
Abstract
Ferroptosis pathway activation is potentially correlated with temporal lobe epilepsy (TLE). However, the diagnostic significance and mechanism of ferroptosis-related genes (FRGs) in TLE require further investigation. A comprehensive analysis of the GSE134697 dataset from the Gene Expression Omnibus (GEO) database using Weighted gene co-expression network analysis (WGCNA) identified 3,212 differentially expressed genes (DEGs) between temporal lobe epilepsy (TLE) and control groups, with a critical focus on the turquoise module. Through intersection of DEGs and key module genes, correlation analyses with functional-related genes (FRG), protein-protein interactions (PPI), least absolute shrinkage and selection operator (LASSO), and machine learning methods, five potential biomarkers of ferroptosis (CBS, SHMT1, RIN3, QDPR, and PLPP4) were isolated. A nomogram was constructed using these markers, and enrichment analyses revealed their links to T-cell activation, allograft rejection, and glial differentiation. Variations in 13 immune cell types were also noted. Upregulation of CBS, RIN3, QDPR, and PLPP4 in TLE was confirmed through RT-qPCR and Western blot assays. Additionally, five SHMT1-targeting and one CBS-targeting drugs were predicted using the Drug-Gene Interaction Database (DGIdb). These findings provide new insights into the potential pathogenesis of TLE and suggest new targets for future research.
Similar content being viewed by others
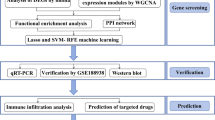

Introduction
Epilepsy is a frequently encountered neurological disorder in the field of clinical medicine1. According to a World Health Organization report, approximately 70 million people worldwide have been diagnosed with epilepsy2. At least 50% of people with epilepsy also have a mental health condition or cognitive dysfunction, which seriously affects their quality of life3. Temporal lobe epilepsy (TLE) is the predominant type of epilepsy and frequently develops into drug-resistant epilepsy4. There are two main syndromes of TLE: mesial temporal lobe epilepsy (mTLE) and neocortical temporal lobe epilepsy (nTLE)5,6. Among them, mTLE mainly originates from lesions in the hippocampus and amygdala7,8. mTLE is the most prevalent epilepsy syndrome in adults. Despite optimal medical management, over 30% of individuals with mTLE continue to experience drug-resistant seizures, indicating an inability to achieve effective seizure control with current anti-seizure medications9,10,11. Recent basic bioinformatics analyses have provided new insights into TLE12,13. However, the exact pathogenesis and effective biomarkers of mTLE remain unclear. Therefore, further studies are needed to enable the early diagnosis and prompt treatment of mTLE, which is imperative to improve the condition and mitigate complications.
In 2012, Dixon et al. first described ferroptosis as a type of cell death distinct from apoptosis, and its characteristic features include an increase in iron ions within cells, depletion of glutathione, and accumulation of lipid reactive oxygen species (ROS)14. Ferroptosis is intricately associated with a spectrum of neurological disorders, including traumatic brain injury, epilepsy, stroke, and Parkinson’s disease15,16,17,18,19,20.Inhibiting ferroptosis can prevent the death of hippocampal neurons and reduce cellular oxidative damage, improving cognitive dysfunction in an epileptic mouse model21. It can be inferred that the activation of the ferroptosis pathway is potentially related to epilepsy and the death of hippocampal neurons. Notably, research on ferroptosis induced by aberrant iron metabolism and distribution has highlighted hippocampal neuronal death as a potential cause of the onset and progression of mTLE with cognitive defect22. In studies investigating the pathological mechanisms of the anti-aging gene Klotho in relation to TLE, it was found that Klotho can mitigate iron overload, reduce reactive oxygen species (ROS) levels, and inhibit hippocampal cognitive defect and neuronal ferroptosis, thereby improving cognitive function in LiCl-pilocarpine-induced TLE rats23. Moreover, iron-dependent lipid peroxidation is a critical mechanism underlying ferroptosis. Lipid accumulation during TLE-associated pathological processes can lead to elevated lipid peroxidation levels in neurons and astrocytes, further indicating a strong association between mTLE and ferroptosis4. These studies suggest that ferroptosis may play a significant role in the development of mTLE, and activation of the ferroptosis pathway may offer novel perspectives for early diagnosis and therapeutic improvement of mTLE. Nevertheless, the diagnostic significance of ferroptosis-related genes (FRGs) in mTLE and their mechanism of action require further investigation.
Given the important role of ferroptosis in the development of mTLE4,24,25, we thoroughly examined how FRGs affect the progression of neuronal cell death, providing a reference for understanding mTLE pathogenesis and diagnosing this disease. We explored the diagnostic value of ferroptosis-related diagnostic markers in mTLE and the mechanism of action of biomarkers, providing new treatment targets to prevent mTLE progression.
Results
Screening of differentially expressed genes (DEGs) and key module genes
This study was based on the GSE134697 dataset obtained from the public database Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) and analyzed fresh hippocampal tissue samples from 17 temporal lobe epilepsy (TLE) patients and fresh neocortical tissue samples from 17 TLE patients. Among these, the 17 hippocampal samples were considered as TLE samples, while the 17 neocortical samples were used as control samples. There were 3,212 differentially expressed genes (DEGs) between the TLE and control samples (Fig. 1a). A heatmap was generated to show the expression of DEGs between the TLE and control samples (Fig. 1b). There were no outlier samples in this study (Fig. 1c). When the soft threshold was 38, the network was closest to the distribution without network scale (Fig. 1d). Three modules were obtained by the hybrid dynamic cut tree algorithm (Fig. 1e). The turquoise gene module (4,843 genes) was selected as the key gene module (R2 = 0.66, p value = 2e-05) (Fig. 1f).

Screening of DEGs and key module genes. (a) Volcano plot of differential gene analysis. Red dots represent upregulated genes, blue dots represent downregulated genes, and black dots represent genes with no significant difference or smallfold changes. (b) The expression heatmap of differential gene analysis, blue for low expression, red for high expression. (c) Hierarchical clustering analysis (d) Selection of the soft threshold power value. (d-1) The left panel represents the scale-free model fit index. (d-2) The right panel represents the mean connectivity of these values (e-1) Clustering of module eigengenes. (e-2) Cluster dendrogram of genes enriched based on dissimilarity measure and assignment modules. (f) Heatmap of the correlation between module genes and TLE.
Protein interactions and enrichment analysis of candidate genes
To further identify genes related to ferroptosis, this study initially identified 1,735 intersecting genes from 3,212 DEGs and 4,843 key module genes (Fig. 2a). Then, 1,106 FRGs were identified by Pearson correlation analysis (Supplementary Tables S1–S2). There were 437 candidate genes with interaction scores > 0.9 (Fig. 2b). Gene Ontology (GO) functional enrichment analysis revealed that the candidate genes were associated with gliogenesis, leukocyte activation involved in the immune response, and positive regulation of cell adhesion (Fig. 2c). Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analysis revealed that the candidate genes participated in platelet activation, the PI3K-Akt signaling pathway, and ether lipid metabolism (Fig. 2d).

