) Immunofluorescence analysis using TSC2 (red) antibody and DAPI (blue) in Bar 15 μm. (C) The western blot of the endogenous TSC2 and the statistics, the originals gels can be seen in the supplementary figure S1. (D) Proliferation rate of fibroblasts. (E) Apoptosis assay of fibroblasts and the statistics.
TSC1-N837fs and TSC2-Q371fs impairs the function of TSC complex
The results of qRT-PCR showed that the VEGF and HIF-1α which were regulated by mTOR signaling pathway in fibroblast with mutation TSC2-Q371fs were elevated as reported before20, suggesting that variation (c.1113delG) in TSC2 impairs the function of TSC complex in fibroblast from patient(Fig. 3A). Western blot results revealed that phosphorylation of p70S6K and 4EBP1 were increased, indicating that mTOR was over activated in mutant cells. Supply of TSC2-WT in mutant cells restored the activity of TSC complex, thus less active mTOR subsequently decreased the p70S6K and 4EBP1 phosphorylation (Fig. 3B).

Functional analysis of the variations (A) Quantification of mRNA level using qRT-PCR, the WT represents the mixture of the equal amount total RNA from the three healthy fibroblasts. Relative expression levels were normalized with GAPDH. (B) Evaluation of the rescue experiment by western blot (the WT group was the WT2). the originals gels can be seen in the supplementary figure S2. (C) The co-ip experiment of TSC1-WT and TSC1-N837fs. The originals gels can be seen in the supplementary figure S3. (D) The measurement of TSC complex using non-denaturing PAGE. The originals gels can be seen in the supplementary figure S3 (E) The co-ip experiment of TSC2-WT and TSC2-Q371fs. The originals gels can be seen in the supplementary figure S4 (F) The measurement of TSC complex using non-denaturing PAGE. The originals gels can be seen in the supplementary figure S4.
To investigate whether the TSC1-N837fs and TSC2-Q371fs influence the interaction with each other, we constructed plasmids containing with the variations. After the transfection, the co-immunoprecipitation assays suggested that TSC1-N837fs and TSC2-Q371fs can’t detected a TSC complex, unlike the overexpressed TSC1-WT and TSC2-WT formed detectable TSC complex respectively, (Fig. 3C and E), and native PAGE showed that the TSC1-N837fs and TSC2-Q371fs failed the formation of TSC complex with the endogenous TSC2 and TSC1 respectively (Fig. 3D and F).
miR-199b-3p directly regulate the expression of mTOR
We found that the expression of mTOR was significantly increased in TSC2-Q371fs fibroblasts compared with TSC2-WT fibroblasts whether in mRNA or protein level (Fig. 4A, B). It has been reported that miR-199b is associated with lower expression of mTOR, we predicted miR-199b-3p at miRbase (http://www.mirbase.org/), which may bind to the 3’ UTR of mTOR and then promote the mRNA degradation of mTOR (Fig. 4C). We performed qRT-PCR to test miR-199b-3p expressing in fibroblasts from either patient or healthy donors. The results indicated that the expression of miR-199b-3p was dramatically decreased in TSC2-Q371fs fibroblasts compared with that in the TSC2-WT fibroblasts (Fig. 4D). To confirm whether miR-199b-3p directly target the 3’-UTR of mTOR, we co‑transfected with miR-199b-3p inhibitor N.C. and mTOR 3’ UTR, the luciferase activity had no obvious difference with that transfected with mTOR 3’ UTR, but the luciferase activity was significantly increased when co‑transfected with miR‑199b-3p inhibitor and mTOR 3’ UTR (Fig. 4E), These results showed that miR-199b-3p directly target the 3’-UTR of mTOR. Furthermore, the expression of mTOR and phosphorylation proteins level of AKT, and p70S6K was significantly decreased in HEK293T cells after the transfection with miR-199b-3p mimics and was significantly increased upon treatment with the inhibitor specific to miR-199b-3p (Fig. 4F, G, H and I). Taken together, these results suggest that miR-199b-3p may directly regulate the expression of mTOR, and the influence the activation of mTORC1 and mTORC2.

