
Abstract
Primary aldosteronism (PA), characterized by autonomous aldosterone overproduction, is a major cause of secondary hypertension with significant cardiovascular complications. Current treatments mainly focus on symptom management rather than addressing underlying mechanisms. This study aims to discover novel therapeutic targets for PA using integrated bioinformatics and experimental validation approaches. We employed a systematic approach combining: gene identification through transcriptome-wide association studies (TWAS); causal inference using summary data-based Mendelian randomization (SMR) and two-sample Mendelian randomization (MR) analyses; additional analyses included phenome-wide association analysis, enrichment analysis, protein-protein interaction (PPI) networks, drug repurposing, molecular docking and clinical validation through aldosterone-producing adenomas (APAs) tissue. Through systematic screening and prioritization, we identified 163 PA-associated genes, of which seven emerged as potential drug targets: CEP104, HIP1, TONSL, ZNF100, SHMT1, and two long non-coding RNAs (AC006369.2 and MRPL23-AS1). SHMT1 was identified as the most promising target, showing significantly elevated expression in APAs compared to adjacent non-tumorous tissues. Drug repurposing analysis identified four potential SHMT1-targeting compounds (Mimosine, Pemetrexed, Leucovorin, and Irinotecan), supported by molecular docking studies. The integration of multiple bioinformatics methods and experimental validation successfully identified novel drug targets for hyperaldosteronism. SHMT1, in particular, represents a promising candidate for future therapeutic development. These findings provide new opportunities for developing causative treatments for PA, though further clinical validation is warranted.
Similar content being viewed by others


Introduction
Primary aldosteronism (PA), the leading cause of secondary hypertension, is characterized by excessive production of aldosterone and suppressed plasma renin activity within the renin-angiotensin-aldosterone system, often accompanied by hypokalemia. While PA affects approximately 10% of individuals with hypertension and up to 20% of those with treatment-resistant hypertension, its clinical significance extends beyond these prevalence rates1. Compared to essential hypertension, PA causes more severe damage to target organs, including the heart, brain, and kidneys. This damage manifests through cardiac hypertrophy, heart failure, and impaired renal function, primarily due to excessive aldosterone production. Notably, even when blood pressure is controlled, prolonged exposure to elevated aldosterone levels continues to cause significant organ damage2. Given these serious complications, effective treatment is crucial for improving patient outcomes. Currently available treatment options, however, remain limited and suboptimal. Unilateral adrenalectomy, the preferred treatment for unilateral PA, generally yields favorable results but requires adrenal venous sampling (AVS), an invasive procedure that is not widely available. For those with bilateral PA or who are not surgical candidates, long-term therapy with mineralocorticoid receptor antagonists (MRAs), such as spironolactone or eplerenone, is common; however, these medications have adverse effects and variable efficacy3. These therapeutic limitations, combined with the significant health burden of PA, underscore the urgent need to investigate its underlying mechanisms and identify novel therapeutic targets.
To address this research need, the advent of multi-omics research offers promising new approaches. Multi-omics research integrates data from diverse biological domains, including transcriptomics and proteomics, enabling comprehensive analysis of biological systems. This integration facilitates cross-validation among different components and provides robust evidence for discovering potential biomarkers and therapeutic targets4. Within this multi-omics framework, the analysis of expression quantitative trait loci (eQTL) and protein quantitative trait loci (pQTL), when combined with genome-wide association study (GWAS) datasets, provides powerful tools for identifying disease-related genetic variations at both gene expression5 and protein levels6. Among these genetic variations, cis-acting variants are particularly informative. These variants, which are located proximal to the transcription gene unit and on the same chromosome as the transcription gene, can more effectively identify phenotype-related functional variations. This proximity allows for direct insight into the genetic mechanisms governing gene expression regulation7,8.
While genetic variants play a crucial role in disease development, studies have indicated that regulatory variants might contribute significantly to disease heritability, yet remain undetected through traditional GWAS. Many of these variants have small effect sizes, making them challenging to identify in standard SNP-based GWAS, even with large sample sizes9. To overcome this limitation, transcriptome-wide association studies (TWAS) were introduced in 2018 as an innovative approach to genetic analysis. TWAS methodology leverages expression reference panels—QTL cohorts containing both expression and genotype data—to integrate GWAS with gene expression data. This integration enables the discovery of gene-trait associations in GWAS datasets, significantly improving the detection of meaningful expression-trait associations10. Importantly, this approach reduces the occurrence of false positives obtained from GWAS and enhances the ability to discover novel disease-related genes11. The effectiveness of this approach has been demonstrated through successful identification of genes linked to various complex diseases and traits, including psychiatric disorders, neurodegenerative diseases, osteoarthritis, and rheumatoid arthritis9,12,13,14. Notably, TWAS has revealed several new risk genes that were not identified by original GWAS analyses12. Despite its proven potential in identifying disease-related genes across multiple conditions, no studies have investigated the association between the genetic component of gene expression and PA to date.
Although TWAS effectively identifies gene associations with PA, these associations alone do not establish causation. To overcome this constraint, Mendelian randomization (MR) offers a robust methodological solution. MR employs genetic variants with robust associations to exposure factors as instrumental variables (IVs). This approach enables a more reliable assessment of the causal relationship between exposures and outcomes of interest, thereby strengthening the validity of causal conclusions15. Building upon the principles of Mendelian randomization, the Summary-data-based Mendelian Randomization (SMR) technique was developed as a refined approach to exploring causal links between gene expression and complex traits. SMR leverages summary data from GWAS and eQTL analyses, allowing for the investigation of pleiotropic effects without requiring individual-level data16. In our study, we implemented an integrated analytical framework combining two-sample MR with SMR methodology to identify key genes and proteins potentially crucial in hyperaldosteronism pathophysiology. This comprehensive approach not only enhances our understanding of disease mechanisms but also facilitates the exploration of shared genetic risk factors.
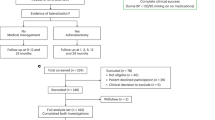
This study presents a comprehensive, multi-phase investigation into hyperaldosteronism-associated genes and their therapeutic implications. Our methodological framework encompassed six key components: (1) initial gene identification through TWAS and conditional analysis in blood and adrenal tissues; (2) causal relationship determination via SMR and two-sample MR analyses; (3) systematic evaluation of pleiotropic effects through phenome-wide association studies (PheWAS); (4) mechanistic investigation through gene ontology (GO), pathway enrichment, and protein-protein interaction (PPI) network analyses; (5) therapeutic target evaluation incorporating drug target databases and molecular docking studies, and (6) clinical specimen validation through immunohistochemistry and western blot analyses. This systematic approach, illustrated in Fig. 1, facilitated the identification and validation of therapeutic targets at the atomic level through computational simulation techniques, thereby providing a robust foundation for understanding the genetic basis of hyperaldosteronism and its potential therapeutic interventions.

Framework of comprehensive research methodology.
Results
TWAS analysis
TWAS revealed 180 significant features derived from 163 distinct genes (p < 0.05) in hyperaldosteronism, utilizing eQTL data from whole blood and adrenal gland tissues (Fig. 2; Supplementary Table S1). Among these features, 94 exhibited upregulation while 86 showed downregulation. Analysis of tissue-specific expression patterns revealed 77 significant genes in the adrenal gland and 103 in whole blood, with 17 genes showing expression in both tissues. Notably, SHMT1 emerged as the most significant feature, displaying strong associations in both GTEx whole blood (Z = 3.46065, p = 0.000539) and GTEx Adrenal Gland (Z = 3.720689, p = 0.000199) (Fig. 2c). These observations suggest that the identified genes may play crucial roles in the genetic architecture of hyperaldosteronism, potentially influencing disease susceptibility.

Transcriptome-wide association study (TWAS) results identifying genes associated with hyperaldosteronism. (a) Manhattan plot of gene-level association statistics in GTEx whole blood samples. The y-axis represents z-scores for gene expression associations, and each point represents a single gene mapped to its chromosomal position on the x-axis. Positive z-scores indicate increased gene expression associated with hyperaldosteronism, while negative z-scores indicate decreased expression. Horizontal blue lines denote the transcriptome-wide significance threshold (z = ± 1.96). Genes that exceed this threshold, such as SHMT1, are labeled, indicating significant associations with hyperaldosteronism in whole blood. These genes may play important roles in hyperaldosteronism pathology and serve as potential therapeutic targets. (b) Manhattan plot for GTEx adrenal gland samples, illustrating gene-level associations similar to (a) but specific to adrenal gland tissue. Genes with significant associations (z-scores > ± 1.96) are labeled, highlighting tissue-specific patterns that may influence hyperaldosteronism through adrenal gland pathways. (c) Heatmap summarizing z-scores for key heritable genes associated with hyperaldosteronism across different SNP weight sets derived from tissue-specific gene expression data. Each column represents a gene, and each row corresponds to a specific SNP weight set. Colored cells represent gene expression changes in hyperaldosteronism: red indicates increased expression (z-score > 3.3), while blue signifies decreased expression (z-score < -3.3). Only genes with z-scores exceeding |3.3| in at least one SNP weight set are shown to highlight genes with strong heritable associations. Empty cells represent genes excluded from TWAS FUSION analysis due to insufficient heritability estimates, meaning that their expression could not be reliably predicted based on the available genetic data. The heatmap provides a comprehensive overview of tissue-specific gene expression changes associated with hyperaldosteronism, highlighting the most significant and consistent alterations across different SNP weight sets.