PPI network and enrichment analysis of differential genes. (a) Venn diagram of turquoise and DEGs genes. (b) PPI network of ferroptosis-related intersection genes. (c) GO enrichment results of candidate genes. (d) KEGG enrichment results of candidate genes.
Identification of five biomarkers in candidate genes
To further analyze the association between the 437 candidate genes and TLE, five key genes were identified through 10-fold cross-validation using LASSO. These genes, with regression coefficients that were not penalized to zero at lambda.min = 0.05, include cystathionine-beta-synthase (CBS), serine hydroxymethyltransferase 1 (SHMT1), rab interactor 3 (RIN3), quinoid dihydropteridine reductase (QDPR), and phospholipid phosphatase 4 (PLPP4) (Fig. 3a). The extreme-gradient boosting (XGB) algorithm, with the lowest residual value and the best evaluation effect, revealed that the five key genes can be used as biomarkers, and the PLPP4 was the most important (Fig. 3b and d). The area under the raw current curves (AUCs) of five biomarkers (CBS, SHMT1, RIN3, QDPR and PLPP4) were all greater than 0.8 (Fig. 3e), indicating that these five biomarkers had a favorable ability to diagnose TLE. This suggests that there may be potential regulatory mechanisms and functional pathways between these biomarkers and TLE.

Identification of five biomarkers. (a-1, 2) LASSO regression coefficient graph and cross-validation graph based on penalty term. (b, c) Residual distribution plots for RF, SVM, GLM, and XGB machine learning. (d) RF, SVM, GLM, and XGB machine learning algorithms to assess genetic significance. (e) ROC curves for biomarkers.
Gene set enrichment analysis (GSEA) of biomarkers
According to the GO functional enrichment analysis, the high expression group of CBS and RIN3 was associated with alpha beta T cell activation, gliogenesis and glial cell differentiation. High expression group of SHMT1, QDPR and PLPP4 was associated with glial cell differentiation, leukocyte proliferation and T cell activation (Supplementary Fig. S1a–e). According to the KEGG functional enrichment analysis, the highly expressed biomarkers (SHMT1, QDPR, PLPP4, CBS and RIN3) were mainly involved in allograft rejection, cell adhesion molecules cams and graft versus host disease (Fig. 4a and e). The similarity scores of RIN3, QDPR and PLPP4 were less than 0.5, and those of CBS and SHMT1 were between 0.5 and 0.6 (Fig. 4f). This indicates that the CBS and SHMT1 genes have similar protein functions, which may help in further understanding the relationship between biomarkers and TLE, as well as their impact on the immune microenvironment in subsequent analyses.

KEGG enrichment analysis of biomarkers. (a) KEGG enrichment analysis of CBS. (b) KEGG enrichment analysis of PLPP4. (c) KEGG enrichment analysis of QDPR. (d) KEGG enrichment analysis of RIN3. (e) KEGG enrichment analysis of SHMT1 The figure shows the top 10 enriched pathways. (f) Semantic similarity scores between biomarker GO terms.
Nomogram and the correlation analysis of biomarkers and immune cells
A nomogram for survival prediction in TLE patients was constructed using CBS, SHMT1, RIN3, QDPR and PLPP4 (Fig. 5a). Calibration, decision, and clinical impact curves were plotted for the above nomogram, indicating that the predictive ability of the model was excellent (Fig. 5b and d). In addition, 13 immune cell types (CD8 + T cells, DCs, pDCs, eosinophils, aDCs, T cells, Tems, macrophages, neutrophils, B cells, natural killer (NK) cells, Th17 cells, and Th2 cells) differed significantly between the TLE and control groups, revealing that the immune microenvironment plays a key role in TLE (Fig. 5e). Our analysis further revealed that the biomarkers positively correlated with most differentially abundant immune cells; for example, neutrophils were positively correlated with QDPR, and CD8 T cells were positively associated with RIN3 (Fig. 5f). The correlations between each biomarker and immune cells are shown in Supplementary Fig. S2. The AUCs of the differentially abundant immune cells (aDCs, macrophages, pDC, neutrophils, CD8 + T cells, DCs, B cells, eosinophils, T cells, Tem, NK cells, Th17 cells and Th2 cells) were greater than 0.69, indicating that cells had a favorable ability to diagnose TLE samples (Fig. 5g). These results suggest that immune cells play a key role in TLE, while also revealing a significant correlation between biomarkers and differences in immune cell presence. This implies that biomarkers may influence the immune microenvironment by regulating immune cells, providing new insights and potential drug targets for the effective control of TLE seizures in the future.

Nomogram and correlation analysis of immune cells of biomarkers. (a) Nomogram for 5 biomarkers. (b) Nomogram calibration curves When S: p > 0.05 indicates that the calibration test is passed; C(ROC) is the ROC area. (c) Nomogram decision curves. (d) Nomogram clinical impact curves. (e) Box plots of the percentage of immune cells in the TLE and normal groups. (f) Correlation network of biomarkers and immune cells. (g) ROC curves of differential immune cells.
Upstream regulatory network and the drug prediction of biomarkers
Four upstream transcription factor biomarkers obtained from the TRRUST database are myelocytomatosis oncogene (MYC), nuclear factor YA (NFYA), specific protein 1 (SP1), and specific protein 3 (SP3). CBS was found to be regulated by NFYA, SP1, and SP3; moreover, SHMT1 was regulated by MYC. In addition, PLPP4 was regulated by 10 multiple microRNAs (miRNAs) such as hsa-miR-205-3p and hsa-miR-4502, and we also found that QDPR was regulated by 10 miRNAs including hsa-miR-449c-5p and hsa-miR-6885-3p (Fig. 6a). A total of six target drugs were obtained, comprising five (fluorouracil, leucovorin, methotrexate, bevacizumab, and irinotecan) for SHMT1 and one target drug (methionine) for CBS. These targeted medications may contribute to the subsequent management of TLE (Fig. 6b, Supplementary Table S3). The results of the molecular docking simulation of potential therapeutic drugs are showed in Fig. 6c and Supplementary Fig. S3. The molecular docking of SHMT1 with the fluorouracil and leucovorin strongly affected the compound-target interactions, providing a potential research direction for the future development of medications targeting TLE.

Upstream regulatory network and the drug prediction of biomarkers. (a) Diagram of the top 10 biomarker miRNA upstream regulation model based on the TF-miRNA regulatory network. (b) Target drugs for SHMT1 and CBS biomarkers. (c-1) Predicted results of molecular docking of the SHMT1 gene with the drug FLUOROURACIL (1 hydrogen bond, binding energy − 5.2 Kcal/mol). (c-2) Predicted results of molecular docking of the SHMT1 gene with the drug LEUCOVORIN (no hydrogen bonding, binding energy − 7.8 Kcal/mol).