miR-199b-3p regulate the expression of mTOR (A) Quantitative analysis of mTOR in fibroblasts using qRT-PCR. (B) Quantitative analysis of mTOR in fibroblasts using western blot and the statistics. The originals gels can be seen in the supplementary figure S5. (C) Predicted miR-199b-3p target sequence of the mTOR 3’UTR using miRBase. (D) Quantification of miR-199b-3p and mRNA using qRT-PCR, the WT represents the mixture of the equal amount total RNA from the two healthy fibroblasts. Relative expression levels were normalized with GAPDH. (E). Luciferase reporter assay. (F) Quantitative analysis of mTOR in HEK293T cells at 48 h post-treatment with miR-199b-3p mimics (low panel) and the statistics (up panel). The originals gels can be seen in the supplementary figure S5. (G) Western bolt analysis of p-AKT, AKT, p-p70S6K, p70S6K in HEK293T cells at 48 h post-treatment with miR-199b-3p mimics,β-actin served as a control of total protein.(H) Quantitative analysis of mTOR in HEK293T cells at 48 h post-treatment with the inhibitor specific to miR-199b-3p (low panel) and the statistics (up panel). (I) Western bolt analysis of p-AKT, AKT, p-p70S6K, p70S6K in HEK293T cells at 48 h post-treatment with the inhibitor specific to miR-199b-3p, β-actin served as a control of total protein. The originals gels can be seen in the supplementary figure S5.
TSC complex regulated the expression of miR-199b-3p
The expression of miR-199b-3p was decreased in TSC2-Q371fs fibroblasts (Fig. 4D). To evaluate whether the TSC complex regulates the expression of miR-199b-3p, we knockdown the expression of either TSC1 or TSC2 for the reduction of TSC complex (Fig. 5A and D), then we performed RT-PCR and found that the expression of miR-199b-3p were down-regulated significantly (p < 0.01) in HEK293T-shTSC2 and HEK293T-shTSC1 cell lines (Fig. 5B and E). However, the expression of miR-199b-3p was significantly up-regulated in HEK293T cells with over-expression TSC1-WT or TSC2-WT (p < 0.001) whereas the expression of miR-199b-3p was significantly decreased in HEK293T cells with overexpression of TSC1-N837fs or TSC2-Q371fs compared to overexpression of TSC1-WT or TSC2-WT (Fig. 5B and E). These results suggest TSC complex regulates the expression of miR-199b-3p. Opposite with the expression of miR-199b-3p, the expression of mTOR was increased in shTSC1-HEK293T and shTSC2-HEK293T cell lines while decreased in TSC1-WT or TSC2-WT overexpressed cell lines (Fig. 5C and F). Taken together, these results indicated that the content of endogenous miR-199b-3p was regulated by TSC complex.

TSC complex regulates the endogenous content of miR199b-3p, *p < 0.05, **p < 0.01,***p < 0.001 comparison with WT. (A) The efficiency of knockdown TSC1 in shTSC1-HEK293T cell line. The originals gels can be seen in the supplementary figure S6. (B) The expression of miR-199b-3p in shTSC1-HEK293T cells and overexpression TSC1-WT or TSC1-N837fs in HEK293T cell line. (C) Quantitative analysis of mTOR in shTSC1-HEK293T cell line and overexpression TSC1-WT or TSC1 N837fs in HEK293T cell line using western blot (low panel) and the statistics (up panel). The originals gels can be seen in the supplementary figure S6. (D) The efficiency of knockdown TSC2 in shTSC2-HEK293T cell line. The originals gels can be seen in the supplementary figure S6. (E) The expression of miR-199b-3p in shTSC2-HEK293T cell line and overexpression TSC2-WT or TSC2- Q371fs in HEK293T cell line. (F) Quantitative analysis of mTOR in shTSC2-HEK293T cell line (low panel) and overexpression TSC2-WT or TSC2- Q371fs in HEK293T cell line using Western blot (low panel) and the statistics analysis (up panel). The originals gels can be seen in the supplementary figure S6.
Discussion
mTORC1 is an important downstream node of TSC complex and the continuous activation of mTORC1 has been reported as the main cause of the disorder Tuberous Sclerosis Complex (TSC). The complex signaling network of mTOR kinase plays a crucial role in cell growth, replication, survival, aging, and metabolism21.
In this study, we investigated two variations that involved in TSC. The variation c.2509_2512del in TSC1which involved in TSC were reported previously22. But the variation in TSC2 (c.1113delG) was a de novo germline variation which was detected in the proband (II-1) of family 1. Unfortunately, her daughter inherited this variation from her and even showed more serious symptom of TSC. The proband in family I have a severe renal angiomyolipoma, this may be because of the abnormal growth of cells. The results of cell proliferation suggested that increased cell growth in TSC2-Q371fs fibroblast was via reduction of cell apoptosis (Fig. 2).
