
Abstract
Protein phosphatase PPM1B, a member of the serine/threonine phosphatase family, has been implicated in various human cancers. In this study, our objective was to investigate the role of PPM1B in GC growth and explore the underlying mechanisms. Our findings revealed that PPM1B expression was downregulated in GC tissues, and higher levels of PPM1B expression were associated with improved overall survival in GC patients. Overexpression of PPM1B significantly inhibited cell proliferation, induced G1 phase cell cycle arrest, and suppressed tumor growth. Conversely, knockdown or knockout of PPM1B yielded opposite effects. Mechanistically, we identified that PPM1B exerted its inhibitory role in GC cell growth and cell cycle regulation through the TRIM25/PPM1B/CDK2 signaling pathway. Specifically, we demonstrated that TRIM25 physically interacts with PPM1B, leading to enhanced degradation of PPM1B and subsequent modulation of CDK2 phosphorylation and GC cell growth. PPM1B emerges as a potential prognostic biomarker and therapeutic target in GC. These findings hold clinical significance by offering opportunities to improve diagnosis and treatment strategies for GC patients. Furthermore, this study provides novel insights into the pathogenesis and progression of GC, expanding our understanding of this disease.
Similar content being viewed by others


Introduction
GC is a significant global health burden, ranking fourth in terms of fatality rate and fifth in terms of incidence rate1. Despite notable advancements in its treatment, the rates of early detection, radical resection, and 5-year survival in GC patients remain inadequate2,3. Consequently, there is an urgent need to identify more effective biomarkers and therapeutic targets for accurate diagnosis and improved therapeutic strategies in GC.
The protein phosphatase 2 C (PP2C) family comprises a set of monomeric enzymes that serve as metal-dependent protein phosphatases, capable of binding to Mn2+ or Mg2 + 4. Within this family, all members share a conserved core structure known as the PPM phosphatase domain. Dysfunction of PPM phosphatase activity has been implicated in the development of various diseases, including tumorigenesis and metabolic disorders5.
PPM1B, also known as PP2C-β, belongs to the PP2C family and was initially cloned in 19986. Previous research has indicated that PPM1B exerts regulatory control over various histopathological processes through the dephosphorylation of downstream substrates. Notably, PPM1B-mediated dephosphorylation of P38 impacts the aging program7, dephosphorylation of PPAR-γ regulates metabolism8, and dephosphorylation of TBK1 inhibits antiviral responses9. Recent studies have established the significant involvement of PPM1B in the development of several malignancies, including osteosarcoma, bladder cancer, and kidney cancer10,11,12. Given the crucial role of PPM1B in these malignancies, it is reasonable to hypothesize that PPM1B may also play a vital role in the progression of GC.
Our study unveils the detailed mechanism by which PPM1B regulates cell cycle progression in GC. We discovered that PPM1B likely exerts its influence by dephosphorylating the key cell cycle regulator CDK2, thereby causing GC cells to accumulate in the G1 phase and significantly impacting their progression. Additionally, we identified for the first time that PPM1B is regulated by the E3 ubiquitin ligase TRIM25, leading to its degradation through the Lys48 (K48) ubiquitin linkage pathway and subsequent reduction in protein levels. This modulation affects the proliferative and migratory abilities of GC cells. Consequently, the promotion of cell proliferation and migration contributes to the progression of GC.
Results
Dysregulated PPM1B expression in GC and its prognostic implications
To investigate the expression of PPM1B in GC patients, we conducted IHC analysis on 100 GC tissues and matched adjacent normal tissues. Our findings revealed a significant downregulation of PPM1B protein expression in the GC tissues (Fig. 1A-B). To further validate these results, we assessed both protein and mRNA expression levels of PPM1B in GC patients, confirming the downregulation of PPM1B (Fig. 1C-D). Additionally, analysis of the GSE26899 dataset from the GEO database provided further support for the decreased expression of PPM1B in GC (Fig. 1E). These results collectively indicate a consistent downregulation of PPM1B in GC patients, highlighting its potential role in the disease.

Reduced PPM1B expression in GC is associated with poor prognosis. (A,B) Expression of PPM1B in GC and adjacent normal tissues was detected by IHC. Representative images and IHC scores are shown. (C) The mRNA expression of PPM1B in 100 GC patients. (D) Expression of PPM1B in GC and adjacent normal tissues was detected by WB. (E) PPM1B mRNA expression in 108 gastric tissue samples from the GEO dataset (GSE26899). (F) Prognostic value of PPM1B was assessed in the GEO cohort. (G) The clinical significance of PPM1B expression on overall survival in 100 GC patients was confirmed by Kaplan-Meier survival analysis in the central cohort of this study. All **P < 0.01 and ***P < 0.001.