Real-time quantitative polymerase chain reaction (RT-qPCR) identification
The RT‒qPCR verification results indicated significant upregulation of CBS, RIN3, QDPR, and PLPP4 expression in the TLE group, which was consistent with our above results (p < 0.05) (Fig. 7, Supplementary Table S4). SHMT1 expression was upregulated in the TLE group, although the difference was not significant (p > 0.05).

Identification of RT-qPCR. (a) RT-qPCR identification results of PLPP4. (b) RT-qPCR identification results of RIN3. (c) RT-qPCR identification results of SHMT1. (d) RT-qPCR identification results of QDPR. (e) RT-qPCR identification results of CBS. The error bars represent the standard deviation (SD), and the overlaid data points in the figure indicate the degree of data dispersion. We used the t-test and Mann-Whitney U test. **: p < 0.01, ***: p < 0.001.
Western blot for quantification of differentially expressed proteins
Western blot analysis revealed a robust and statistically significant upregulation of CBS, SHMT1,RIN3, QDPR, and PLPP4 protein expression in the TLE group, corroborating the above findings (Fig. 8, Supplementary Table S5). The consistency between the RT-qPCR and Western blot analysis results indicates the reliability of this study, providing a valuable reference for research into new therapeutic targets for TLE and the development of medications based on biomarkers (CBS, SHMT1, RIN3, QDPR, and PLPP4).

Identification of Western blot. (a) Western blot results of RIN3, CBS, SHMT1, PLPP4, and QDPR in rats hippocampus. Uncropped images are presented in Supplementary Fig. S4. (b) Western blot identification results of RIN3. (c) Western blot identification results of CBS. (d) Western blot identification results of SHMT1. (e) Western blot identification results of PLPP4. (f) Western blot identification results of QDPR. The error bars represent the standard deviation (SD), and the overlaid data points in the figure indicate the degree of data dispersion. We used the t-test and Mann-Whitney U test. *: p < 0.05, **: p < 0.01, ***: p < 0.001.
Discussion
Diagnosing and treating TLE has long been a major focus of neurological research26. Researchers have recently found that the pathogenesis of TLE overlaps with the occurrence of ferroptosis in neurons27. The accumulation of ROS exacerbates the neuronal loss caused by epileptic seizures in TLE28. Oxidative stress is one of the main pathophysiological causes of TLE29. Moreover, oxidative stress, increased ROS, and iron overload lead to severe lipid peroxidation, a hallmark of ferroptosis27. However, the connection between TLE and ferroptosis has not been extensively explored in previous studies. Therefore, exploring diagnostic biomarkers of ferroptosis in TLE can help clarify the mechanisms underlying this disease and identify potential therapeutic targets.
In the central nervous system, CBS catalyzes the production of hydrogen sulfide and is expressed in neurons; changes in CBS expression affect neuronal growth, differentiation, and function30,31. CBS activation of CBS in the brain increases the production of hydrogen sulfide during epileptic seizures in newborn pigs32. CBS has a significant effect on regulating neuronal death regulation and is also associated with ferroptosis, which is involved in iron homeostasis regulation33,34. SHMT1 is folic acid–dependent enzyme expressed in the hippocampus, and the changes in its expression affect neuronal and cognitive function in adult mice35. The expression level of S-adenosylmethionine, which is synthesized by serine hydroxymethyltransferase, is increased in some epilepsy patients and in the brain tissue of animals36,37. When S-adenosylmethionine is highly expressed, lipid peroxidation and ferroptosis occur gradually in cells38. RIN3 is a gene that is expressed in neuronal cells and a reliable risk gene for neurological diseases39. An increase in RIN3 expression may modify the axonal transport of neurons, control neuronal signal transduction, and increase the level of the phosphorylated Tau protein level, potentially contributing to the development of neurodegenerative disorders39. Some scholars have shown that RIN3 is associated with neuronal cell death, metabolic dysfunction, and inflammatory processes in neurological diseases40. The close association between RIN3 and initial cognitive impairment in neurological disorders has been verified41. The enzyme QDPR plays a crucial role in energy metabolism and is important in the cellular respiratory chain42. This genetic sequence is responsible for producing a crucial protein involved in the growth and function of neurons, including regulating connections between neurons and affecting the response of neurons to electrical signals43. One study revealed that high QDPR expression promotes seizures in rats44. Some researchers have reported that the activity and expression level of QDPR may serve as a crucial indicator for severity of TLE and predicting seizures45. Additionally, abnormal QDPR activity can affect the balance of iron ions in the cell and induce oxidative stress, thereby interfering with ferroptosis46. Moreover, PLPP4 also referred to as phosphatidic acid phosphatase type 2 domain containing 1 A (PPAPDC1A), can degrade phospholipid molecules on the cell membrane47. PLPP4 plays a vital role in facilitating the transfer of signals inside cells, dynamic changes in membrane structure, and intracellular material transport48. PLPP4 is highly expressed in the brain, especially in the hippocampus, substantia nigra, and other brain regions, and is a driving factor in neurological diseases49,50. Studies have suggested that increased PLPP4 activity and expression may indicate the onset of neurological disease48. Previous studies have not elucidated the genes dependent on ferroptosis in TLE neurons. In this study, random forest (RF), support vector machine (SVM), and XGB were used to screen for TLE-related biomarkers, and five biomarkers were ultimately identified (CBS, SHMT1,RIN3, QDPR, and PLPP4). The gene and protein expression levels were confirmed in animal tissue samples. In addition, the AUCs of the receiver operating characteristic (ROC) curves of CBS (AUC: 0.9689), SHMT1 (AUC: 0.9689), RIN3 (AUC: 0.9654), QDPR (AUC: 0.8789) and PLPP4 (AUC: 1) indicated that they had a strong diagnostic effect. These findings indicate the potential of these biomarkers as a novel focus for identifying TLE, and targeted therapy against these genes could effectively improve TLE symptoms.
Interestingly, GO functional enrichment analysis revealed that elevated levels of CBS and RIN3 were associated with T-cell activation, gliogenesis, and differentiation, and high expression of SHMT1, QDPR, and PLPP4 was involved in glial cell differentiation, leukocyte proliferation, and T-cell activation. Moreover, KEGG functional enrichment analysis revealed that the five significantly differentially expressed biomarkers were primarily involved in allograft rejection, cell adhesion molecules, and graft-versus-host disease. In addition, the model constructed using the five biomarkers obtained from the above analysis was validated using the rms package. The model demonstrated excellent predictive ability, as indicated by the calibration, decision, and clinical impact curves.