Joint/conditional analysis
Since TWAS typically identifies multiple genes associated with individual risk loci, we conducted conditional and joint analyses to evaluate the independence of transcriptome-wide association signals and assess GWAS signal significance after excluding TWAS expression weights. Our analysis identified several significant features clustered within identical loci (defined as regions spanning 1.5 Mb ± 0.5 Mb), yielding 18 distinct genomic regions (Table 1). The Conditional analysis revealed 18 jointly significant features (18 unique genes) and 23 marginally significant features (21 unique genes).
We also evaluated the impact of gene expression adjustment on GWAS-identified SNP-trait associations. The explained variance in GWAS associations at specific loci ranged from 8.4 to 100%. SHMT1 in the GTEx.Adrenal_Gland dataset demonstrated complete explanation of variance (100%) at its locus (rs4925159 lead SNP pGWAS = 0.00162, conditioned on SHMT1 lead SNP pGWAS = 0.978) (Fig. 3a). Similarly, TONSL in the GTEx.Adrenal_Gland dataset accounted for 90.3% of the signal at its locus (rs12682512 lead SNP pGWAS = 0.000806, conditioned on TONSL lead SNP pGWAS = 0.297) (Fig. 3b). These conditional analyses successfully identified independent genes driving the transcriptome-wide association signals.

Conditional and joint analysis of TWAS-significant loci on chromosomes 17 and 8 associated with hyperaldosteronism. (a) Conditional and joint analysis of the TWAS-significant locus on chromosome 17. (b) Conditional and joint analysis of the TWAS-significant locus on chromosome 8. In both panels, the top section shows all genes within each locus (gray), with marginally significant TWAS-associated genes highlighted in blue and jointly significant genes highlighted in green. The bottom section presents Manhattan plots of GWAS summary statistics: gray points represent the original GWAS data, while blue points indicate results after conditioning on the jointly significant genes (green). The x-axis denotes genomic coordinates, and the y-axis represents -log10(p-values) of GWAS associations.
cis-eQTL SMR analysis
SMR analysis, utilizing data from the eQTLGen Consortium, GTEx whole blood, and GTEx adrenal gland eQTL datasets, identified eight genes (HIP1, VPS28, TONSL, SHMT1, ZNF100, AC006369.2, MRPL23-AS1, and CEP104) with pleiotropic associations to hyperaldosteronism. Four of these genes (ZNF100, HIP1, SHMT1, and TONSL) demonstrated consistent associations across multiple datasets, highlighting their robust relationship with the condition (Fig. 4).

Notable SMR results for the association between TWAS-significant genes and hyperaldosteronism risk. The analysis includes data from GTEx (whole blood and adrenal gland) and the eQTLGen Consortium. Each row lists a gene, its source data, and its odds ratio (OR) with 95% confidence interval (CI) for hyperaldosteronism risk. In the forest plots, OR values greater than 1 indicate an increased risk, while values below 1 suggest a protective effect. Genes are highlighted in color-coded rows to differentiate individual genes and enhance readability. The FDR_P_SMR column presents false discovery rate (FDR)-adjusted p-values for SMR significance, and the P_HEIDI column provides heterogeneity statistics to assess the robustness of each association.
cis-eQTL two-sample MR analysis
Two-sample MR analysis revealed seven significant gene-hyperaldosteronism associations (Fig. 5). Enhanced expression of CEP104, HIP1, TONSL, and ZNF100 correlated with reduced hyperaldosteronism risk, whereas increased expression of SHMT1, AC006369.2, and MRPL23-AS1 was linked to elevated risk. Steiger filtering validated these directional relationships. The analysis showed no evidence of heterogeneity or pleiotropy among the examined genes.

Causal relationships between eight potential genes and hyperaldosteronism in Two-Sample Mendelian Randomization (MR) analysis. Genes are listed in alternating blue and white backgrounds to aid differentiation. Each row represents a gene, with data sources from the eQTLGen Consortium and GTEx (Whole Blood and Adrenal). In the forest plot, line colors indicate the source of the data: black (eQTLGen Consortium), blue (GTEx Whole Blood), and red (GTEx Adrenal). The size of each square represents the standard error of the estimate, reflecting the precision of each association. The “method” column specifies the MR approach used, such as inverse variance weighted and wald ratio methods. Each gene’s odds ratio (OR) and 95% confidence interval (CI) for hyperaldosteronism risk are presented; OR values above 1 denote an increased risk, while values below 1 indicate a protective effect. Statistically significant MR p-values (P_MR) are reported in the relevant column, and the “steiger_dir” column indicates the direction of causality, with “TRUE” confirming a consistent causal direction.
pQTL Mendelian randomization analysis
To further establish causality between the eQTL-significant genes and hyperaldosteronism, we performed pQTL Mendelian randomization analysis. Among the identified genes, CEP104, HIP1, SHMT1, TONSL, and ZNF100 were protein-coding, while AC006369.2 and MRPL23-AS1 were classified as long non-coding RNAs (lncRNAs). Within the three pQTL databases examined, only SHMT1 had available protein data. Analysis using both SMR and two-sample MR methods across multiple plasma protein databases revealed a significant association between SHMT1 protein levels and hyperaldosteronism risk (Fig. 6).

Causal associations between SHMT1 protein and hyperaldosteronism in Two-Sample MR and SMR analyses. The blue background represents MR results using data from different sources (Zheng J et al., Ferkingstad E et al., and Sun BB et al.), with methods like wald ratio and inverse variance weighting. Odds ratios (ORs) and 95% confidence intervals (CIs) indicate each association, with OR > 1 implying increased risk and OR < 1 indicating a protective effect. The P_MR column shows significance, while “steiger_dir” indicates causal direction consistency (TRUE if consistent). The beige background represents SMR results from Ferkingstad E et al. and Sun BB et al., showing ORs, CIs, P_SMR for significance, and P_HEIDI for horizontal pleiotropy.
PheWAS
To evaluate the potential impact of the identified drug target genes on other traits and uncover any pleiotropic effects not detected by the MR-Egger intercept test, we performed a PheWAS using data from the AstraZeneca PheWAS Portal17. Comprehensive results are available in Supplementary Table S2 and Supplementary Figs. S1-S10. Remarkably, with the sole exception of SHMT1, none of the four drug targets (CEP104, HIP1, TONSL, and ZNF100) exhibited statistically significant associations with other traits at the gene level (P < 1 × 10− 8 for genomic association). This finding suggests that pharmacological interventions targeting these genes are unlikely to elicit substantial off-target effects or be confounded by horizontal pleiotropy, thereby bolstering the reliability of our study’s conclusions. Conversely, SHMT1 demonstrated a negative correlation with inflammation markers in the OLINK proteomics dataset. This finding necessitates careful interpretation of the Mendelian randomization results for SHMT1, given the possibility of pleiotropic mechanisms influencing the observed associations.
Gene ontology and pathway enrichment analysis
To identify the biological processes and pathways associated with hyperaldosteronism, we performed GO and pathway enrichment analyses using the five protein-coding genes (CEP104, HIP1, SHMT1, TONSL, and ZNF100) identified as potential therapeutic targets in our study. Two additional identified targets (AC006369.2 and MRPL23-AS1) were long non-coding RNAs and were excluded from the GO and pathway analyses due to the limitations of current enrichment analysis tools for non-coding RNAs. The GO enrichment analysis results (Fig. 7a) revealed that the most significantly enriched biological processes were related to nucleotide and amino acid metabolism, including pyrimidine deoxyribonucleoside monophosphate metabolism, L-serine metabolism, serine family amino acid catabolism, and deoxyribonucleotide biosynthesis. In the cellular component category, genes associated with the minichromosome maintenance (MCM) complex, a critical regulator of DNA replication and cell cycle progression, were notably enriched18. Moreover, the molecular function category showed enrichment of genes involved in mRNA regulatory element binding, translation repression, histone reader activity, clathrin adaptor activity, and cargo adaptor activity.

Results of Gene ontology and pathway enrichment analysis. (a) Top 30 Gene Ontology (GO) terms enriched for the target genes, categorized into three types: biological process (BP, red), molecular function (MF, green), and cellular component (CC, blue). Each bar represents the -log10(p-value) for a specific GO term, with longer bars indicating greater significance. Key processes include nucleotide metabolism and cellular assembly. (b) Pathway enrichment analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, displaying pathways with significant gene involvement, ranked by -log10(p-value). Higher fold enrichment is indicated by darker colors. Key pathways include “One carbon pool by folate,” “Antifolate resistance,” and amino acid metabolism, suggesting roles in metabolic and disease-related processes.
Pathway enrichment analysis using the KEGG database identified several key pathways in which the potential therapeutic target genes play significant roles (Fig. 7b). These pathways included those related to folate metabolism (such as the one-carbon pool by folate and antifolate resistance), amino acid metabolism (including glycine, serine, and threonine metabolism, and the biosynthesis of amino acids), and carbon metabolism (like glyoxylate and dicarboxylate metabolism, and carbon metabolism). Interestingly, the analysis also highlighted the involvement of these genes in pathways related to antifolate resistance, which could be particularly relevant for cancer therapy.
PPI networks
We constructed and analyzed PPI networks using two complementary approaches to elucidate the functional relationships among our identified drug targets.
Initially, we generated a PPI network through the STRING database, which was subsequently visualized using Cytoscape (Fig. 8a). This network comprised 23 nodes connected by 134 edges, revealing intricate interactions between the three drug target proteins and their associated partners. SHMT1 emerged as a central node, displaying robust connections with several proteins, including SHMT2, AGXT, GNMT, BHMT, and MTR. These interactions strongly indicate SHMT1’s crucial role in amino acid and one-carbon metabolism pathways. A separate functional cluster was also identified, connecting TONSL and HIP1 with proteins such as MMS22L, IFT57, and CLTB, suggesting involvement in DNA repair mechanisms.