The clinical-pathological analysis revealed a significant association between the expression of PPM1B and TNM staging (p = 0.008), lymph node metastasis (p = 0.035), degree of differentiation (p = 0.001), and tumor size (p = 0.015) (Table 1). Survival analysis of 100 GC patients showed that high PPM1B expression was associated with extended overall survival (p < 0.01) (Fig. 1G). Consistent with these findings, analysis of data from the GEO database demonstrated a significant correlation between upregulated PPM1B expression and prolonged survival in GC patients (p < 0.001) (Fig. 1F).
PPM1B functions as a potent suppressor of proliferation and migration in GC cells
Based on the aforementioned findings regarding the association between PPM1B and GC tumor characteristics, we aimed to further explore the functional impact of PPM1B on the behavior of GC cells. To investigate this, we established stable PPM1B-overexpressing cell lines in AGS and MGC-803 cells (Fig. 2A). Notably, the overexpression of PPM1B resulted in a significant inhibition of cell proliferation (Fig. 2B-C) and migration (Fig. 2D) in these cells. Conversely, in PPM1B knockdown cells (Fig. 2E), we observed a substantial promotion of cell proliferation (Fig. 2F-G) and migration (Fig. 2H). Furthermore, using the CRISPR-Cas9 system, we generated PPM1B knockout (PPM1B-KO) MGC-803 cells (Fig. 2I). Notably, compared to the mock control group, the PPM1B-KO group exhibited significantly increased cell proliferation and migration in GC cells (Fig. 2J-L). Collectively, these findings provide compelling evidence that PPM1B functions as a cancer suppressor, exerting inhibitory effects on the malignant behavior of GC cells.

PPM1B suppresses the proliferative and migratory capabilities of GC cells. (A) AGS and MGC-803 cells were stably transfected with Myc-PPM1B plasmid or mock control plasmid and screened with puromycin, and effective stable overexpression of PPM1B was verified by WB in the transfected cells. The impact of PPM1B overexpression on the proliferation (B-C) and migration (D) of GC cells was investigated and analyzed in stable PPM1B overexpressing cell lines. (E) Lentivirus-mediated knockdown of PPM1B in AGS and MGC-803 cells was confirmed by WB analysis, validating effective PPM1B suppression. Stable PPM1B knockdown cells were subsequently generated and utilized for further experimental analysis. The effect of PPM1B downregulation on the proliferation (F-G) and migration (H) of GC cells was examined and analyzed in stable PPM1B low-expressing cell lines. (I) PPM1B knockout MGC-803 cells were generated using CRISPR-Cas9, confirmed by WB analysis, and selected for stable PPM1B knockout clones. PPM1B-KO cells were further examined and analyzed for proliferation (J-K) and migration (L). All *P < 0.05,**P < 0.01, ****P < 0.001. Figures presented are representative of data from at least three independent experiments.
PPM1B-dependent dephosphorylation of CDK2 suppresses cell proliferation in GC
To investigate the mechanisms underlying the inhibitory effects of PPM1B on tumor growth, we performed flow cytometry analysis to assess cell cycle distribution. Overexpression of PPM1B in AGS and MGC-803 cells resulted in an accumulation of cells in the G1 phase (Fig. 3A), while knockdown or knockout of PPM1B led to a decrease in G1 phase cells (Fig. 3C, E). To further understand the molecular mechanisms involved, we examined the expression of G1/S phase-related proteins, including CDK2, CDK4, cyclin D1, and cyclin E1. Our results revealed that PPM1B overexpression reduced the phosphorylation and expression levels of CDK2, CDK4, cyclin D1, and cyclin E1 in AGS and MGC-803 cells (Fig. 3B). Conversely, knockdown or knockout of PPM1B had the opposite effect (Fig. 3D, F). These findings suggest that PPM1B exerts its inhibitory effects on cell proliferation and growth in GC through modulation of the CDK2-mediated signaling pathway.

PPM1B-mediated dephosphorylation of CDK2 inhibits cell proliferation. (A) Analysis of the cell cycle distribution of AGS and MGC-803 cells overexpressing PPM1B and mock control plasmids by flow cytometry. (B) The protein levels of overexpressed PPM1B GC cell cycle-related markers, such as p-CDK2, CDK2, CDK4, Cyclin D1, Cyclin E1, were detected by WB. (C, E) Flow cytometry analysis of the cell cycle distribution of AGS and MGC-803 cells with knockdown, knockout PPM1B and mock control plasmids. (D, F) The protein levels of cell cycle-related markers of knockdown and knockout PPM1B GC cells were detected by WB.