Hydrogen sulfide produced mainly by CBS is believed to be an endogenous signaling gasotransmitter in mammals that regulates the production and function of diverse immune cells (e.g., neutrophils, macrophages, DCs, NK cells, eosinophils, T cells, and B cells)51. In an inflammatory state, the depletion of SHMT1 leads to a reduction in the production of IL-10 by B cells52. In addition, SHMT1 can regulate progression through natural killer cells53. In recent years, many scholars have discovered that RIN3 is involved in impacting immune responses in the pathogenesis of neurological diseases54. QDPR deficiency and PLPP4 downregulation are also inevitably related to the immune response55,56. Unfortunately, the relationship between biomarkers of ferroptosis associated with temporal lobe epilepsy and immune infiltration remains unclear. Our investigation revealed notable differences in 13 immune cell types (aDCs, B cells, CD8 + T cells, DCs, eosinophils, macrophages, neutrophils, NK cells, pDCs, T cells, T cells, Th17 cells, and Th2 cells). CBS, SHMT1, RIN3, QDPR, and PLPP4 are associated with these immune cells. An increasing number of researchers have shown that various immune cells infiltrate the hippocampus of TLE patients and that immune response induction is a pathogenic mechanism of TLE57. Hence, we believe that the immune microenvironment is a significant factor in epilepsy and contributes to its development.
Additionally, our results revealed that NFYA, SP1, and SP3 are involved in regulating CBS; MYC regulates SHMT1; hsa-miR-205-3p and hsa-miR-4502, among others, regulate PLPP4; and hsa-miR-449c-5p and hsa-miR-6885-3p, among others, regulate QDPR. Many studies have shown that NFYA, SP1, and SP3 are transcription factors (TFs) for CBS58. They exert a significant influence on the regulation of cell growth, differentiation, and gene expression33,59. MYC is crucial for regulating cell cycle progression and managing cellular proliferation. MYC activates or inhibits gene expression by binding to DNA, promoting cell proliferation and growth. MYC plays a role in regulating SHMT1, which may be an important disease treatment target60. In many diseases, miRNAs target PLPP4 and QDPR, regulating gene expression and controlling cell growth, differentiation, apoptosis, and other biological processes61,62. Previous TLE studies did not clearly identify pathways associated with ferroptosis-related diagnostic biomarkers. We hypothesize that these upstream transcription factor biomarkers control cellular biological processes by regulating the expression levels of CBS, SHMT1,RIN3, QDPR, and PLPP4 in cells, thereby achieving the effect of improving TLE. This provides a new perspective for the future new treatment methods of TLE from the level of gene regulation.
Moreover, we used the Drug–Gene Interaction Database (DGIdb) database to predict drug candidates for biomarkers and analyzed their interactions. Fortunately, we identified six potential drugs for TLE treatment: 5-fluorouracil, leucovorin, methotrexate, bevacizumab, irinotecan, and methionine. Some scholars have also confirmed that fluorouracil, leucovorin, methotrexate, bevacizumab and irinotecan target the SHMT1 gene63,64,65,66, and that methionine targets CBS. CBS is a key regulatory factor in cysteine metabolism that transfers homocysteine from the methionine cycle for cysteine biosynthesis through the reverse sulfation pathway67,68. Therefore, the above six compounds have the potential as novel targeted therapy to inhibit the progression of the disease in patients with TLE.
In this study, we innovatively discovered biomarkers related to ferroptosis in TLE, and identified correlations between these biomarkers and specific immune cells. Moreover, we newly explored the upstream regulatory networks of these biomarkers and predicted biomarker-targeting drugs. However, our study has some limitations. Although we have used RT‒qPCR and Western blot assays to authenticate the expression of candidate genes in TLE rat samples, future studies are required to further refine the expression and localization of these genes using immunohistochemistry or immunofluorescence assays. In addition, future research endeavors will systematically investigate the molecular mechanisms underlying the biomarkers identified in this study. We will investigate the mechanisms by which biomarkers influence the pathophysiology of TLE via genetic modification or regulation of gene expression. We will also combine the upstream regulatory network of biomarkers to explore the potential pathways for biomarkers to participate in and regulate the pathogenesis of TLE. We hope to effectively translate the research achievements of these biomarkers into clinical applications, providing new perspectives and research foundations for new treatment strategies of TLE.
Conclusion
Our investigation revealed that the differentially expressed FRGs (CBS, SHMT1, RIN3, QDPR, and PLPP4) in TLE have high diagnostic value and could serve as new biological markers for early TLE diagnosis, offering fresh insights into its pathogenesis and treatment.
Materials and methods
Date sources
The RNA sequencing (RNA‒seq) dataset GSE134697 of hippocampus and neocortex samples from 17 patients with TLE was used in this study; the 17 hippocampal samples were used as the disease (TLE) samples, and the 17 neocortical samples were used as the controls (controls). Moreover, 79 FRGs were identified through the intersection of FRGs in the FerrDb database (http://www.zhounan.org/ferrdb/current/), FRGs in the reported literature, and FRGs in the BioGPS database (http://biogps.org/#goto=welcome).
Analysis of differential expression and weighted gene coexpression network analysis (WGCNA)
Using the limma (v 3.48.3) package69, DEGs between TLE and control samples were identified using the thresholds of |log2fold change (FC)| > 1 and an adjusted p-value (i.e., False Discovery Rate (FDR) p-value) < 0.05. Next, ggplot2 (v 3.3.5)70 was used to generate volcano plots, and heatmaps were created using the pheatmap (v 1.0.12) package. In addition, the TLE and control samples from GSE134697 were used for the studied traits, and a coexpression network was created using the WGCNA (v 1.70-3) package71 to identify the key module genes. Based on the clinical information in the GSE134697 dataset, TLE was used as the trait, and sample clustering was performed to remove samples that deviated from the normal range to ensure the accuracy of the analysis. Subsequently, to construct a co-expression network with a scale-free topology, the scale-free network evaluation coefficient (R²) was set to 0.85, and the optimal soft threshold (β) was selected to ensure that the co-expression network closely approximated a scale-free distribution. A scale-free network was constructed based on the β value, and the hybrid dynamic tree-cutting algorithm was applied, with the minimum number of genes in each gene module set to 200, resulting in co-expression modules. Finally, the correlation between TLE and each module was calculated (P < 0.05, |correlation (cor)| > 0.3), and the module with the highest correlation to TLE was selected as the key module, with genes in this module defined as key module genes.
Screening and enrichment analysis of candidate genes
DEGs between the TLE and control samples and key module genes were crossed to acquire intersecting genes. Then, the Pearson correlation coefficient between FRGs and intersecting genes was calculated (|cor| >0.9 and p < 0.01). Next, based on the intersecting genes, a protein‒protein interaction (PPI) network was constructed with the STRING online database (http://stringdb.org), and strongly interacting genes with interaction scores > 0.9 were selected as candidate genes. In addition, to further explore some interrelated signaling pathways and functional items of the candidate genes, GO and KEGG72,73,74 enrichment analyses were performed with the clusterProfiler (v 4.0.2) package75 (adjusted p-value < 0.05 and count > 1).