Protein-Protein Interaction (PPI) network analysis. (a) PPI network built with STRING. Node colors represent the degree of interactions, with darker colors indicating higher degrees of connectivity. Node sizes are proportional to betweenness centrality, highlighting nodes critical for linking different parts of the network. SHMT1, HIP1, and TONSL appear as key hub proteins. (b) PPI network created using GeneMANIA. Nodes are color-coded based on functional pathways, with different edge colors representing types of associations (e.g., physical interactions, co-expression). Functional pathways, such as folate metabolism and amino acid processing, are prominently represented, underscoring the interconnected roles of SHMT1 and associated proteins.
To validate and extend these findings, we performed a secondary analysis using GeneMANIA (Fig. 8b). This approach incorporated our five drug targets along with 20 predicted interacting partners, yielding 145 distinct interactions. The network connections were supported by multiple lines of evidence: physical interactions (44.83%), co-expression patterns (19.31%), and shared protein domains (8.27%). Functional enrichment analysis through GeneMANIA highlighting the involvement of these gene networks in clathrin binding and metabolism of both folic acid-containing and pteridine-containing compounds.
Drug prediction and molecular docking analysis
Following our Mendelian randomization analysis, which revealed causal relationships between molecular alterations and disease progression, we employed a systematic approach to identify potential therapeutic compounds. We interrogated three comprehensive pharmacological databases (DrugBank, ChEMBL, and DGIdb) to discover existing drugs that might target our identified genetic loci.
Our screening identified four promising candidates targeting SHMT1: Mimosine, Pemetrexed, Leucovorin, and Irinotecan. To evaluate their binding potential, we performed molecular docking simulations using Autodock Vina. The binding energies for each drug-target interaction were calculated and are summarized in Table 2. All candidates demonstrated binding energies below our predetermined threshold of -5 kcal/mol, indicating thermodynamically favorable interactions with SHMT1. Lower binding energy values suggest more stable ligand-receptor conformations, thus predicting stronger therapeutic potential. Figure 9 provides visual representations of the most energetically favorable conformations for these molecular interactions, offering insights into their potential binding modes and mechanisms of action. The consistent achievement of favorable binding energies across all candidates supports their potential efficacy as SHMT1 modulators, warranting further experimental validation.

Docking results of available proteins with small molecules. (a) SHMT1 docking Mimosine; (b) SHMT1 docking Pemetrexed; (c) SHMT1 docking Leucovorin; (d) SHMT1 docking Irinotecan. (The PDB number of SHMT1 is 1bj4)
Validation of SHMT1 expression in clinical specimens
To further validate the bioinformatics findings, we examined SHMT1 expression in aldosterone-producing adenomas (APAs) and adjacent non-tumorous adrenal tissues (NATs) using immunohistochemistry (IHC) and western blot analysis. IHC staining revealed consistently higher SHMT1 expression in APAs compared to NATs across all three cases. Quantitative analysis of IHC scores revealed significantly higher SHMT1 expression in APAs (mean score: 8) than in NATs (mean score: 1.67; p < 0.05; Fig. 10a). Western blot analysis corroborated these findings, demonstrating a two-fold increase in SHMT1 protein expression in APAs compared to NATs, with GAPDH as a loading control (p < 0.05; Fig. 10b). These findings demonstrate that SHMT1 is upregulated in APAs and may play a critical role in the pathogenesis of hyperaldosteronism, supporting its potential as a therapeutic target.

SHMT1 expression in aldosterone-producing adenomas (APAs) and adjacent non-tumorous adrenal tissues (NATs). (a) Immunohistochemical (IHC) staining of SHMT1 in NAT and APA tissues. Representative micrographs show significantly increased SHMT1 protein expression in APA compared to NAT. The scale bar represents 100 μm. Quantification of IHC scores is presented in the bar graph, with error bars indicating standard deviation. (b) Western blot analysis of SHMT1 protein expression in NAT and APA tissues. The upper panel shows representative western blot bands for SHMT1 and GAPDH, the loading control. The bar graph below quantifies relative SHMT1 expression levels, normalized to GAPDH. These data suggest that SHMT1 is upregulated in APAs and may contribute to the pathogenesis of hyperaldosteronism. *p < 0.05 compared to NAT.
Discussion
This study aimed to identify novel drug targets for hyperaldosteronism using a comprehensive bioinformatics approach. We integrated GWAS data on hyperaldosteronism with eQTL data from the GTEx project, specifically from adrenal gland and whole blood tissues. TWAS identified 180 significant features across 163 unique genes, and subsequent conditional analysis refined these findings, revealing 18 significant and 23 marginally significant features. To increase the specificity of these results and identify promising therapeutic targets, we applied SMR methods, complemented by the HEIDI test to assess phenotype-causative relationships. Using eQTL data and SMR methods, we identified eight potential therapeutic targets, seven of which (CEP104, HIP1, TONSL, ZNF100, SHMT1, AC006369.2, and MRPL23-AS1) were validated through two-sample MR analysis. Among these, SHMT1 emerged as a particularly promising candidate due to the availability of protein-level data, which allowed for a more comprehensive analysis. Notably, pQTL SMR and two-sample MR analyses revealed a significant association between elevated SHMT1 expression and an increased risk of hyperaldosteronism, highlighting SHMT1 as a key target for future therapeutic interventions. Five potential drug targets (CEP104, HIP1, TONSL, ZNF100, and SHMT1) were then subjected to functional characterization, including PheWAS, enrichment analysis, PPI network analysis, drug repurposing. In contrast, AC006369.2 and MRPL23-AS1, as lncRNAs, were not included in these analyses. To assess the specificity of the drug targets, we performed PheWAS to evaluate potential side effects. Four of the five targets (CEP104, HIP1, TONSL, ZNF100) showed no significant associations with other traits, suggesting minimal pleiotropic effects. However, SHMT1 was found to be negatively associated with inflammation-related proteins, a result that warrants further investigation. GO and pathway enrichment analyses revealed that these targets are involved in crucial processes such as nucleotide and amino acid metabolism, DNA replication, and cell cycle regulation. To further elucidate the functional roles of these targets, we examined their PPI and involvement in biological pathways. PPI networks highlighted strong interactions among the targets, particularly SHMT1’s central role in amino acid and one-carbon metabolism. Drug repurposing and molecular docking analysis revealed four potential drug candidates targeting SHMT1: mimosapine, pemetrexed, folinic acid, and irinotecan. Experimental validation using IHC and western blot analyses confirmed significantly higher SHMT1 expression in APAs compared to NATs, further supporting its role in PA pathogenesis. By integrating bioinformatics-driven target identification with experimental validation, this study demonstrates the power of this approach in uncovering novel therapeutic targets for complex disorders like hyperaldosteronism. These findings open new avenues for therapeutic intervention in the management of hyperaldosteronism.
The findings of this study highlight the potential roles of CEP104, HIP1, TONSL, and ZNF100 in the pathogenesis of hyperaldosteronism. The increased expression of these genes was associated with a decreased risk of developing this condition, suggesting their protective effects against the disease. However, the lack of available pQTL data and druggable information for these proteins limits further analysis. Additional research is required to clarify the mechanistic roles of these genes and their potential implications for the diagnosis and treatment of PA.
CEP104, a microtubule-plus-end tracking protein, is essential for ciliogenesis and ciliary tip formation, and functional cilia are involved in key signaling pathways, including those that respond to extracellular signals such as angiotensin II and potassium, which regulate aldosterone secretion19,20. By maintaining ciliary tip integrity and supporting calcium signaling and Hedgehog pathway function, increased CEP104 expression may help maintain the balance of aldosterone synthesis and secretion, preventing the dysregulation seen in hyperaldosteronism21,22. Specifically, calcium signaling is directly involved in the activation of aldosterone synthase (CYP11B2), and Hedgehog signaling regulates the structural and functional maintenance of zona glomerulosa cells23,24. Moreover, CEP104’s direct binding to α/β-tubulin heterodimers and its phosphorylation by Nek1 kinase, a regulator of ciliary length and function, further support primary ciliary stability and cytoskeletal dynamics, which are important for cellular responses to mechanical and chemical stimuli25,26. Therefore, upregulation of CEP104 expression likely acts as a protective mechanism against PA, reducing the risk of aldosterone dysregulation and the development of hyperaldosteronism.
HIP1 (Huntingtin interacting protein 1) is a critical regulator of clathrin-mediated endocytosis and membrane trafficking, processes essential for maintaining cellular homeostasis27,28. In PA, HIP1 may influence aldosterone production through its impact on the trafficking of angiotensin II type 1 receptors (AT1R) and voltage-gated calcium channels (VGCCs), which are pivotal regulators of aldosterone synthesis in adrenal cells29. Through the formation and recycling of clathrin-coated vesicles, HIP1 facilitates the internalization and surface regulation of these receptors, directly affecting calcium influx and angiotensin II signaling30. Although direct evidence linking HIP1 to aldosterone production is limited, studies in pancreatic beta cells suggest a broader role for HIP1 in hormone secretion. For instance, HIP1 influences insulin release in beta cells, hinting at a similar mechanism in aldosterone-producing adrenal cells31. Beyond its role in receptor trafficking, HIP1’s regulation of growth factor signaling pathways, including Hedgehog and fibroblast growth factor (Fgf), may limit the proliferative signals that contribute to adenoma formation32. This underscores HIP1’s dual role in both hormone secretion and the pathophysiology of adenoma formation. Future studies are needed to delineate HIP1’s role in adrenal cell function and its contribution to PA pathophysiology.