We found that the expression of mTOR in TSC2-WT fibroblasts was less than in TSC2-Q371fs fibroblasts (Fig. 4). It was reported that micro199b might influence the expression of mTOR12. Online analysis through miRBase (http://www.mirbase.org/), we predicted that miR-199b-3p potentially targets 3’ UTR of mTOR so that regulates the expression of mTOR post-transcription. To evaluate our hypothesis, we detected the expression of miR-199b-3p in fibroblast, and the results indicated that the expression of miR-199b-3p and mTOR had negative correlation in fibroblasts, at the same time, the activation of the mTORC1 had negative correlation with the expression of miR-199b-3p. The results of dual-luciferase reporter assay suggested that miR-199b-3p directly regulates the expression of mTOR (Fig. 4).
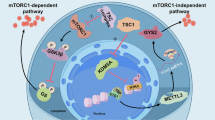
TSC2 regulates mTOR signaling to control cell growth and survival. To test if this process is mediated by miR-199b-3p, we detected the expression of miR-199b-3p and found that TSC1-N837fs and TSC2-Q371fs overexpression could decrease the expression of miR-199b-3p compared with overexpression TSC1-WT or TSC2-WT, respectively. Furthermore, knockdown of either TSC1 or TSC2 also decreased the expression of miR-199b-3p (Fig. 5). These results from both overexpression and knockdown of TSC1 or TSC2 suggest there is an axis that TSC complex regulates the expression of miR-199b-3p, and the low level of miR-199b-3p introduces the higher level expression of mTOR, causing abnormally active mTOR signal pathways which involves many cellular biological processes, in this axis miR-199b-3p acts a regulator for mTOR expression and then mTOR signaling pathway (Fig. 6).

The molecular mechanism model of TSC complex regulate mTOR signal pathway through miR-199b-3p.
It seems that the expression of miR-199b-3p was decreased more when knockdown TSC1 than knockdown TSC2. As the same time, the expression of mTOR was increased more when knockdown TSC1(Figure 5). These results may suggest that TSC1 has a larger effect on the expression of miR-199b-3p and mTOR. But in fact, the efficiency of knockdown is the determining factor of this question. The efficiency of knockdown TSC1 was higher than knockdown TSC2 in Fig. 5. The difference in effect of regulation the expression of miR-199b-3p and mTOR by TSC1 and TSC2 can’t get a clean conclussion. But from the difference in Fig. 5, it confirmed further that the negative relationship between miR-199b-3p and mTOR, when miR-199b-3p decreased the expression of mTOR is increase, when miR-199b-3p increased the expression of mTOR is decrease. The results showed that overexpression of TSC2 has a larger effect on the expression on miR-199b-3p, but not on the expression of mTOR in Fig. 5. It’s very funny to see these phenomenons, but we can’t get a certain conclusion about the difference between TSC1 and TSC2 in the regulation on miR-199b-3p just according to few experiments. We will further explore the difference regulation between TSC1 and TSC2 on miR-199b-3p to get a confirmed conclusion in future.
In summary, our studies revealed a novel molecular mechanism of TSC that the failure of the formation of physiological functional TSC complex causes the decrease of miR-199b-3p expression by which introduces the increase of mTOR expression and changes of mTOR signal pathway, followed the effects of changed pathway on all related cellular biological processes (Fig. 6). The significance of our studies on variations in TSC1 and one de novo variation in TSC2 is that we not only defined pathogenesis of those two variations in either TSC1 or TSC2, we also elucidated a novel molecular mechanism of TSC, and discovered the therapeutic potentials of miR-199b-3p for TSC.
Data availability
All primary data are available upon reasonable request from the corresponding author.
Change history
03 June 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-03849-0
References
Salussolia, C. L., Klonowska, K., Kwiatkowski, D. J. & Sahin, M. Genetic etiologies, diagnosis, and treatment of Tuberous Sclerosis Complex. Annu. Rev. Genomics Hum. Genet. 20, 217–240. https://doi.org/10.1146/annurev-genom-083118-015354 (2019).
Washko, G. R. & Parraga, G. Imaging biomarkers in lymphangioleiomyomatosis clinical trials. A Wolf in Sheep’s Clothing? Ann. Am. Thorac. Soc. 13, 307–308. https://doi.org/10.1513/AnnalsATS.201512-818ED (2016).
Jones, A. T., Narov, K., Yang, J., Sampson, J. R. & Shen, M. H. Efficacy of dual inhibition of Glycolysis and glutaminolysis for therapy of renal lesions in Tsc2(+/-) mice. Neoplasia 21, 230–238. https://doi.org/10.1016/j.neo.2018.12.003 (2019).
Henske, E. P. et al. Loss of heterozygosity in the tuberous sclerosis (TSC2) region of chromosome band 16p13 occurs in sporadic as well as TSC-associated renal angiomyolipomas. Genes Chromosomes Cancer. 13, 295–298. https://doi.org/10.1002/gcc.2870130411 (1995).
van Slegtenhorst, M. et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–808. https://doi.org/10.1126/science.277.5327.805 (1997).