Screening of biomarkers
We used the least absolute shrinkage and selection operator (LASSO) regression algorithm to analyze the candidate genes according to their expression levels using the glmnet (v 4.1-4) package76. Moreover, to further assess the value of key genes in TLE, four machine learning methods, namely, RF, SVM, GLM, and XGB, were used to calculate the importance of key genes. Then, four models were analyzed using the DALEX (v 2.4.0) package to construct the residual distribution map and obtain the best model. In addition, to study the ability of biomarkers to distinguish the TLE and control groups, the biomarkers were verified in the GSE134697 dataset. The pROC (v 1.18.0) package77 was used to plot ROC curves for the biomarkers.
GSEA
We divided the samples into high- and low-expression groups according to the median value of biomarker expression and then performed GSEA on all genes in the high- and low-expression groups (|NES|>1, NOM P < 0.05 and q < 0.25) using the clusterProfiler (v 4.0.2)75 and org.Hs.eg.db (v 3.13.0) packages. In addition, we used the GOSemSim (v 2.18.1) package78 to calculate the semantic similarity between biomarkers, and the geometric mean of the semantic similarity of biomarkers was used to determine the similarity of protein functions.
Construction of a nomogram and immune microenvironment analysis
A nomogram of the obtained biomarkers was constructed using the rms (v 6.1-0) package to determine the relationship between biomarker expression and TLE. Then, calibration, decision, and clinical impact curves were drawn to evaluate the precision of the prediction. In addition, the ssGSEA algorithm was used to calculate the rank value of each gene from the expression profile for subsequent statistical analysis. Using 24 immune cell sets, we used the ssGSEA algorithm to calculate the percentage of infiltrating immune cells in each sample. The Wilcoxon test was used to analyze the difference in the infiltration of immune cells between the TLE and control groups (p < 0.05). Moreover, correlations between biomarkers and differential immune cells were determined by Spearman correlation analysis. Furthermore, to verify the ability of differential immune cells to distinguish the TLE and control groups, we plotted the receiver operating characteristic (ROC) curve of differential immune cells using the GSE134697 dataset.
Construction of upstream regulatory network and the drug prediction
Biomarker TFs were predicted using the TRRUST database (https://www.grnpedia.org/trrust/). Then, the miRNAs corresponding to biomarkers were obtained through the miRWalk (http://mirwalk.umm.uni-heidelberg.de/) and miRDB (https://mirdb.org/) databases, and the top 10 miRNAs with the highest accessibility predicted in the miRWalk and miRDB databases were selected as the upstream miRNAs of the biomarkers. The upstream regulatory network was constructed using the TFs and miRNAs. In addition, we predicted biomarkers through the DGIdb (https://www.dgidb.org/) and analyzed the interactions between the predicted drugs and biomarkers to identify potential drugs for treating TLE. In addition, the three-dimensional structures of the candidate drugs were imported into the PubChem (https://pubchem.ncbi.nlm.nih.gov/) database. The biomarkers were imported into the universal protein resource Universal Protein (UniProt) database (https://www.uniprot.org/) to obtain receptor structures with the highest resolution. Molecular docking simulations of potential therapeutic drugs were performed using the AutoDock (https://autodock.scripps.edu/) online database.
Ethics declarations
In the study, all rat experiments were conducted in accordance with the ARRIVE guidelines, U.K. Animals (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments, and the National Research Council’s Guide for the Care and Use of Laboratory Animals. Approval was granted by the Animal Care Welfare Committee of GuiZhou Medical University, China (Approval No 2200896, Supplementary Fig. S5).
Rat model of TLE
Twenty-five male Sprague Dawley rats (weight: 180–220 g) aged 8 weeks were provided by the Experimental Animal Center of Guizhou Medical University. Fifteen male SD rats, all of whom were healthy adults, were included in this study. The rats were fasted for 10 h and intraperitoneally injected with 127 mg/kg lithium chloride. The rats were then intraperitoneally injected with 10 mg/kg pilocarpine 18–20 h later. Rats with three or more seizures of grade IV or higher in succession, according to the Racine method, were observed. Long-term video monitoring for 30 days was used to determine whether spontaneous recurrent seizures were considered successful induction of kindling in rats79,80. Ultimately, TLE was successfully induced in 10 rats.
RT-qPCR
The rats were deeply anesthetized with 1% pentobarbital sodium (50 mg/kg) via intraperitoneal injection, followed by rapid skull separation, hippocampal extraction on ice, and blood cleaning. Ten pairs of rat hippocampal tissue samples were divided into two groups: 10 normal samples and 10 TLE samples. Then, total RNA was isolated from the samples and purified with 1 mL of TRIzol (Ambion) reagent following the manufacturer’s instructions. Then, the concentration of the extracted RNA was tested with a NanoPhotometer N50. Next, reverse transcription was performed via the SureScript First Strand cDNA Synthesis Kit (Servicebio) with an ordinary PCR instrument. The reverse transcription product cDNA was diluted 15 times with ddH2O (RNase/DNase free). Subsequently, PCR amplification was performed on a CFX96 fluorescent PCR instrument. The reaction proceeded for 1 min at 95 °C (predenaturation), followed by 95 °C for 20 s (denaturation), 55 °C for 20 s (annealing), and 72 °C for 30 s (elongation). The above reactions were conducted for 40 cycles. The primer sequences are shown in Supplementary Table S6. RT‒qPCR was performed to verify the expression levels of the significantly different hub genes in the TLE patients’ validation set.
Western blot
Western blot analysis was used to compare the protein expression levels of CBS, SHMT1,RIN3, QDPR, and PLPP4 in the TLE hippocampal tissue samples with those in the normal samples. Total cell protein was extracted on ice using RIPA buffer (Beijing Solarbio Technology Co., Ltd.) and PMSF (Wuhan Servicebio Technology Co., Ltd.). The protein was transferred to polyvinylidene fluoride membrane by wet transfer method after SDS-PAGE gel electrophoresis. Then, seal the membrane with 5% skimmed milk. The membrane was incubated with the first antibodies, such as primary β-actin antibody (1:1000; Wuhan Servicebio Technology Co., Ltd.), CBS antibody (1:1000; Wuhan Servicebio Technology Co., Ltd.), SHMT1 antibody (1:1000; Wuhan Servicebio Technology Co., Ltd.),RIN3 antibody (1:1000; Wuhan Sanying Biotechnology Co., Ltd.), QDPR antibody (1:1000; Wuhan Sanying Biotechnology Co., Ltd.), and PLPP4 antibody (1:1000; ThermoFisher), at 4℃ overnight. After washing, goat anti-rabbit antibody (1:5000; Wuhan Servicebio Technology Co., Ltd.) was incubated at room temperature for two hours. After washing again, the protein bands were visualized using enhanced chemiluminescence reagent (Dalian Meilunbio Biotechnology Co., Ltd.). Finally, the relative protein expression levels of CBS, SHMT1, RIN3, QDPR, and PLPP4 were calculated using β-actin as an internal control.