PPM1B interacts directly with TRIM25 protein to form a protein complex
To elucidate the underlying molecular mechanisms of PPM1B-mediated regulation in cancer cells, we employed Myc-tagged pulldown assays combined with liquid chromatography and tandem mass spectrometry (LC-MS/MS) in AGS cells to identify interacting proteins (Fig. 4A, B). Among the identified proteins, the E3 ubiquitin ligase TRIM25 drew our attention. To validate the interaction between PPM1B and TRIM25, we performed endogenous and exogenous co-immunoprecipitation (co-IP) assays. Our results demonstrated that both exogenous and endogenous PPM1B proteins interacted with TRIM25 (Fig. 4C, D). Furthermore, endogenous TRIM25 was found to bind with endogenous PPM1B (Fig. 4E, F), indicating their natural interaction. Additionally, we utilized an in vitro transcription and translation system to generate PPM1B protein, further confirming its direct interaction with TRIM25 protein (Fig. 4G). Confocal microscopy images of HEK293T, AGS, and MGC-803 cells provided visual evidence of the interaction between TRIM25 and PPM1B proteins (Fig. 4H).

PPM1B binds directly to TRIM25 protein. (A) Coomassie brilliant blue staining confirmed that PPM1B interacts with various proteins via Myc-tagged pulldown. (B) Combined with LC-MS/MS, it was found that TRIM25 could interact with PPM1B. (C) Co-transfection of Flag-TRIM25 plasmid and Myc-PPM1B plasmid in HEK 293T and AGS cells, Co-IP analysis of the binding between exogenous TRIM25 and exogenous PPM1B. (D) HEK 293T and AGS cells were transfected with Flag-TRIM25 plasmid or mock control plasmid, Co-IP analysis of the binding of exogenous TRIM25 and endogenous PPM1B in HEK293T and AGS cells. (E,F) Co-IP analysis of the binding of endogenous TRIM25 and endogenous PPM1B in AGS cells. (G) Co-IP analysis of direct binding between GST-PPM1B and Flag-TRIM25 proteins obtained from an in vitro transcription and translation system. (H) The position of TRIM25 and PPM1B in HEK 293T, AGS, MGC-803 cells observed by confocal fluorescence microscope. (I,J) Schematic representation of TRIM25 and PPM1B, showing wild-type and truncating mutants of TRIM25 and PPM1B. (K,L) HEK293T cells were co-transfected with Myc-PPM1B plasmid or Myc-PPM1B truncated mutant plasmid and Flag-TRIM25 plasmid, and the interaction between TRIM25 and PPM1B or PPM1B truncated mutant was analyzed by Co-IP. (M,N) HEK 293T cells were co-transfected with Flag-TRIM25 plasmid or Flag-TRIM25 truncated mutant plasmid and Myc-PPM1B plasmid, and the interaction between PPM1B and TRIM25 or TRIM25 truncated mutant was analyzed by Co-IP. (O) The Flag-TRIM25 truncated mutant plasmid (RING structure and CC domain) and PPM1B truncated mutant (phosphatase domain and C-terminal domain) were co-transfected into HEK 293T cells, and Co-IP analysis of the interaction between the TRIM25 truncating mutant plasmid and the PPM1B truncating mutant.
To investigate the molecular basis of the PPM1B-TRIM25 interaction, we utilized functional domain information obtained from NCBI and GeneCards database to generate schematic diagrams of the respective proteins (Fig. 4I-J). Our findings revealed that the PPM domain of PPM1B (Fig. 4K-L) and the RING domain with the CC domain of TRIM25 (Fig. 4M-N) are crucial for their interaction. Co-ip assays involving the PPM domain and C-terminal domain of PPM1B along with the RING domain and CC domain of TRIM25 demonstrated that the interaction between PPM1B and TRIM25 requires the PPM domain of PPM1B and the RING domain of TRIM25 (Fig. 4O).
TRIM25 facilitates the ubiquitin-mediated degradation of PPM1B
We investigated the regulatory role of TRIM25 in the content and stability of PPM1B protein, given its direct interaction with PPM1B. Transient transfection of TRIM25 expression plasmids in GC cells resulted in a significant decrease in PPM1B protein levels (Fig. 5A), indicating translational regulation by TRIM25. Blocking de novo protein synthesis with cycloheximide (CHX) in GC cells overexpressing TRIM25 led to a gradual reduction in PPM1B protein levels over time (Fig. 5B). Notably, treatment with the proteasome inhibitor MG132 rescued the negative regulation of PPM1B by TRIM25 (Fig. 5C), suggesting proteasomal-mediated degradation.