TONSL (Tonsoku-like DNA repair protein) may play a protective role in adrenal physiology, including in the context of hyperaldosteronism, by maintaining genomic stability through the homologous recombination (HR) DNA repair pathway33. This function could potentially prevent the accumulation of DNA damage that might lead to somatic mutations in various genes, including those implicated in PA such as KCNJ5 and ATP1A1. By repairing DNA damage, TONSL reduces mutation risk, stabilizes replication forks, and enhances cellular resilience under stress. Interactions with MMS22L and histone chaperones like ASF1 and CAF-1 facilitate this stress response, protecting adrenal cells from damage34,35. Given its role in DNA repair, stress response, and genomic stability, TONSL could potentially be explored as a biomarker or therapeutic target for hyperaldosteronism. However, its function is context-dependent; in some cancers, TONSL overexpression contributes to tumorigenesis, highlighting the importance of considering tissue-specific effects when evaluating its role in adrenal disorders36,37,38. Future research integrating pQTL data with experimental validation is crucial for elucidating the precise mechanisms by which this gene contribute to the pathogenesis of hyperaldosteronism.
ZNF100 (zinc finger protein 100) appears to play a protective role in PA through mechanisms related to its regulatory functions in gene expression. Zinc finger proteins, including ZNF100, are known for their critical roles in transcriptional regulation, epigenetic silencing, and cellular homeostasis39. Recent studies, such as those by Zuo (2023), have highlighted ZNF100’s involvement in silencing retrotransposons, which may prevent genomic instability—a potential factor in the pathogenesis of PA40. By regulating retrotransposon activity, ZNF100 might influence the transcriptional landscape of aldosterone-producing cells, potentially limiting aberrant signaling pathways that drive excessive aldosterone synthesis. Additionally, ZNF100 has been implicated in the regulation of lncRNAs, which play essential roles in transcriptional control and gene expression. ZNF100-associated lncRNAs, such as ZNF100-6:2, show diagnostic potential, reflecting the gene’s broader impact on transcriptional networks41. This suggests that ZNF100’s regulatory functions extend beyond protein-coding genes to encompass a wide range of molecular interactions relevant to cellular processes involved in PA. Future research should focus on elucidating the molecular interactions between ZNF100 and the signaling networks implicated in PA, as well as exploring its potential as a biomarker or therapeutic target.
In addition to the protein-coding genes discussed earlier, elevated levels of AC006369.2 and MRPL23-AS1, two lncRNAs, are associated with an increased risk of PA, suggesting their substantial contribution to the development of this condition. Although direct evidence linking AC006369.2 to PA is limited, its role in regulating gene expression, modulating the immune microenvironment, and influencing metabolic pathways indicates its potential significance in PA pathophysiology. AC006369.2 may influence aldosterone production by modulating the expression of key transcription factors and enzymes involved in adrenal steroidogenesis, such as NR5A1 (steroidogenic factor 1, SF-1), which regulates CYP11A1 (converting cholesterol to pregnenolone) and CYP11B2 (converting 11-deoxycorticosterone to aldosterone)42. Additionally, AC006369.2 may shape the immune microenvironment in APAs by modulating immune cell infiltration and immune checkpoints, facilitating immune evasion43. Its influence on metabolic pathways, including lipid metabolism and oxidative stress, may impact ferroptosis, a process critical to cell survival and proliferation, thus supporting tumor growth and excessive aldosterone production44. Similarly, MRPL23-AS1, known for its role in cancer, has the potential to influence aldosterone-producing cell dynamics by mediating the transcriptional silencing of E-cadherin through the formation of an RNA-protein complex with EZH245,46. This interaction promotes tumor progression and carcinogenesis by activating the Wnt/β-catenin signaling pathway47. MRPL23-AS1’s involvement in epithelial-mesenchymal transition (EMT) suggests it could enhance adrenal cell plasticity, favoring abnormal aldosterone synthesis48,49. Furthermore, MRPL23-AS1’s interaction with miRNAs, such as miR-30b, and its effects on genes like MYH9, imply a complex regulatory network affecting both adrenal cell behavior and steroidogenesis47. These mechanisms highlight the therapeutic potential of targeting AC006369.2 and MRPL23-AS1 to modulate adrenal function and improve treatment outcomes in PA. Further research is needed to unravel the precise pathways involved and to develop targeted interventions, such as RNA interference or small molecule inhibitors, to counteract their effects.
The most significant finding of this study is the identification of SHMT1 as a promising drug target for hyperaldosteronism through its role in metabolic reprogramming. SHMT1, a key enzyme in one-carbon metabolism, catalyzes the reversible conversion of serine to glycine, producing essential one-carbon units that support nucleotide biosynthesis, redox balance, and DNA methylation50. Our findings demonstrate that SHMT1 is upregulated in APAs, similar to its elevated expression observed in certain cancer cells, where it meets the high biosynthetic demands of proliferation by providing crucial metabolic intermediates51,52. SHMT1 overactivity increases the production of 5,10-methylene-THF, a crucial one-carbon donor that are essential for generating NADPH and NADH, which act as electron donors for CYP11B2, a rate-limiting enzyme in aldosterone synthesis53. Although there is no direct evidence, we speculate that through this pathway, enhanced SHMT1 activity promotes CYP11B2 function by ensuring adequate cofactor supply, ultimately resulting in increased aldosterone production54. Additionally, SHMT1 may support glutathione synthesis, protecting aldosterone-producing cells from oxidative stress and enabling their survival under metabolic stress55,56. SHMT1’s involvement in lipid metabolism and glycolysis further amplifies its contribution to tumor growth and aldosterone production57,58. Its role in various cancers underscores its broader impact on cellular proliferation and survival59. For instance, SHMT1 promotes cell growth and migration in ovarian cancer, and its knockout in lung cancer induces cell cycle arrest and apoptosis through p53 activation60,61. The inverse correlation of SHMT1 expression with prognostic factors in breast cancer further supports its oncogenic role62.
Given the pivotal role of SHMT1 in cellular metabolism and its link to hyperaldosteronism, targeting SHMT1 or its downstream pathways could present novel therapeutic opportunities. Inhibiting SHMT1 may suppress adrenal cell proliferation, normalize aldosterone production, and alleviate the symptoms associated with hyperaldosteronism. A pharmacological analysis identified several potential SHMT1 inhibitors, including Mimosine, Pemetrexed, Leucovorin, and Irinotecan, all of which demonstrated strong binding affinity to SHMT1. Notably, Pemetrexed, Leucovorin, and Irinotecan are already approved drugs, while Mimosine is currently experimental. The mechanisms of action for these compounds, which involve modulating folate metabolism, DNA synthesis, and cell cycle progression, align with key aspects of hyperaldosteronism pathophysiology, such as increased cell proliferation and metabolic hyperactivity, highlighting their promise as candidates for drug repurposing in this condition. Pemetrexed is a multitargeted antifolate that inhibits key enzymes in folate metabolism and DNA synthesis, including thymidylate synthase, dihydrofolate reductase, and glycinamide ribonucleotide formyltransferase63. These enzymes are crucial for purine and pyrimidine synthesis, processes that are often upregulated in rapidly proliferating cells, such as APAs. By inhibiting these pathways, Pemetrexed may reduce aldosterone production in hyperaldosteronism, potentially alleviating symptoms of the condition64. Additionally, Pemetrexed may directly inhibit SHMTs, further disrupting one-carbon metabolism65. Leucovorin, known for its role in modulating fluorouracil (5-FU) in colorectal cancer66, enhances the stability and function of reduced folate pools, which are crucial for maintaining nucleotide synthesis and DNA repair processes67. By restoring and maintaining folate metabolism, Leucovorin may normalize these pathways, potentially reducing the excessive cell proliferation and hormone secretion characteristic of adenomatous tissue. Irinotecan, a topoisomerase I inhibitor, disrupts DNA synthesis by stabilizing the cleavable complex between DNA and topoisomerase I, leading to replication stress and cell cycle arrest68,69. This mechanism of action has potential implications for hyperaldosteronism, where targeted inhibition of cell cycle progression could mitigate the overgrowth of adrenal tissue. Mimosine, known for its capacity to arrest DNA synthesis at replication forks by inhibiting deoxyribonucleotide metabolism, specifically targets pathological cell proliferation pathways70. By curbing the overactive cell cycle progression characteristic of adrenal hyperplasia, Mimosine may help regulate the excessive growth associated with hyperaldosteronism. Preclinical studies highlight Mimosine’s protective effects on postmitotic cells and its inhibition of enzymes involved in cell growth and angiogenesis, which could mitigate the excessive cell proliferation and vascularization seen in APAs71. Although direct clinical evidence for these drugs in hyperaldosteronism is limited, clinical trials in oncology provide valuable insights for their potential repurposing.
It is worth noting that this study finds a negative correlation between SHMT1 expression and inflammatory markers, suggesting a potential role for SHMT1 in maintaining a low-inflammatory environment. This finding aligns with PA’s nature as a functionally benign tumor characterized by a relatively ordered microenvironment, contrasting sharply with the cellular heterogeneity and immune activation typically observed in malignant tumors72,73. Previous research has found that SHMT1 contributes to NADPH generation, a critical reducing agent in cellular antioxidant defense systems53,74. Through NADPH-dependent enzymes like glutathione reductase and thioredoxin reductase, SHMT1 helps maintain cellular redox balance by neutralizing reactive oxygen species (ROS)75. This regulation prevents ROS-mediated activation of inflammatory pathways, such as the NF-kB76. Based on these findings, we propose that in PA, elevated SHMT1 activity enhances NADPH production, which supports cellular antioxidant defense and facilitates ROS neutralization, thereby suppressing inflammation-driven pathways. Drawing parallels with SHMT-mediated antioxidant defense observed in cancer cells77, SHMT1 in PA likely protects aldosterone-producing cells from oxidative stress, thereby indirectly suppressing inflammation. The stable microenvironment of PA, maintained through SHMT1’s regulation of redox balance and inflammation, aligns with its benign nature and contributes to its controlled, orderly development. It is plausible that, SHMT1’s observed effects on inflammation are likely an indirect consequence of its metabolic functions. SHMT1 appears to play a critical role in the pathophysiology of PA, primarily by driving metabolic reprogramming rather than directly modulating inflammatory pathways.