Statistical analysis
Statistical analyses were conducted using R (version 4.2.2), SPSS 26.0 software and GraphPad Prism 8.0 software. A significance level of p < 0.05 was considered to indicate statistical significance.
Data availability
The datasets analysed during the current study are available in the GSE134697 dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE134697), FerrDb database (http://www.zhounan.org/ferrdb/current/), and BioGPS database (http://biogps.org/#goto=welcome).
References
Thijs, R. D., Surges, R., O’Brien, T. J. & Sander, J. W. Epilepsy in adults. Lancet 393, 689–701. https://doi.org/10.1016/S0140-6736(18)32596-0 (2019).
Karabacak, M., Jagtiani, P., Jain, A., Panov, F. & Margetis, K. Tracing topics and trends indrug-resistant epilepsy research using a natural languageprocessing–based topic modeling approach. Epilepsia 65, 861–872. https://doi.org/10.1111/epi.17890 (2024).
McDonald, C. R. et al. Development and application of the International classification of Cognitive disorders in Epilepsy (IC-CoDE): Initial results from a Multi-center study of adults with temporal lobe Epilepsy. Neuropsychology 37, 301–314. https://doi.org/10.1037/neu0000792 (2023).
Su, Y., Cao, N., Zhang, D. & Wang, M. The effect of ferroptosis-related mitochondrial dysfunction in the development of temporal lobe epilepsy. Ageing Res. Rev. 96, 102248. https://doi.org/10.1016/j.arr.2024.102248 (2024).
Pascual, M. R. Temporal lobe epilepsy: Clinical semiology and neurophysiological studies. Semin. Ultrasound CT MRI 28, 416–423. https://doi.org/10.1053/j.sult.2007.09.004 (2007).
Zhu, H. Y. et al. Metabolic profiles and correlation with surgical outcomes in mesial versus neocortical temporal lobe epilepsy. CNS Neurosci. Ther. 29, 2656–2665. https://doi.org/10.1111/cns.14209 (2023).
Modarres, M. S. et al. Clinical correlation of altered molecular signatures in epileptic human hippocampus and amygdala. Mol. Neurobiol. 61, 725–752. https://doi.org/10.1007/s12035-023-03583-6 (2024).
Mivalt, F. et al. Impedance rhythms in human limbic system. J. Neurosci. 43, 6653–6666. https://doi.org/10.1523/JNEUROSCI.0241-23.2023 (2023).
Asadi-Pooya, A. A., Brigo, F., Lattanzi, S. & Blumcke, I. Adult epilepsy. Lancet 402, 412–424. https://doi.org/10.1016/S0140-6736(23)01048-6 (2023).
Perucca, E., Perucca, P., White, H. S. & Wirrell, E. C. Drug resistance in epilepsy. Lancet Neurol. 22, 723–734. https://doi.org/10.1016/S1474-4422(23)00151-5 (2023).
Kjaer, C. et al. Differential expression of the beta3 subunit of voltage-gated Ca2+ channel in mesial temporal lobe epilepsy. Mol. Neurobiol. 60, 5755–5769. https://doi.org/10.1007/s12035-023-03426-4 (2023).
Wang, W. et al. Identification of hub genes significantly linked to temporal lobe epilepsy and apoptosis via bioinformatics analysis. Front. Mol. Neurosci. 17, 1300348. https://doi.org/10.3389/fnmol.2024.1300348 (2024).
Zhang, L. M. et al. Identification of key potassium channel genes of temporal lobe epilepsy by bioinformatics analyses and experimental verification. Front. Neurol. 14, 1175007. https://doi.org/10.3389/fneur.2023.1175007 (2023).
Dixon, S. J. et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. https://doi.org/10.1016/j.cell.2012.03.042 (2012).
Chen, Y. et al. Mapping the Research of Ferroptosis in Parkinson’s Disease from 2013 to 2023: A scientometric review. Drug Des. Dev. Ther. 18, 1053–1081. https://doi.org/10.2147/DDDT.S458026 (2024).
Thal, D. R., Gawor, K. & Moonen, S. Regulated cell death and its role in Alzheimer’s disease and amyotrophic lateral sclerosis. Acta Neuropathol. 147, 69. https://doi.org/10.1007/s00401-024-02722-0 (2024).
Huang, L. Y. et al. Remote ischemic postconditioning-mediated neuroprotection against stroke by promoting Ketone Body-Induced ferroptosis inhibition. ACS Chem. Neurosci. 15, 2223–2232. https://doi.org/10.1021/acschemneuro.4c00014 (2024).
Lv, B. et al. DMT1 ubiquitination by Nedd4 protects against ferroptosis after intracerebral hemorrhage. CNS Neurosci. Ther. 30, e14685. https://doi.org/10.1111/cns.14685 (2024).
Song, Y. et al. Mitochondrial ferritin alleviates ferroptosis in a kainic acid-induced mouse epilepsy model by regulating iron homeostasis: Involvement of nuclear factor erythroid 2‐related factor 2. CNS Neurosci. Ther. 30, e14663. https://doi.org/10.1111/cns.14663 (2024).
Xu, Y., Jia, B., Li, J., Li, Q. & Luo, C. The interplay between ferroptosis and neuroinflammation in central neurological disorders. Antioxidants 13. https://doi.org/10.3390/antiox13040395 (2024).
Xie, R. et al. Quercetin alleviates kainic acid-induced seizure by inhibiting the Nrf2-mediated ferroptosis pathway. Free Radic. Biol. Med. 191, 212–226. https://doi.org/10.1016/j.freeradbiomed.2022.09.001 (2022).
Ye, Q. et al. Inhibition of ferroptosis processes ameliorates cognitive impairment in kainic acid-induced temporal lobe epilepsy in rats. Am. J. Transl. Res. 11, 875–884 (2019).
Xiang, T. et al. Klotho ameliorated cognitive deficits in a temporal lobe epilepsy rat model by inhibiting ferroptosis. Brain Res. 1772, 147668. https://doi.org/10.1016/j.brainres.2021.147668 (2021).
Ye, Q., Zeng, C., Luo, C. & Wu, Y. Ferrostatin-1 mitigates cognitive impairment of epileptic rats by inhibiting P38 MAPK activation. Epilepsy Behav. 103, 106670. https://doi.org/10.1016/j.yebeh.2019.106670 (2020).
Wang, F. et al. The clock gene regulates kainic acid-induced seizures through inhibiting ferroptosis in mice. J. Pharm. Pharmacol. 74, 1640–1650. https://doi.org/10.1093/jpp/rgac042 (2022).
Wagstyl, K. et al. WONOEP 2022: Neurotechnology for the diagnosis of epilepsy. Epilepsia 65, 2238–2247. https://doi.org/10.1111/epi.18028 (2024).