TRIM25 mediates the ubiquitinated degradation of PPM1B. (A) WB analysis of PPM1B protein levels in AGS and MGC-803 cells transiently transfected with Flag-TRIM25 plasmid or mock control plasmid. (B) AGS and MGC-803 cells were transient transfected and cultured with Flag-TRIM25 plasmid or simulated control plasmid for 24 h, and then treated with CHX for 3, 6, 9 h, respectively. WB was used to detect the protein level of PPM1B in the transfected cells. (C) AGS cells were transfected with Flag-TRIM25 plasmid or mock control plasmid and cultured for 24 h, then further incubated with MG132 (10 µM) for 4 h or with chloroquine (25 µM) for 6 h, WB analysis of PPM1B protein levels in transfected cells. (D) Ubiquitination of PPM1B in HEK293T cells co-transfected with Flag-TRIM25, HA-UB, and Myc- PPM1B plasmids by Co-IP analysis. (E) Co-ip analysis of ubiquitination of PPM1B in HEK293T cells co-transfected with Flag-TRIM25 plasmid, Myc-PPM1B plasmid and HA-UB-K48 or HA-UB-K63 plasmid. (F) Co-IP analysis of PPM1B ubiquitination in AGS cells co-transfected with Flag-TRIM25 plasmid or Flag-TRIM25 truncated mutant (RING domain) plasmid, Myc-PPM1B plasmid and HA- UB plasmid.
As TRIM25 is a RING-type E3 ubiquitin ligase13,14, we examined whether its E3 ligase activity is involved in the negative regulation of PPM1B. Our results revealed that TRIM25 can attach polyubiquitin chains to PPM1B protein (Fig. 5D), indicating direct degradation of PPM1B by TRIM25 through polyubiquitination. Furthermore, TRIM25 was found to link Lys48 (K48)-linked polyubiquitin chains to PPM1B protein (Fig. 5E), suggesting the induction of K48-linked polyubiquitination of PPM1B by TRIM25. Overexpression of TRIM25’s RING domain significantly increased PPM1B polyubiquitination compared to the control (Fig. 5F), further supporting TRIM25’s role in promoting PPM1B ubiquitination through its RING domain. Collectively, these findings demonstrate that TRIM25 directly interacts with PPM1B protein and induces Lys48-linked polyubiquitination of PPM1B via its RING domain.
Regulation of cell proliferation by PPM1B through the TRIM25/PPM1B/CDK2 signaling pathway
We conducted additional investigations to examine the impact of TRIM25 on CDK2 phosphorylation through the regulation of PPM1B. To accomplish this, we transfected TRIM25 and PPM1B plasmids into AGS cells. WB analysis revealed that the overexpression of TRIM25 resulted in an increase in CDK2 phosphorylation. However, when PPM1B levels were partially reduced, the effect of TRIM25 on CDK2 phosphorylation was attenuated (Fig. 6A). We confirmed a positive correlation between TRIM25 mRNA expression and CDK2 levels using the TCGA dataset. This strengthens the link between TRIM25 and CDK2 in our study (Fig. 6B). Moreover, functional studies involving cellular biology demonstrated that PPM1B partially inhibited the promoting effect of TRIM25 on GC cell proliferation (Fig. 6C, D). These findings highlight the intricate interplay between TRIM25, PPM1B, and CDK2, shedding light on their roles in GC progression.

Regulation of cell proliferation by PPM1B through the TRIM25/PPM1B/CDK2 signaling pathway. (A) TRIM25, PPM1B and p-CDK2 were analyzed by WB in AGS cells transfected with mock control plasmid, Flag-TRIM25, Myc-PPM1B or both Flag-TRIM25 and Myc-PPM1B. (B) The association between TRIM25 mRNA expression and the cell cycle-related protein CDK2 was validated using data from the TCGA dataset. (C) Validation of the effect of TRIM25 and PPM1B on the growth of AGS cells by colony formation. (D) The effects of TRIM25 and PPM1B on the proliferation of AGS cells were observed by CCK-8 assay.
PPM1B suppresses tumor growth via TRIM25/PPM1B/CDK2 signaling in a mouse model of GC
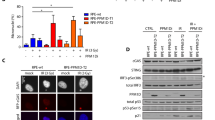
The effects of PPM1B overexpression and knockdown, as well as TRIM25 overexpression, on xenograft tumor growth were examined. The results revealed that overexpression of PPM1B in AGS cells significantly inhibited xenograft tumor growth, as evidenced by reduced tumor volume and weight (Fig. 7A-D). Conversely, PPM1B knockdown and TRIM25 overexpression led to a significant promotion of xenograft tumor growth compared to the control group (Fig. 7E-G). IHC analysis confirmed a negative correlation between TRIM25 and PPM1B in xenograft tumors, supporting their regulatory relationship (Fig. 7H). WB analysis further demonstrated that TRIM25 influenced CDK2 phosphorylation through PPM1B, with PPM1B partially mitigating the effect of TRIM25 on CDK2 phosphorylation (Fig. 7I).