While SHMT1 upregulation appears to support adrenal cell proliferation in PA, its therapeutic inhibition must be approached cautiously to minimize off-target effects. Given SHMT1’s involvement in essential biosynthetic processes, its inhibition could disrupt not only tumor growth but also normal cell function if not carefully regulated. However, studies on SHMT1 inhibitors in cancer suggest that targeting SHMT1 could selectively affect proliferating cells dependent on one-carbon metabolism, potentially sparing normal tissues that rely less on SHMT1 due to alternative metabolic pathways, such as mitochondrial SHMT278,79. Consequently, SHMT1 inhibitors could enable selective drug targeting of tumors, potentially reducing side effects, including the risk of widespread inflammatory reactions. The therapeutic potential of targeting SHMT1 is supported by multiple lines of evidence. Inhibition of SHMT1 has been shown to induce apoptosis and cell cycle arrest in cancer cells. Small molecule inhibitors of SHMT1/2 have demonstrated efficacy in reducing cancer cell viability, particularly in c-Myc positive tumors and diffuse large B-cell lymphoma80,81,82. These findings, combined with our understanding of SHMT1’s role in hyperaldosteronism, suggest that targeting this enzyme could be a viable therapeutic strategy for both cancer and endocrine disorders.
Several limitations should be considered when interpreting the findings. Firstly, the publicly available pQTL datasets are not comprehensive, and information for some proteins, such as CEP104, HIP1, TONSL, and ZNF100, is lacking. Secondly, the research primarily focused on cis-QTLs, potentially overlooking the influence of other regulatory elements and environmental factors. Thirdly, reproducibility is a crucial principle in assessing the validity of association study findings83. However, we currently lack other large-scale European-only summary statistics for hyperaldosteronism beyond the Finngen GWAS data to replicate our association findings. Additionally, due to the unavailability of relevant data, our pleiotropy analysis primarily focused on the European population. Given the transethnic diversity in the genetic architecture of hyperaldosteronism84, it remains uncertain whether our findings are applicable to other ancestral groups, such as East Asians. Future studies in multiethnic cohorts may help validate our findings and explore the genetic basis of hyperaldosteronism in a broader context. Incorporating such data will also allow us to explore potential population-specific therapeutic targets and evaluate the clinical relevance of SHMT1 as a therapeutic candidate on a global scale. Lastly, it’s important to note that while our Mendelian randomization and pharmacological studies offer promising insights, they do not ensure clinical efficacy. The complex nature of disease states can lead to alterations in gene expression patterns that may not be fully captured in our analyses. Therefore, these findings should be viewed as a starting point for further investigation rather than definitive proof of therapeutic success. Additional experimental validation and clinical studies are essential to verify their therapeutic potential.
Collectively, our analysis suggests that SHMT1 and the other identified genes contribute to PA pathogenesis through distinct yet interconnected mechanisms, including (1) metabolic reprogramming, (2) cellular structural modifications, (3) genomic stability, and (4) epigenetic regulation. These genes appear to function in complementary pathways: SHMT1 affects metabolic regulation, CEP104 and HIP1 influence cellular structure and trafficking, while TONSL and ZNF100 maintain genomic stability. The long non-coding RNAs AC006369.2 and MRPL23-AS1 add additional layers of regulation - AC006369.2 regulates steroidogenesis and immune responses, while MRPL23-AS1 promotes cell plasticity. Understanding these multiple pathways could lead to more effective therapeutic strategies, potentially combining SHMT1-targeted therapy with modulators of these additional pathways for enhanced treatment outcomes.
Conclusion
Our findings highlight potential targets for future treatment of PA, necessitating further research to evaluate the feasibility of these seven potential drug targets (CEP104, HIP1, TONSL, ZNF100, SHMT1, AC006369.2, and MRPL23-AS1) as therapeutic drug targets for PA. These identified drug targets, particularly SHMT1, provide a foundation for future research aimed at developing targeted therapeutic interventions for this complex endocrine disorder. Our findings offer new insights into the etiology of PA and support further mechanistic studies and future drug development efforts.
Methods
Hyperaldosteronism GWAS summary data source
The summary-level data on PA utilized in this study were obtained from the R10 release of the FinnGen GWAS results, available as of December 18, 2023 (https://r10.finngen.fi/). The specific phenotypic code employed to identify PA cases was “E4_HYPERALDO” (Hyperaldosteronism). The dataset encompassed 744 cases (409 females and 335 males) and 395,289 controls. Detailed information can be found in the corresponding published studies85.
TWAS analysis
We performed a tissue-specific TWAS for hyperaldosteronism using the FUSION software package. This analysis integrated hyperaldosteronism GWAS summary statistics with pre-established gene expression weights from adrenal gland and whole blood tissues, obtained from the TWAS FUSION website86. TWAS FUSION analysis was performed on autosomal chromosomes using the default settings from the TWAS FUSION protocol. The FUSION methodology employs multiple statistical approaches to generate TWAS expression weights that capture SNP-gene expression relationships. These approaches include Best Linear Unbiased Prediction (BLUP), Bayesian Sparse Linear Mixed Model (BSLMM), LASSO, Elastic Net, and a model based on top SNPs from reference panels such as GTEx. To optimize predictive accuracy, FUSION implements a fivefold cross-validation process, evaluating out-of-sample R2 values to determine the most effective predictive model for each gene. The resulting optimized expression weights are then integrated with GWAS data to estimate associations between predicted gene expression and the phenotype of interest86. We computed a TWAS p-value for each gene in the adrenal gland and whole blood for European populations, considering genes with a p-value less than 0.05 as significant87.
Joint/conditional analysis
To distinguish between genetic and expression-driven associations, we conducted joint and conditional analyses using FUSION to investigate TWAS-identified signals. These analyses incorporated two key components: primary TWAS association statistics and a matrix representing genetic correlations and linkage disequilibrium. Our primary objective was to determine whether GWAS associations maintained their significance after accounting for TWAS-detected expression associations.
Using an iterative permutation approach, we systematically evaluated each SNP from the hyperaldosteronism GWAS by conditioning it on the multi-gene model. The analysis allowed up to 100,000 permutations per SNP, with a significance threshold of p < 0.05. To visualize and interpret the results, we utilized the “FUSION.post_process.R” script, which generated conditional output plots and summary statistics. This approach enabled us to effectively assess the independence of GWAS signals from gene expression-driven effects.
cis-eQTL SMR analysis
cis-eQTLs data were obtained from two databases: (1) Genotype Tissue Expression (GTEx, version 8) for whole blood and adrenal gland (available at https://yanglab.westlake.edu.cn/software/smr/#eQTLsummarydata); (2) cis-eQTL summary statistics from eQTLGen Consortium (available at https://www.eqtlgen.org/cis-eqtls.html). cis-eQTLs were defined as SNPs and gene probes with a distance of less than 1 Mb between them.
We performed SMR analysis to test whether shared causal variants at specific loci influence both gene expression and hyperaldosteronism, as previously described16. To assess the validity of the observed associations, we implemented the heterogeneity in dependent instruments (HEIDI) test. A significant HEIDI test result (p_HEIDI < 0.05) suggests that the association may be driven by two distinct genetic variants in high linkage disequilibrium. The analysis was conducted using SMR software version 1.3.1 with default settings. We adjusted p-values using the Benjamini-Hochberg method and considered associations significant at a FDR threshold of < 0.05.
cis-eQTL two-sample MR analysis
To further investigate the causal relationship between SMR-significant genes and hyperaldosteronism, we conducted a two-sample MR analysis using the ‘TwoSampleMR’ package (https://github.com/MRCIEU/TwoSampleMR). For this analysis, we used cis-eQTLs of SMR-significant genes as exposure variables, derived from three sources: the eQTLGen Consortium data, GTEx whole blood eQTL data, and GTEx adrenal gland eQTL data. Hyperaldosteronism served as the outcome variable.
The selection of independent SNPs associated with SMR-significant genes was based on two criteria: genome-wide significance (p < 5 × 10− 8) and linkage disequilibrium assessment. We excluded SNPs that showed linkage disequilibrium (r2 > 0.001) or were located within a 1 Mb clumping window of other SNPs with higher p-values. For the statistical analysis, we employed the Wald ratio when only one genetic instrument was available for a given gene. In cases with two or more genetic instruments, we applied inverse variance weighted MR (MR-IVW), followed by heterogeneity and pleiotropy analysis. To ensure correct causal direction, we implemented Steiger filtering to verify the directionality of associations between genes and hyperaldosteronism. Statistical significance was established at p < 0.05.
pQTL Mendelian randomization analysis
To further verify the causal relationship between significant genes and hyperaldosteronism at the protein level, a MR analysis of pQTL was performed. The plasma pQTL data were retrieved from three sources: (1) the study by Zheng et al., which integrated five previously published GWAS88; (2) the UK Biobank Pharma Proteomics Project (UKB-PPP), which conducted proteomic profiling on blood plasma samples from 54,219 participants using the Olink platform and collected data on 2,923 protein89; and (3) the deCODE Health study, where 4,907 aptamers were measured among 35,559 Icelanders using the SomaScan platform90. Cis-SNPs were defined as SNPs within 1 Mb from the gene encoding the protein, and linkage disequilibrium was estimated based on 1000 Genomes European panel. The specific methods for SMR and two-sample MR were the same as those used for the eQTL analysis.