Yi, T. T., Zhang, L. M. & Huang, X. N. Glycyrrhizic acid protects against temporal lobe epilepsy in young rats by regulating neuronal ferroptosis through themiR -194‐5p/PTGS2 axis. Kaohsiung J. Med. Sci. 39, 154–165. https://doi.org/10.1002/kjm2.12642 (2023).
Kishore, M. et al. Regulation of Keap1-Nrf2 axis in temporal lobe epilepsy—hippocampal sclerosis patients may limit the seizure outcomes. Neurol. Sci. 44, 4441–4450. https://doi.org/10.1007/s10072-023-06936-0 (2023).
Yue, J. et al. Negative effects of brain regulatory T cells depletion on epilepsy. Prog. Neurobiol. 217, 102335. https://doi.org/10.1016/j.pneurobio.2022.102335 (2022).
Fujimoto, S. et al. Hydrogen sulfide potently promotes neuronal differentiation of adipose tissue-derived stem cells involving nitric oxide-mediated signaling cascade with the aid of cAMP-elevating agents. Nitric Oxide Biol. Chem. 127, 10–17. https://doi.org/10.1016/j.niox.2022.07.003(2022).
Yuan, M. et al. Vitamin B(6) alleviates chronic sleep deprivation-induced hippocampal ferroptosis through CBS/GSH/GPX4 pathway. Biomed. Pharmacother. 174, 116547. https://doi.org/10.1016/j.biopha.2024.116547 (2024).
Liu, J., Pourcyrous, M., Fedinec, A. L. & Parfenova, H. Cerebroprotective actions of hydrogen sulfide in the epileptic brain in newborn pigs. Pediatr. Res. 94(495). https://doi.org/10.1038/s41390-023-02486-5 (2023).
Park, J. M. et al. A case-control study in Taiwanese cohort and meta-analysis of serum ferritin in pancreatic cancer. Sci. Rep. 11. https://doi.org/10.1038/s41598-021-00650-7 (2021).
Zhou, P. et al. A novel link between circPDE3B and ferroptosis in esophageal squamous cell carcinoma progression. Genomics 116, 110761. https://doi.org/10.1016/j.ygeno.2023.110761 (2024).
Abarinov, E. V. et al. Disruption of Shmt1 impairs hippocampal neurogenesis and mnemonic function in Mice1–3. J. Nutr. 143, 1028–1035. https://doi.org/10.3945/jn.113.174417 (2013).
Staufner, C. et al. Adenosine kinase deficiency: Expanding the clinical spectrum and evaluating therapeutic options. J. Inherit. Metab. Dis. 39, 273–283. https://doi.org/10.1007/s10545-015-9904-y (2016).
Farrar, C. E. & Clarke, S. Diet-dependent survival of protein repair-deficient mice. J. Nutr. Biochem. 16, 554–561. https://doi.org/10.1016/j.jnutbio.2005.02.004 (2005).
Homma, T., Kobayashi, S. & Fujii, J. Methionine deprivation reveals the pivotal roles of cell cycle progression in ferroptosis that is Induced by cysteine starvation. Cells 11, 1603. https://doi.org/10.3390/cells11101603 (2022).
Shen, R. et al. Upregulation of RIN3 induces endosomal dysfunction in Alzheimer’s disease. Transl. Neurodegener. 9. https://doi.org/10.1186/s40035-020-00206-1 (2020).
Pathak, G. A. et al. Genome-wide methylation of mild cognitive impairment in Mexican americans highlights genes involved in synaptic transport, Alzheimer’s disease-precursor phenotypes, and metabolic morbidities. J. Alzheimers Dis. 72, 733–749. https://doi.org/10.3233/JAD-190634 (2019).
Miao, M. et al. Methylation of the RIN3 promoter is Associated with transient ischemic Stroke/Mild ischemic stroke with early cognitive impairment. Neuropsychiatr. Dis. Treat. 17, 2587–2598. https://doi.org/10.2147/NDT.S320167 (2021).
Miyajima, K. et al. Perturbation of monoamine metabolism and enhanced fear responses in mice defective in the regeneration of tetrahydrobiopterin. J. Neurochem. 161, 129–145. https://doi.org/10.1111/jnc.15600 (2022).
Girish, A. et al. Comprehensive bioinformatics analysis of structural and functional consequences of deleterious missense mutations in the human QDPR gene. J. Biomol. Struct. Dyn. 42, 5485–5501. https://doi.org/10.1080/07391102.2023.2226740 (2023).
Damasceno, S. et al. Transcriptome of the Wistar audiogenic rat (WAR) strain following audiogenic seizures. Epilepsy Res. 147, 22–31. https://doi.org/10.1016/j.eplepsyres.2018.08.010 (2018).
Ray, S. et al. Disorders of Tetrahydrobiopterin Metabolism: experience from South India. Metab. Brain Dis. 37, 743–760. https://doi.org/10.1007/s11011-021-00889-z (2022).
Wang, H. et al. Protective effect of Phellinus linteus polysaccharide extracts against thioacetamide-induced liver fibrosis in rats: A proteomics analysis. Chin. Med. 7(23). https://doi.org/10.1186/1749-8546-7-23 (2012).
Zhang, X. et al. Phospholipid phosphatase 4 promotes proliferation and tumorigenesis, and activates Ca2+-permeable Cationic Channel in lung carcinoma cells. Mol. Cancer 16. https://doi.org/10.1186/s12943-017-0717-5 (2017).
Tian, W. et al. Phospholipid phosphatase 4 as a driver of malignant glioma and pancreatic adenocarcinoma. Front. Oncol. 11, 790676. https://doi.org/10.3389/fonc.2021.790676 (2021).
Sequeros, C. B. et al. A genome-wide association study of social trust in 33,882 Danish blood donors. Sci. Rep. 14. https://doi.org/10.1038/s41598-024-51636-0 (2024).
Brolin, K. et al. Insights on genetic and environmental factors in Parkinson’s disease from a regional Swedish case-control cohort. J. Parkinsons Dis. 12, 153–171. https://doi.org/10.3233/JPD-212818 (2022).
Dilek, N., Papapetropoulos, A., Toliver-Kinsky, T. & Szabo, C. Hydrogen sulfide: An endogenous regulator of the immune system. Pharmacol. Res. 161, 105119. https://doi.org/10.1016/j.phrs.2020.105119 (2020).
Fu, Y. et al. Oxidative stress-initiated one-carbon metabolism drives the generation of interleukin-10-producing B cells to resolve pneumonia. Cell. Mol. Immunol. 21, 19–32. https://doi.org/10.1038/s41423-023-01109-7 (2024).
Yang, Q. et al. MiR-218-5p suppresses the killing effect of natural killer cell to lung adenocarcinoma by targeting SHMT1. Yonsei Med. J. 60, 500–508. https://doi.org/10.3349/ymj.2019.60.6.500 (2019).