PPM1B suppresses tumor growth via TRIM25/PPM1B/CDK2 signaling in a mouse model of GC. (A) A BALB/c nude mouse subcutaneous tumor model was established using PPM1B-overexpressing cells. Tumor growth was monitored every 3 days. (B) The images show tumor formation in mice with PPM1B overexpression in AGS cells compared to the control tumor group. (C-D) Statistical analysis of the volume and weight of tumor tissue. (E) A subcutaneous tumor model was established in BALB/c nude mice with high protein levels of TRIM25 or stably knockdown PPM1B cells. Tumors were measured every 3 days. Images show tumor formation in mice with TRIM25 overexpression or stable knockdown of PPM1B in AGS cells and control cells. (F-G) Statistical analysis of tumor volume and weight. (H) Immunohistochemical staining was performed on consecutive paraffin sections of AGS cells expressing TRIM25, showing protein expression levels of TRIM25 and PPM1B. (I) The protein expression levels of PPM1B and p-CDK2 in the xenograft tumors were detected by western blot analysis. (J) Schematic representation of the mechanism by which PPM1B inhibits tumor growth through the TRIM25/PPM1B/CDK2 signaling axis. Data shown as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
In this study, we conducted a comprehensive analysis of clinical samples and publicly available databases, confirming a significant downregulation of PPM1B expression in GC. Importantly, we found a strong correlation between high PPM1B expression and improved prognosis in GC patients. To experimentally validate these findings, functional assays were performed using GC cell lines. The results unequivocally demonstrated that PPM1B overexpression remarkably suppressed both proliferation and migration of GC cells, providing robust evidence supporting its tumor-suppressive role.
Importantly, our findings align with previous research indicating a substantial downregulation of PPM1B in GC15, reinforcing the reliability and consistency of our observations. Notably, Yang et al. have reported low expression of PPM1B in bladder cancer cells, emphasizing its role in cell cycle regulation10. Moreover, other scholars have documented the involvement of PPM1B in regulating tumor cell migration and invasion16,17,18. In this study, our results also confirmed that PPM1B can significantly inhibit cell proliferation and migration in gastric cancer and may play an essential role in cell cycle progression, further substantiating the critical function of PPM1B in tumor progression.
The functional role of PPM1B primarily revolves around dephosphorylation of downstream target proteins. Previous studies have demonstrated its ability to dephosphorylate various proteins, including CDK6, p53, PPAR-γ, and IKK-β8,9,19,20,21. Notably, in tumor-related investigations, PPM1B has been reported to dephosphorylate RhoGDI1, thereby exerting negative regulation on the proliferation and migration of breast cancer cells18. Wang et al. uncovered the negative regulation of the IKKβ/NF-κB signaling pathway by PPM1B, leading to the inhibition of migration and invasion in colorectal cancer cells22. In our study, we found that PPM1B had no significant effect on the baseline protein levels of CDK2. Further examination with a specific phosphorylation antibody revealed a significant decrease in p-CDK2 protein expression. These findings highlight the important role of PPM1B in influencing the phosphorylation levels of downstream protein CDK2, which may in turn regulate the cell cycle progression of gastric cancer cells.
In this study, we provided the first evidence that TRIM25, a member of the TRIM family, functions as a novel negative regulator of PPM1B. The TRIM family encompasses approximately 100 human TRIM genes, and emerging evidence suggests their involvement in various pathologies, including carcinogenesis23,24,25. Notably, many TRIM proteins act as E3 ubiquitin ligases, directly interacting with their substrates to induce ubiquitination and modulate diverse biological processes, such as protein stability, transcriptional control, signal transduction, and cell cycle progression26,27,28,29. Specifically, TRIM25 has been implicated in promoting survival and growth of liver cancer cells through its targeting of the Keap1-Nrf2 pathway30 and enhancing EZH2 stability, thereby regulating resistance to Oxaliplatin in colorectal cancer31. In our study, we confirmed the physical interaction between TRIM25 and PPM1B at both in vitro and in cells. The direct binding of TRIM25 to PPM1B relies on the RING domain of TRIM25 and the PPM domain of PPM1B. Furthermore, we discovered that TRIM25 induces polyubiquitination of PPM1B at the Lys48 linkage via its RING domain. These findings introduce a novel perspective for investigating the molecular mechanisms of PPM1B in tumors, offering new avenues for future research.
Finally, through in vitro and in vivo experiments, we confirmed that TRIM25 can influence the phosphorylation of CDK2 by degrading PPM1B. Importantly, the overexpression of PPM1B partially attenuated the impact of TRIM25 on cell proliferation and CDK2 phosphorylation levels. These findings collectively suggest that the TRIM25/PPM1B/CDK2 axis may represent a potential oncogenic signaling pathway in GC.