Phenome-wide association analysis
To evaluate the potential pleiotropic effects of the identified drug targets and assess possible side effects, a PheWAS was conducted utilizing the AstraZeneca PheWAS Portal (https://azphewas.com/)17. This analysis leveraged exome sequencing data from approximately 470,000 participants from the UK Biobank, encompassing around 10,000 binary and 3,500 continuous phenotypes. The detailed methodology for constructing the PheWAS portal is described in the original publication cited17. To minimize false-positive findings, we implemented multiple testing corrections, adhering to the stringent significance threshold of 1 × 10− 8 set by the AstraZeneca PheWAS Portal.
Functional enrichment analysis
To explore the functional attributes and biological significance of the identified potential therapeutic target genes, we utilized the R package clusterProfiler to conduct Gene Ontology and pathway enrichment analyses91. The input gene list for these analyses consisted of the potential therapeutic target genes identified through our integrative bioinformatics approach. GO enrichment analysis was performed to determine the overrepresentation of these genes in specific biological processes, molecular functions, and cellular components. Additionally, pathway enrichment analysis was conducted using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database to identify the significantly enriched biological pathways associated with the target genes92.
Protein-protein interaction network construction
Evaluating and analyzing PPI networks can provide a better understanding of how proteins interact with each other intracellularly, offering insights into cellular processes and potential therapeutic targets. To construct and analyze PPI networks in this study, we utilized multiple complementary tools. First, we employed the STRING database (version 12.0, https://string-db.org/) to construct the initial PPI network. This network was then visualized and further analyzed using Cytoscape (version 3.10.2), a popular open-source software platform for network visualization and analysis93,94. To complement and validate our findings, we also used GeneMANIA (https://genemania.org/), another widely-used tool for PPI analysis and functional prediction95. By integrating these tools, we were able to comprehensively examine the protein interactions relevant to our study.
Candidate drug prediction
Following our MR analysis, we identified several druggable genes significantly associated with hyperaldosteronism GWAS signals. To explore potential therapeutic interventions targeting these genes, we conducted a comprehensive gene-drug search using three complementary databases: DrugBank (version 5.1.12, https://go.drugbank.com), ChEMBL (version 34, https://www.ebi.ac.uk/chembl), and DGIdb (version 5.0.7, https://www.dgidb.org/). These databases are valuable resources that connect drugs and various chemical compounds to their target genes, facilitating the identification of existing drugs that might be repurposed for hyperaldosteronism treatment.
Molecular docking analysis
To explore the potential therapeutic implications of our findings, we employed molecular docking simulations. This computational approach allowed us to examine the atomic-level interactions between promising drug candidates and the protein targets implicated in hyperaldosteronism. By doing so, we aimed to assess the feasibility of these targets for drug development and gain insights into the mechanisms of potential therapeutic interventions. The protein structures of the target genes were retrieved from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (PDB) (http://www.pdb.org/). The chemical structures for the small molecules were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/). Before conducting the docking studies, the protein structures were prepared using AutoDock 1.5.6, which included steps such as removing ligands and water molecules, adding hydrogen atoms, adjusting charges, and refining the conformations of amino acids96. Molecular docking simulations were carried out with AutoDock Vina, and the docking results were visualized using PyMOL (Version 3.0.3, https://pymol.org/).
Clinical validation of SHMT1 expression in APAs
Tissue collection and processing: Fresh tissue samples from three cases were collected post-surgery, with each case providing paired samples of both APAs and NATs. APAs and NATs were fixed in 10% neutral buffered formalin for IHC. For western blot analysis, an additional set of three APAs and NATs, which had been stored at -80℃ previously, was used.
Immunohistochemistry: IHC procedures were performed according to the manufacturer’s instructions for the IHC Detect Kit for Rabbit/Mouse Primary Antibody (ProteinTech, Cat No: PK10006). The primary anti-SHMT1 antibody (1:2000; Proteintech, 30192-1-AP) was used for the analysis. SHMT1 expression was evaluated by a professional pathologist as well as ImageJ software (version 1.54, IHC Profiler) and scored via a scoring system97,98. The intensity of staining (no staining = 0, weak staining = 1, moderate staining = 2, strong staining = 3) and the percentage of stained cells (0–5%=0, 5–25%=1, 26–50%=2, 51–75%=3, 76–100%=4) were scored, and these scores were multiplied to obtain the final score99.
Western blot analysis: Frozen tissue samples were homogenized in RIPA buffer (Thermo Fisher Scientific, 89900) containing protease and phosphatase inhibitors. Protein concentrations were determined using the BCA protein assay kit (Thermo Fisher Scientific, 23227). Equal amounts of protein lysates (30 µg per lane) were separated on 10% SDS-polyacrylamide gels and were electro-transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, CA). After a blocking incubation with 5% milk in 1X TBST, menbranes were incubated with primary antibodies overnight at 4℃, followed by 1 h incubations in a secondary antibody. Visualized with the enhanced chemiluminescence (ECL), images were obtained with a chemiluminescence imager (AMERSHAM Imagequant 800, America). All antibodies were as follows: anti-SHMT1 antibody (1:2000, Proteintech, 30192-1‐AP), anti-GAPDH (1:5000, Cell Signaling Technology, 2118) and anti-rabbit IgG, HRP-linked (1:5000, CST, no.7074). The GAPDH used as an internal control.
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 8.0 (GraphPad Software, San Diego, CA, USA). All data are presented as mean ± standard error of the mean (SEM). Statistical significance was determined using Student’s t-test. Differences were considered statistically significant at p < 0.05.
Data availability
Hyperaldosteronism summary data used in this study are available at: https://r10.finngen.fi/; FUSION software, weights/models, and reference LD are available at http://gusevlab.org/projects/fusion/; SMR software and eQTL summary data are available at https://yanglab.westlake.edu.cn/software/smr; cis-eQTL summary statistics from eQTLGen Consortium are available at https://www.eqtlgen.org/cis-eqtls.html. All data sources used in this study were thoroughly detailed within the manuscript. The results of this study can be obtained by contacting the corresponding author.
References
Huang, M. et al. Global and regional prevalence and cardiovascular risk of primary aldosteronism: A systematic review and meta-analysis. Curr. Probl. Cardiol. 49, 102791. https://doi.org/10.1016/j.cpcardiol.2024.102791 (2024).
Turcu, A. F., Yang, J. & Vaidya, A. Primary aldosteronism—A multidimensional syndrome. Nat. Rev. Endocrinol. 18, 665–682. https://doi.org/10.1038/s41574-022-00730-2 (2022).
Dogra, P., Bancos, I. & Young, W. F. Primary aldosteronism: A pragmatic approach to diagnosis and management. Mayo Clinic Proc. 98, 1207–1215. https://doi.org/10.1016/j.mayocp.2023.04.023 (2023).
Hasin, Y., Seldin, M. & Lusis, A. Multi-omics approaches to disease. Genome Biol. 18, 83. https://doi.org/10.1186/s13059-017-1215-1 (2017).
Emilsson, V. et al. Genetics of gene expression and its effect on disease. Nature 452, 423–428. https://doi.org/10.1038/nature06758 (2008).
Chick, J. M. et al. Defining the consequences of genetic variation on a proteome-wide scale. Nature 534, 500–505. https://doi.org/10.1038/nature18270 (2016).
Nica, A. C. & Dermitzakis, E. T. Expression quantitative trait loci: Present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20120362. https://doi.org/10.1098/rstb.2012.0362 (2013).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195. https://doi.org/10.1126/science.1222794 (2012).
Yao, S. et al. A transcriptome-wide association study identifies susceptibility genes for Parkinson’s disease. NPJ Parkinsons Dis. 7, 79. https://doi.org/10.1038/s41531-021-00221-7 (2021).
Wainberg, M. et al. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 51, 592–599. https://doi.org/10.1038/s41588-019-0385-z (2019).
Barfield, R. et al. Transcriptome-wide association studies accounting for colocalization using Egger regression. Genet. Epidemiol. 42, 418–433. https://doi.org/10.1002/gepi.22131 (2018).
Li, X. et al. Transcriptome-wide association study identifies new susceptibility genes and pathways for depression. Transl Psychiatry. 11, 306. https://doi.org/10.1038/s41398-021-01411-w (2021).
Qi, X. et al. Integration of transcriptome-wide association study and messenger RNA expression profile to identify genes associated with osteoarthritis. Bone Jt. Res. 9, 130–138. https://doi.org/10.1302/2046-3758.93.Bjr-2019-0137.R1 (2020).
Wu, C. et al. Transcriptome-wide association study identifies susceptibility genes for rheumatoid arthritis. Arthritis Res. Ther. 23. https://doi.org/10.1186/s13075-021-02419-9 (2021).
Sanderson, E. et al. Mendelian randomization. Nat. Rev. Methods Primers 2. https://doi.org/10.1038/s43586-021-00092-5 (2022).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487. https://doi.org/10.1038/ng.3538 (2016).
Wang, Q. et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature 597, 527–532. https://doi.org/10.1038/s41586-021-03855-y (2021).
Lei, M. The MCM complex: Its role in DNA replication and implications for cancer therapy. Curr. Cancer Drug Targets 5, 365–380. https://doi.org/10.2174/1568009054629654 (2005).