Saxton, R. A. & Sabatini, D. M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 169, 361–371. https://doi.org/10.1016/j.cell.2017.03.035 (2017).
Pal, R., Xiong, Y. & Sardiello, M. Abnormal glycogen storage in tuberous sclerosis complex caused by impairment of mTORC1-dependent and -independent signaling pathways. Proc. Natl. Acad. Sci. U S A. 116, 2977–2986. https://doi.org/10.1073/pnas.1812943116 (2019).
Corradetti, M. N. & Guan, K. L. Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene 25, 6347–6360. https://doi.org/10.1038/sj.onc.1209885 (2006).
Sarbassov, D. D., Ali, S. M. & Sabatini, D. M. Growing roles for the mTOR pathway. Curr. Opin. Cell. Biol. 17, 596–603. https://doi.org/10.1016/j.ceb.2005.09.009 (2005).
Guo, H., Ingolia, N. T., Weissman, J. S. & Bartel, D. P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840. https://doi.org/10.1038/nature09267 (2010).
Lin, S. & Gregory, R. I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer. 15, 321–333. https://doi.org/10.1038/nrc3932 (2015).
Shao, S. et al. Tumor-triggered personalized microRNA cocktail therapy for hepatocellular carcinoma. Biomater. Sci. 8, 6579–6591. https://doi.org/10.1039/d0bm00794c (2020).
Gholipour, A. et al. Significance of microRNA-targeted ErbB signaling pathway genes in cardiomyocyte differentiation. Mol. Cell. Probes. 69, 101912. https://doi.org/10.1016/j.mcp.2023.101912 (2023).
Ogórek, B. et al. TSC2 regulates microRNA biogenesis via mTORC1 and GSK3β. Hum. Mol. Genet. 27, 1654–1663. https://doi.org/10.1093/hmg/ddy073 (2018).
Xiong, Q. et al. Functional evidence for a de novo mutation in WDR45 leading to BPAN in a Chinese girl. Mol. Genet. Genomic Med. 7, e858. https://doi.org/10.1002/mgg3.858 (2019).
De Falco, E. et al. A standardized laboratory and surgical method for in vitro culture isolation and expansion of primary human Tenon’s fibroblasts. Cell. Tissue Bank. 14, 277–287. https://doi.org/10.1007/s10561-012-9325-1 (2013).
Li, P. et al. A novel α-Galactosidase A splicing mutation predisposes to Fabry Disease. Front. Genet. 10, 60. https://doi.org/10.3389/fgene.2019.00060 (2019).
Cao, J. et al. Tuberous sclerosis complex inactivation disrupts melanogenesis via mTORC1 activation. J. Clin. Invest. 127, 349–364. https://doi.org/10.1172/jci84262 (2017).
Korotchkina, L. G. et al. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2, 344–352. https://doi.org/10.18632/aging.100160 (2010).
Brugarolas, J. & Kaelin, W. G. Jr. Dysregulation of HIF and VEGF is a unifying feature of the familial hamartoma syndromes. Cancer Cell. 6, 7–10. https://doi.org/10.1016/j.ccr.2004.06.020 (2004).
Szwed, A., Kim, E. & Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 101, 1371–1426. https://doi.org/10.1152/physrev.00026.2020 (2021).
van Slegtenhorst, M. et al. Mutational spectrum of the TSC1 gene in a cohort of 225 tuberous sclerosis complex patients: no evidence for genotype-phenotype correlation. J. Med. Genet. 36, 285–289 (1999).
Acknowledgements
We thank all patients and their family members and the volunteers for their participation in this study.
Funding
This study was supported by the National Natural Science Foundation of China (No.82070691) and the Grant of Natural Science Foundation of Shanxi Province in China(No.202203021212057).
Author information
Authors and Affiliations
Contributions
Conceptualization and designed the experiments: C.W., H.X.; formal analysis and investigation: N.Z.; original draft preparation: N.Z., Q.X.; review and editing, C.W. and P.L.; resources: H.X.; supervision, H.X. and C.W.; funding acquisition: C.W. and P.L. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of Shanxi University.
Informed consent
was obtained from all subjects involved in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained errors in the order of the Figures and in the Funding section. Full information regarding the corrections made can be found in the correction for this Article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, N., Xiong, Q., Li, P. et al. TSC complex decrease the expression of mTOR by regulated miR-199b-3p. Sci Rep 15, 1892 (2025). https://doi.org/10.1038/s41598-025-85706-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-85706-8