Conclusion
In summary, our study demonstrates the downregulation of PPM1B in GC and its role in suppressing cell proliferation and migration. We identify TRIM25 as a novel inhibitor of PPM1B, leading to increased phosphorylated CDK2 expression. These findings highlight the potential of PPM1B as a prognostic biomarker and therapeutic target in GC through the TRIM25/PPM1B/CDK2 signaling pathway (Fig. 7J).
Methods
Patient samples and data sources
A total of 100 human GC tissue samples and corresponding non-tumor tissue samples were collected from patients who underwent GC surgery at the First Affiliated Hospital of Fujian Medical University (Fuzhou, China) between 2018 and 2020. Inclusion criteria for patient samples included a histopathological diagnosis of GC and no history of prior chemotherapy or radiotherapy before surgery. All patients provided informed consent, and the study protocol was approved by the Institutional Review Board (IRB) of the hospital. In addition to this, we confirm that all methods were performed in accordance with the relevant guidelines and regulations.
To comprehensively assess the clinical significance of PPM1B, we conducted an extensive data analysis utilizing the publicly available GEO dataset. Our study employed Kaplan-Meier plotter database (http://kmplot.com/analysis/) to examine the relationship between PPM1B expression levels and overall survival rates.
Antibodies, and reagents
Antibody specific for PPM1B (ab244353) was sourced from Abcam. Antibodies targeting HA-tag (5017 S), Myc-tag (2276 S), Flag-tag (14793 S), and GAPDH (5174 S) were obtained from Cell Signaling Technology. TRIM25 (DF3276), CDK2 (AF6237), Phospho-CDK2 (Tyr15) (AF7274), CDK4 (AF4034), Cyclin D1 (AF0931), Cyclin E1 (AF0144) antibodies were procured from Affinity Biosciences. Unless otherwise specified, all antibodies were used at a dilution of 1:1000. Additionally, cycloheximide (CHX) and MG132 were purchased from MedChemExpress. All other reagents not listed were procured from Sigma-Aldrich and Thermo Fisher Scientific.
Tissue IHC and evaluation
The IHC experiment utilized the ElivisionTM plus Polymer HP (mouse/rabbit) IHC kit from MXB Biotechnologies. Tissue sections were acquired from formalin-fixed and paraffin-embedded specimens following a previously established protocol32. Consecutive sections of GC tissues were subjected to incubation with a rabbit monoclonal antibody against PPM1B (diluted 1:200) and a mouse monoclonal antibody against TRIM25 (diluted 1:200). The tissue protein levels were assessed using Image-Pro Plus 6.0 software (Media Cybernetics, Inc).
To ensure representative analysis, three slides were randomly selected from each group, and five regions of equal size were randomly chosen within each slide. Immunohistochemical data was evaluated utilizing a semi-quantitative scoring approach. The scoring was determined by multiplying the percentage of tumor-positive cells (P) by the staining intensity (I). The P values were assigned as follows: 0 (no staining), 1 (0–25% staining), 2 (26–50% staining), 3 (51–75% staining), and 4 (76–100% staining). The I values were defined as 0 (negative), 1 (weak positive), 2 (positive), and 3 (strong positive).
Cell culture and transfection
AGS and MGC-803 human gastric carcinoma cell lines were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai). AGS cells were cultured in F12 medium with 10% FBS, streptomycin (100 µg/ml), and penicillin (100 IU/ml). Similarly, MGC-803 cells were cultured in DMEM medium with 10% FBS, streptomycin (100 µg/ml), and penicillin (100 IU/ml). HEK293T cells were obtained from the Key Laboratory of Gastrointestinal Cancer, Ministry of Education, Fujian Medical University, and cultured in DMEM medium with 10% FBS, streptomycin (100 µg/ml), and penicillin (100 IU/ml). All cell lines were maintained under 5% CO2 in a humidified incubator.
In this study, gene primers and cloning vectors were synthesized by Shangya Biotechnology (Shanghai) following standard protocols widely employed in scientific research. Small interfering RNA (siRNA) specifically designed to target PPM1B was also synthesized by Shangya Biotechnology (Shanghai). To establish stable PPM1B knockout cell lines, the CRISPR-Cas9 genome editing system from GeneCopoeia was utilized, following the recommended manufacturer’s instructions. For DNA transfection, Lipoectamine™3000 reagent (Invitrogen, Thermo Fisher Scientific) was employed in either 6-well or 10-cm plates, in accordance with the manufacturer’s instructions.