Yamazoe, T., Nagai, T., Umeda, S., Sugaya, Y. & Mizuno, K. Roles of TOG and jelly-roll domains of centrosomal protein CEP104 in its functions in cilium elongation and hedgehog signaling. J. Biol. Chem. 295, 14723–14736. https://doi.org/10.1074/jbc.RA120.013334 (2020).
Legal, T. et al. CEP104/FAP256 and associated cap complex maintain stability of the ciliary tip. J. Cell. Biol. 222. https://doi.org/10.1083/jcb.202301129 (2023).
Frikstad, K. M. et al. A CEP104-CSPP1 complex is required for formation of primary cilia competent in hedgehog signaling. Cell Rep. 28, 1907-1922.e1906. https://doi.org/10.1016/j.celrep.2019.07.025 (2019).
Moore, B. S. et al. Cilia have high cAMP levels that are inhibited by Sonic hedgehog-regulated calcium dynamics. Proc. Natl. Acad. Sci. U S A 113, 13069–13074. https://doi.org/10.1073/pnas.1602393113 (2016).
Yamashiro, T., Kuge, H., Zhang, J. & Honke, K. Calcineurin mediates the angiotensin II-induced aldosterone synthesis in the adrenal glands by up-regulation of transcription of the CYP11B2 gene. J. Biochem. 148, 115–123. https://doi.org/10.1093/jb/mvq037 (2010).
Gomes, D. C. et al. Sonic hedgehog signaling is active in human adrenal cortex development and deregulated in adrenocortical tumors. J. Clin. Endocrinol. Metab. 99, E1209–1216. https://doi.org/10.1210/jc.2013-4098 (2014).
Al-Jassar, C. et al. The ciliopathy-associated Cep104 protein interacts with tubulin and Nek1 kinase. Structure 25, 146–156. https://doi.org/10.1016/j.str.2016.11.014 (2017).
Rezabkova, L., Kraatz, S. H., Akhmanova, A., Steinmetz, M. O. & Kammerer, R. A. Biophysical and structural characterization of the Centriolar protein Cep104 Interaction Network. J. Biol. Chem. 291, 18496–18504. https://doi.org/10.1074/jbc.M116.739771 (2016).
Waelter, S. et al. The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum. Mol. Genet. 10, 1807–1817. https://doi.org/10.1093/hmg/10.17.1807 (2001).
Metzler, M. et al. Disruption of the endocytic protein HIP1 results in neurological deficits and decreased AMPA receptor trafficking. Embo J. 22, 3254–3266. https://doi.org/10.1093/emboj/cdg334 (2003).
Bollag, W. B. Regulation of aldosterone synthesis and secretion. Compr. Physiol. 4, 1017–1055. https://doi.org/10.1002/cphy.c130037 (2014).
Inoue, K. et al. Inhibition of endocytosis of clathrin-mediated angiotensin II receptor type 1 in Podocytes augments glomerular Injury. J. Am. Soc. Nephrol. 30, 2307–2320. https://doi.org/10.1681/asn.2019010053 (2019).
Buenaventura, T. et al. A targeted RNAi screen identifies endocytic trafficking factors that control GLP-1 receptor signaling in pancreatic β-cells. Diabetes 67, 385–399. https://doi.org/10.2337/db17-0639 (2018).
Chuang, P. T., Kawcak, T. & McMahon, A. P. Feedback control of mammalian hedgehog signaling by the hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev. 17, 342–347. https://doi.org/10.1101/gad.1026303 (2003).
Li, X. & Heyer, W. D. Homologous recombination in DNA repair and DNA damage tolerance. Cell. Res. 18, 99–113. https://doi.org/10.1038/cr.2008.1 (2008).
Piwko, W. et al. The MMS22L-TONSL heterodimer directly promotes RAD51-dependent recombination upon replication stress. Embo J. 35, 2584–2601. https://doi.org/10.15252/embj.201593132 (2016).
Huang, T. H. et al. The histone chaperones ASF1 and CAF-1 promote MMS22L-TONSL-Mediated Rad51 loading onto ssDNA during homologous recombination in human cells. Mol. Cell. 69, 879–892e875. https://doi.org/10.1016/j.molcel.2018.01.031 (2018).
Yu, B. et al. Overexpression of TONSL might be an independent unfavorable prognostic indicator in hepatocellular carcinoma. Pathol. Res. Pract. 215, 939–945. https://doi.org/10.1016/j.prp.2019.01.044 (2019).
Khatpe, A. S. et al. TONSL is an immortalizing oncogene and a therapeutic target in breast cancer. Cancer Res. 83, 1345–1360. https://doi.org/10.1158/0008-5472.Can-22-3667 (2023).
Lee, H. et al. Oncogenic impact of TONSL, a homologous recombination repair protein at the replication fork, in Cancer Stem cells. Int. J. Mol. Sci. 24. https://doi.org/10.3390/ijms24119530 (2023).
Kamaliyan, Z. & Clarke, T. L. Zinc finger proteins: Guardians of genome stability. Front. Cell. Dev. Biol. 12, 1448789. https://doi.org/10.3389/fcell.2024.1448789 (2024).
Zuo, Z. THE1B may have no role in human pregnancy due to ZNF430-mediated silencing. Mob. DNA. 14, 6. https://doi.org/10.1186/s13100-023-00294-6 (2023).
Huang, S. et al. Neutrophil lncRNA ZNF100-6:2 is a potential diagnostic marker for active pulmonary tuberculosis. Eur. J. Med. Res. 29, 162. https://doi.org/10.1186/s40001-024-01755-1 (2024).
Li, F. & Xue, X. Identification and characterization of an ageing-associated 13-lncRNA signature that predicts prognosis and immunotherapy in hepatocellular carcinoma. J. Oncol. 2023, 4615297. https://doi.org/10.1155/2023/4615297 (2023).
Yang, S. et al. The prognostic value of an autophagy-related lncRNA signature in hepatocellular carcinoma. BMC Bioinform. 22, 217. https://doi.org/10.1186/s12859-021-04123-6 (2021).
Xu, Z. et al. Construction of a ferroptosis-related Nine-lncRNA signature for predicting prognosis and immune response in hepatocellular carcinoma. Front. Immunol. 12, 719175. https://doi.org/10.3389/fimmu.2021.719175 (2021).
Li, Y. R., Fu, M., Song, Y. Q., Li, S. L. & Ge, X. Y. Long non-coding RNA MRPL23-AS1 suppresses anoikis in salivary adenoid cystic carcinoma in vitro. Oral Dis. 29, 1588–1601. https://doi.org/10.1111/odi.14156 (2023).
Chen, C. W. et al. Long noncoding RNA MRPL23-AS1 promotes adenoid cystic carcinoma lung metastasis. Cancer Res. 80, 2273–2285. https://doi.org/10.1158/0008-5472.Can-19-0819 (2020).
Zhang, H., Liu, S., Tang, L., Ge, J. & Lu, X. Long non-coding RNA (LncRNA) MRPL23-AS1 promotes tumor progression and carcinogenesis in osteosarcoma by activating Wnt/β-catenin signaling via inhibiting microRNA miR-30b and upregulating myosin heavy chain 9 (MYH9). Bioengineered 12, 162–171. https://doi.org/10.1080/21655979.2020.1863014 (2021).
Zhang, C., Gu, H., Liu, D., Fang, J. & Yang, Y. The role of MRPL23 antisense RNA 1 (MRPL23-AS1) in the pre-metastatic microenvironment of malignancy during the process of epithelial-mesenchymal transition. J. Biomaterials Tissue Eng. 11, 864–871. https://doi.org/10.1166/jbt.2021.2638 (2021).
Tan, X. et al. Comprehensive analysis of lncRNA-miRNA-mRNA regulatory networks for microbiota-mediated colorectal cancer associated with immune cell infiltration. Bioengineered 12, 3410–3425. https://doi.org/10.1080/21655979.2021.1940614 (2021).
Newman, A. C. & Maddocks, O. D. K. One-carbon metabolism in cancer. Br. J. Cancer 116, 1499–1504. https://doi.org/10.1038/bjc.2017.118 (2017).
Giardina, G. et al. The catalytic activity of serine hydroxymethyltransferase is essential for de novo nuclear dTMP synthesis in lung cancer cells. Febs J. 285, 3238–3253. https://doi.org/10.1111/febs.14610 (2018).
Monti, M. et al. Modelling of SHMT1 riboregulation predicts dynamic changes of serine and glycine levels across cellular compartments. Comput. Struct. Biotechnol. J. 19, 3034–3041. https://doi.org/10.1016/j.csbj.2021.05.019 (2021).
Fan, J. et al. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510, 298–302. https://doi.org/10.1038/nature13236 (2014).
Lewis, C. A. et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell. 55, 253–263. https://doi.org/10.1016/j.molcel.2014.05.008 (2014).
Lionaki, E., Ploumi, C. & Tavernarakis, N. One-carbon metabolism: Pulling the strings behind aging and neurodegeneration. Cells 11. https://doi.org/10.3390/cells11020214 (2022).
Gong, S. et al. Primary aldosteronism: Spatial multiomics mapping of genotype-dependent heterogeneity and tumor expansion of aldosterone-producing adenomas. Hypertension 80, 1555–1567. https://doi.org/10.1161/hypertensionaha.123.20921 (2023).
Yang, Y. et al. HOXD8 suppresses renal cell carcinoma growth by upregulating SHMT1 expression. Cancer Sci. 114, 4583–4595. https://doi.org/10.1111/cas.15982 (2023).
Jain, M. et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–1044. https://doi.org/10.1126/science.1218595 (2012).
Dou, C. et al. SHMT1 inhibits the metastasis of HCC by repressing NOX1-mediated ROS production. J. Exp. Clin. Cancer Res. 38, 70. https://doi.org/10.1186/s13046-019-1067-5 (2019).