Tesi, N. et al. Cognitively healthy centenarians are genetically protected against Alzheimer’s disease. Alzheimers Dement. 20, 3864–3875. https://doi.org/10.1002/alz.13810 (2024).
Liu, J. et al. QDPR deficiency drives immune suppression in pancreatic cancer. Cell. Metab. 36, 984–999. https://doi.org/10.1016/j.cmet.2024.03.015 (2024).
Tan, M., Schaffalitzky, D. M. O. & Joergensen, M. T. Gene expression network analysis of precursor lesions in familial pancreatic cancer. J. Pancreat. Cancer 6, 73–84. https://doi.org/10.1089/pancan.2020.0007 (2020).
Yang, X. et al. Cuproptosis-related genes signature and validation of differential expression and the potential targeting drugs in temporal lobe epilepsy. Front. Pharmacol. 14, 1033859. https://doi.org/10.3389/fphar.2023.1033859 (2023).
Ge, Y., Jensen, T. L., Matherly, L. H. & Taub, J. W. Synergistic regulation of human cystathionine-beta-synthase-1b promoter by transcription factors NF-YA isoforms and Sp1. Biochim. Biophys. Acta. 1579, 73–80. https://doi.org/10.1016/s0167-4781(02)00509-2 (2002).
Ge, Y., Jensen, T. L., Matherly, L. H. & Taub, J. W. Transcriptional regulation of the cystathionine-beta -synthase gene in Down syndrome and non-down syndrome megakaryocytic leukemia cell lines. Blood 101, 1551–1557. https://doi.org/10.1182/blood-2002-07-2337 (2003).
Situ, Y. et al. SHMT as a potential therapeutic target for renal cell carcinoma. Front. Biosci. 28, 196. https://doi.org/10.31083/j.fbl2809196 (2023).
Guo, X. et al. MiR-598-5p inhibits breast cancer tumor growth and lung metastasis by targeting PPAPDC1A. Chin. J. Physiol. 66, 103. https://doi.org/10.4103/cjop.CJOP-D-22-00089 (2023).
Xie, X., Zhang, G. & Liu, N. Comprehensive analysis of abnormal methylation modification differential expression mRNAs between low-grade and high-grade intervertebral disc degeneration and its correlation with immune cells. Ann. Med. 56, 2357742. https://doi.org/10.1080/07853890.2024.2357742 (2024).
Zhao, M. et al. Drosha-independent miR-6778–5p strengthens gastric cancer stem cell stemness via regulation of cytosolic one-carbon folate metabolism. Cancer Lett. 478, 8–21 ; https://doi.org/10.1016/j.canlet.2020.02.040(2020).
Chen, Y. et al. Gancao Nourishing-Yin decoction combined with methotrexate in treatment of aging CIA mice: A study based on DIA proteomic analysis. Chin. Med. 18, 9 (2023).
Misselbeck, K., Marchetti, L., Priami, C., Stover, P. J. & Field, M. S. The 5-formyltetrahydrofolate futile cycle reduces pathway stochasticity in an extended hybrid-stochastic model of folate-mediated one-carbon metabolism. Sci. Rep. 9, 4322. https://doi.org/10.1038/s41598-019-40230-4 (2019).
Budai, B. et al. Impact of SHMT1 polymorphism on the clinical outcome of patients with metastatic colorectal cancer treated with first-line FOLFIRI + bevacizumab. Pharmacogenet. Genomics 22, 69–72. https://doi.org/10.1097/FPC.0b013e32834d8376 (2012).
Tu, Y. et al. Crystal structures of cystathionine β-synthase from Saccharomyces cerevisiae: One enzymatic step at a time. Biochemistry 57, 3134–3145. https://doi.org/10.1021/acs.biochem.8b00092 (2018).
Besen, S., Ozkale, Y., Ceylaner, S., Noyan, A. & Erol, I. Clinical and laboratory findings and etiologies of genetic homocystinemia: A single-center experience. Acta Neurol. Belg. 124, 213–222. https://doi.org/10.1007/s13760-023-02356-1 (2024).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Ito, K. & Murphy, D. Application of ggplot2 to pharmacometric graphics. CPT Pharmacomet Syst. Pharmacol. 2, e79. https://doi.org/10.1038/psp.2013.56 (2013).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. https://doi.org/10.1093/nar/gkac963( (2023).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141. https://doi.org/10.1016/j.xinn.2021.100141 (2021).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized Linear models via Coordinate Descent. J. Stat. Softw. 33, 1–22 (2010).
Robin, X. et al. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform. 12. https://doi.org/10.1186/1471-2105-12-77 (2011).
Yu, G. et al. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 26, 976–978. https://doi.org/10.1093/bioinformatics/btq064 (2010).
Tang, W. et al. The role of hippocampal neurogenesis in ANT-DBS for LiCl-Pilocarpine-Induced epileptic rats. Stereotact. Funct. Neurosurg. 99, 55–64. https://doi.org/10.1159/000509314 (2021).
Racine, R. J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294. https://doi.org/10.1016/0013-4694(72)90177-0 (1972).
Acknowledgements
We are grateful to the Center for Tissue Englneering and Stem Cell Reserch of of Guizhou Medical University for assistance in molecular biology analysis experiments. We are thankful to LetPub (www.letpub.com) for linguistic assistance during the preparation of this manuscript.
Funding
This study was funded by the Natural Science Foundation of China [82001380], Guizhou Provincial Science and Technology Projects Qiankehe platform talent general [2021] 5612 and General Fund Cultivation Program of National Natural Science Foundation of Guizhou Medical University Affiliated Hospital [gyfynsfc-2021-8].
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. The conception and design of the project were completed by nine experts, namely J.L., Z.N., G.W., L.W., S.R., W.G., H.Y., H.X., and D.S. J.L., Z.N., and D.S. collected, analyzed, and interpreted the data. J.L. and W.G. were responsible for the experiments in rats. The first draft of this manuscript was written by J.L., Z.N., and D.S. G.W., L.W., S.R., H.Y., and H.X. were responsible for investigation and guidance. All authors made revisions to the previous manuscript and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shi, D., Li, J., Niu, Z. et al. Identification and validation of diagnostic biomarkers for temporal lobe epilepsy related to ferroptosis and potential therapeutic targets. Sci Rep 15, 4908 (2025). https://doi.org/10.1038/s41598-025-89390-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-89390-6
Keywords
This article is cited by
-
The miR-34a-5p Promotes Hippocampal Neuronal Ferroptosis in Epilepsy by Regulating SIRT1
Neurochemical Research (2025)
-
SAT1 Knockdown Decreases Glutamate-Induced Oxidative Stress, Ferroptosis, and Apoptosis in HT22 Cells via Activating the Nrf2/ARE Pathway
NeuroMolecular Medicine (2025)