Immunoprecipitation (IP) and immunoblotting
The protein extraction procedure was conducted on ice, where cell samples were lysed on ice using IP lysis buffer (Thermo Fisher Scientific) and protease inhibitor cocktail (Beyotime) for a duration of 30 min. Subsequently, the lysate was centrifuged at 12,000 × g for 10 min, and the resulting supernatant was collected. The total protein concentration was quantified using the BCA assay kit (Thermo Fisher Scientific). For IP, HEK293T, AGS, or MGC-803 cells were transiently transfected with Myc-PPM1B or Myc-PPM1B fragments, along with Flag-TRIM25, and used for protein IP. Cell lysates were mixed with equal amounts of IgG, anti-Flag affinity beads, or anti-Myc affinity beads (Pierce), followed by overnight incubation at 4 °C. The magnetic beads were washed thrice with IP lysis buffer, and the prepared samples were subjected to WB. The immunoblotting procedure was performed using the Amersham™ ImageQuant™ 800 instrument as previously described32.
Immunofluorescence staining
Immunofluorescence staining was conducted on cells plated onto chamber slides. The cells were fixed with 4% paraformaldehyde for a duration of 10 min, followed by three washes with PBS. Subsequently, permeabilization was performed using 0.2% Triton X-100 for 10 min. The cells were then subjected to overnight incubation at 4 °C with the appropriate primary antibody (Flag, diluted 1:800; Myc, diluted 1:1000). After washing with PBS buffer, the cells were incubated with a FITC-conjugated secondary antibody (Life Technologies) in the dark at 37 °C for 2 h. To visualize the cell nucleus, 4’,6-diamidino-2-phenylindole (DAPI, ab285390) staining was performed for 5 min. Finally, images were captured using a Leica SP5 confocal microscope (Leica, Germany).
Ubiquitination assay
To investigate polyubiquitination, IP of ubiquitinated proteins was conducted using HEK293T cells transiently overexpressing Flag-TRIM25 or its fragments, along with HA-Ub or specific ubiquitin mutants (Ub-K48 or Ub-K63). Equivalent amounts of cell lysate were added to anti-IgG and anti-Myc affinity beads (Pierce) and incubated overnight at 4 °C. The beads were subsequently washed thrice with IP lysis buffer, followed by WB using HA-Ub.
Quantitative polymerase chain reaction (qPCR)
Total cellular or tissue RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) following the manufacturer’s instructions. Subsequently, reverse transcription was performed using an RT kit (Takara) as per the recommended protocol. Quantitative PCR was conducted to assess the expression levels of PPM1B and GAPDH using a SYBR premixed EX Taq kit (Takara). The relative gene expression was determined using the 2−ΔΔCt method, with normalization to the endogenous GAPDH level. All gene-specific primers were synthesized by Shangya Biotechnology (Shanghai).
Cell proliferation and colony-forming assay
For each group of stably expressed cells, a seeding density of 1 × 10^3 AGS cells and 800 MGC-803 cells was used to inoculate 96-well plates. Five replicate wells were included for each cell group. Cell proliferation was assessed at specific time points (6, 24, 48, and 72 h) using the CCK-8 from Dojindo (Japan). A volume of 10 µl CCK-8 solution was added to each well, followed by incubation in an incubator (Thermo Fisher Scientific) for 2 h. Subsequently, the absorbance at 450 nm was measured using Gen5 software (BioTek). Each experiment was independently repeated three times.
Each group of stably expressed cells, comprising 800 AGS cells and 600 MGC-803 cells, was plated into six-well plates and cultured in the incubator for 14 days. Clonogenic colony-forming units were obtained by washing the cells with PBS, fixing them with methanol, and staining with 1% crystalline violet (Beyotime Biotechnology). The number of colonies was calculated using ImageJ software. Each experiment was independently repeated three times.
Cell migration assay
A total of 2.5 × 104 AGS and MGC-803 cells were seeded in the upper chamber (8 μm, 24-well format) of a transwell system. Serum-free medium was used in the upper chamber, while the lower chamber contained medium supplemented with 10% fetal bovine serum. After 24 h of incubation at 37 °C with 5% CO2, cells that migrated to the lower membrane were fixed and stained. Cell counting was performed by examining five random microscopic fields (100×) in each experiment. The entire procedure was repeated three times independently to ensure accuracy and reproducibility.
Cell cycle analysis
Cell cycle analysis was performed by staining the cells with PI staining solution following the protocol provided in the cell cycle kit (Bestbio). The distribution of cells across the cell cycle phases was assessed using a flow cytometer LSRFortessaX-20.
Tumor formation assay in nude mice
Animal experiments were approved by the Medical Ethics Committee of Fujian Medical University following the UK Animals (Scientific Procedures) Act, 1986 and Animal Research Reporting In Vivo Experiments (ARRIVE) guidelines.
Female BALB/c nude mice, approximately 5 weeks old, were procured from the Shanghai Laboratory Animal Center of the Chinese Academy of Sciences. To establish the subcutaneous tumor models, 5 × 106 stably high-expressing PPM1B AGS cells were injected into the experimental group, while control group received cells with simulated plasmids. Similarly, to further investigate the functional roles of PPM1B knockdown and TRIM25 overexpression, stable cell lines with PPM1B knockdown and TRIM25 overexpression were established. Subcutaneous tumor models were generated by injecting these cell lines, while the control group received an equal number of cells.