Gupta, R., Yang, Q., Dogra, S. K. & Wajapeyee, N. Serine hydroxymethyl transferase 1 stimulates pro-oncogenic cytokine expression through sialic acid to promote ovarian cancer tumor growth and progression. Oncogene 36, 4014–4024. https://doi.org/10.1038/onc.2017.37 (2017).
Paone, A. et al. SHMT1 knockdown induces apoptosis in lung cancer cells by causing uracil misincorporation. Cell. Death Dis. 5, e1525. https://doi.org/10.1038/cddis.2014.482 (2014).
Noh, S., Kim, D. H., Jung, W. H. & Koo, J. S. Expression levels of serine/glycine metabolism-related proteins in triple negative breast cancer tissues. Tumour Biol. 35, 4457–4468. https://doi.org/10.1007/s13277-013-1588-z (2014).
Adjei, A. A. Pemetrexed (Alimta): A novel multitargeted antifolate agent. Expert Rev. Anticancer Ther. 3, 145–156. https://doi.org/10.1586/14737140.3.2.145 (2003).
Adjei, A. A. Pharmacology and mechanism of action of pemetrexed. Clin. Lung Cancer. 5(Suppl 2), 51–55. https://doi.org/10.3816/clc.2004.s.003 (2004).
Daidone, F. et al. In silico and in vitro validation of serine hydroxymethyltransferase as a chemotherapeutic target of the antifolate drug pemetrexed. Eur. J. Med. Chem. 46, 1616–1621. https://doi.org/10.1016/j.ejmech.2011.02.009 (2011).
Grogan, L., Sotos, G. A. & Allegra, C. J. Leucovorin modulation of fluorouracil. Oncology 7, 63–72 (1993). Discussion 75–66.
Wang, S. et al. Leucovorin enhances the anti-cancer effect of Bortezomib in colorectal cancer cells. Sci. Rep. 7, 682. https://doi.org/10.1038/s41598-017-00839-9 (2017).
Fujita, K., Kubota, Y., Ishida, H. & Sasaki, Y. Irinotecan, a key chemotherapeutic drug for metastatic colorectal cancer. World J. Gastroenterol. 21, 12234–12248. https://doi.org/10.3748/wjg.v21.i43.12234 (2015).
de Man, F. M., Goey, A. K. L., van Schaik, R. H. N., Mathijssen, R. H. J. & Bins, S. Individualization of irinotecan treatment: A review of pharmacokinetics, pharmacodynamics, and pharmacogenetics. Clin. Pharmacokinet. 57, 1229–1254. https://doi.org/10.1007/s40262-018-0644-7 (2018).
Park, S. Y. et al. Mimosine arrests the cell cycle prior to the onset of DNA replication by preventing the binding of human Ctf4/And-1 to chromatin via Hif-1α activation in HeLa cells. Cell. Cycle 11, 761–766. https://doi.org/10.4161/cc.11.4.19209 (2012).
Nguyen, B. C. & Tawata, S. The chemistry and biological activities of mimosine: A review. Phytother. Res. 30, 1230–1242. https://doi.org/10.1002/ptr.5636 (2016).
Tavares Murta, B. M., Cunha Fde, Q., Miranda, R., Adad, S. J. & Murta, E. F. Differential tumor microenvironment in human ovarian cystic tumors. Tumori 90, 491–497. https://doi.org/10.1177/030089160409000509 (2004).
Toki, M. I., Kumar, D., Ahmed, F. S., Rimm, D. L. & Xu, M. L. Benign lymph node microenvironment is associated with response to immunotherapy. Precis Clin. Med. 3, 44–53. https://doi.org/10.1093/pcmedi/pbaa003 (2020).
Mattaini, K. R., Sullivan, M. R. & Vander Heiden, M. G. The importance of serine metabolism in cancer. J. Cell. Biol. 214, 249–257. https://doi.org/10.1083/jcb.201604085 (2016).
Sánchez-Castillo, A. & Kampen, K. R. Understanding serine and glycine metabolism in cancer: A path towards precision medicine to improve patient’s outcomes. Discov Oncol. 15, 652. https://doi.org/10.1007/s12672-024-01544-6 (2024).
Yu, Y. et al. Roles of reactive oxygen species in inflammation and cancer. MedComm (2020) 5, e519. https://doi.org/10.1002/mco2.519 (2024).
Lee, H. M. et al. Concurrent loss of LKB1 and KEAP1 enhances SHMT-mediated antioxidant defence in KRAS-mutant lung cancer. Nat. Metab. 6, 1310–1328. https://doi.org/10.1038/s42255-024-01066-z (2024).
Lee, W. D. et al. Tumor Reliance on Cytosolic versus mitochondrial one-Carbon Flux depends on Folate availability. Cell. Metab. 33, 190–198.e196. https://doi.org/10.1016/j.cmet.2020.12.002 (2021).
MacFarlane, A. J. et al. Cytoplasmic serine hydroxymethyltransferase regulates the metabolic partitioning of methylenetetrahydrofolate but is not essential in mice. J. Biol. Chem. 283, 25846–25853. https://doi.org/10.1074/jbc.M802671200 (2008).
Ducker, G. S. et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. U S A. 114, 11404–11409. https://doi.org/10.1073/pnas.1706617114 (2017).
García-Cañaveras, J. C. et al. SHMT inhibition is effective and synergizes with methotrexate in T-cell acute lymphoblastic leukemia. Leukemia 35, 377–388. https://doi.org/10.1038/s41375-020-0845-6 (2021).
Pandey, S. et al. Nucleotide biosynthesis arrest by silencing SHMT1 function via vitamin B6-coupled vector and effects on tumor growth inhibition. Biomaterials 35, 9332–9342. https://doi.org/10.1016/j.biomaterials.2014.07.045 (2014).
McCarthy, M. I. et al. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 9, 356–369. https://doi.org/10.1038/nrg2344 (2008).
Lu, H. et al. Evaluating marginal genetic correlation of associated loci for complex diseases and traits between European and east Asian populations. Hum. Genet. 140, 1285–1297. https://doi.org/10.1007/s00439-021-02299-8 (2021).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518. https://doi.org/10.1038/s41586-022-05473-8 (2023).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252. https://doi.org/10.1038/ng.3506 (2016).
Xu, J. et al. A cross-tissue transcriptome-wide Association Study identifies Novel susceptibility genes for juvenile idiopathic arthritis in Asia and Europe. Front. Immunol. 13. https://doi.org/10.3389/fimmu.2022.941398 (2022).
Zheng, J. et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet. 52, 1122–1131. https://doi.org/10.1038/s41588-020-0682-6 (2020).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 622, 329–338. https://doi.org/10.1038/s41586-023-06592-6 (2023).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 53, 1712–1721. https://doi.org/10.1038/s41588-021-00978-w (2021).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Szklarczyk, D. et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646. https://doi.org/10.1093/nar/gkac1000 (2023).
Otasek, D., Morris, J. H., Bouças, J., Pico, A. R. & Demchak, B. Cytoscape automation: Empowering workflow-based network analysis. Genome Biol. 20, 185. https://doi.org/10.1186/s13059-019-1758-4 (2019).
Warde-Farley, D. et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38, W214–220. https://doi.org/10.1093/nar/gkq537 (2010).
Forli, S. et al. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 11, 905–919. https://doi.org/10.1038/nprot.2016.051 (2016).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 9, 671–675. https://doi.org/10.1038/nmeth.2089 (2012).
Varghese, F., Bukhari, A. B., Malhotra, R. & De, A. I. H. C. Profiler: An open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS ONE 9, e96801. https://doi.org/10.1371/journal.pone.0096801 (2014).
Xie, P. et al. The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat. Commun. 5, 3733. https://doi.org/10.1038/ncomms4733 (2014).
Acknowledgements
The authors thank the participants and investigators for providing publicly available summary statistics.
Funding
This work was supported by The National Key R&D Program of China [2021YFC2501603 and 2021YFC2501600], Zhejiang Provincial Medical and Health Technology Project [2020380946, 2022KY172, and 2024KY1024], Science and technology of Zhejiang Province Project [LGF21H070003], Zhejiang Province Traditional Chinese Medicine Science and Technology Project [2024ZL565]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
MJ conceptualized the study, analyzed and interpreted all data, and wrote the manuscript. LL contributed to data collection, conducted the immunohistochemistry and western blot experiments, and performed bioinformatics analyses. HY was involved in the transcriptome-wide association studies (TWAS) and the Mendelian randomization (MR) analyses. ZD conducted the phenome-wide association studies (PheWAS), enrichment analysis, and protein-protein interaction (PPI) network construction, as well as collected aldosterone-producing adenoma (APA) samples. XP contributed to literature searches and APA sample collection. XS conceptualized the study and revised the manuscript. All authors reviewed the manuscript and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All procedures involving human tissue samples were approved by the Institutional Review Board (IRB) of the Second Affiliated Hospital of Zhejiang University School of Medicine, and written informed consent was obtained from all participants. The study was conducted in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jia, M., Lin, L., Yu, H. et al. Integrative bioinformatics approach identifies novel drug targets for hyperaldosteronism, with a focus on SHMT1 as a promising therapeutic candidate. Sci Rep 15, 1690 (2025). https://doi.org/10.1038/s41598-025-85900-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-85900-8
Keywords
This article is cited by
-
Challenges and opportunities of wearable molecular sensors in endocrinology and metabolism
Nature Reviews Endocrinology (2025)
-
Comprehensive genome-wide analysis of retinal vessel caliber reveals microvascular-blood pressure pathways: advancing predictive, preventive, and personalized medicine
EPMA Journal (2025)