Tumor growth was monitored every three days by measuring tumor volume using a vernier caliper: tumor volume (cm3) = 0.5 (cm) × tumor length (cm) × tumor width (cm). After approximately three weeks, the mice were euthanized by cervical dislocation method. And the subcutaneous xenografts were removed, photographed, weighed, and measured. Tumor tissue (50 mg) was lysed with RIPA buffer, and proteins were extracted using ultrasonic cell disruption. The remaining transplant tumors were fixed in formalin for immunohistochemical analysis.
Statistical analysis
Statistical analyses were conducted using SPSS 22.0 software and GraphPad Prism 8 software. The data were presented as mean ± standard deviation (SD) and analyzed using one-way ANOVA or Student’s t-test. Survival analysis was performed using Kaplan-Meier analysis. A significance level of p < 0.05 was considered statistically significant.
Data availability
The datasets generated and/or analyzed during the current study are available in the GEO repository, GSE26899.
References
Sung, H. et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. Cancer J. Clin. 71, 209–249. https://doi.org/10.3322/caac.21660 (2021).
Sexton, R. E., Hallak, A., Diab, M. N., Azmi, A. S. & M. & Gastric cancer: a comprehensive review of current and future treatment strategies. Cancer Metastasis Rev. 39, 1179–1203. https://doi.org/10.1007/s10555-020-09925-3 (2020).
Russo, A. E. & Strong, V. E. Gastric Cancer etiology and management in Asia and the West. Annu. Rev. Med. 70, 353–367. https://doi.org/10.1146/annurev-med-081117-043436 (2019).
Li, Z. et al. A comprehensive overview of PPM1B: from biological functions to diseases. Eur. J. Pharmacol. 947, 175633. https://doi.org/10.1016/j.ejphar.2023.175633 (2023).
Kamada, R. et al. Metal-dependent Ser/Thr protein phosphatase PPM family: evolution, structures, diseases and inhibitors. Pharmacol. Ther. 215, 107622. https://doi.org/10.1016/j.pharmthera.2020.107622 (2020).
Marley, A. E., Kline, A., Crabtree, G., Sullivan, J. E. & Beri, R. K. The cloning expression and tissue distribution of human PP2Cbeta. FEBS Lett. 431, 121–124. https://doi.org/10.1016/s0014-5793(98)00708-x (1998).
Park, J. H., Hale, T. K., Smith, R. J. & Yang, T. PPM1B depletion induces premature senescence in human IMR-90 fibroblasts. Mech. Ageing Dev. 138, 45–52. https://doi.org/10.1016/j.mad.2014.03.003 (2014).
Tasdelen, I. et al. The serine/threonine phosphatase PPM1B (PP2Cβ) selectively modulates PPARγ activity. Biochem. J. 451, 45–53. https://doi.org/10.1042/bj20121113 (2013).
Zhao, Y. et al. PPM1B negatively regulates antiviral response via dephosphorylating TBK1. Cell. Signal. 24, 2197–2204. https://doi.org/10.1016/j.cellsig.2012.06.017 (2012).
Yang, J., Yuan, D., Li, J., Zheng, S. & Wang, B. miR-186 downregulates protein phosphatase PPM1B in bladder cancer and mediates G1-S phase transition. Tumour Biol. 37, 4331–4341. https://doi.org/10.1007/s13277-015-4117-4 (2016).
Hu, L. et al. TBK1 is a synthetic Lethal Target in Cancer with VHL loss. Cancer Discov. 10, 460–475. https://doi.org/10.1158/2159-8290.Cd-19-0837 (2020).
Zhang, S., Ding, L., Li, X. & Fan, H. Identification of biomarkers associated with the recurrence of osteosarcoma using ceRNA regulatory network analysis. Int. J. Mol. Med. 43, 1723–1733. https://doi.org/10.3892/ijmm.2019.4108 (2019).
Martin-Vicente, M., Medrano, L. M., Resino, S., Garcia-Sastre, A. & Martinez, I. TRIM25 in the regulation of the antiviral innate immunity. Front. Immunol. 8, 1187. https://doi.org/10.3389/fimmu.2017.01187 (2017).
Choudhury, N. R., Heikel, G. & Michlewski, G. TRIM25 and its emerging RNA-binding roles in antiviral defense. Wiley Interdiscip Rev. RNA. 11, e1588. https://doi.org/10.1002/wrna.1588 (2020).
Wang, G. et al. NAT10-mediated mRNA N4-acetylcytidine modification promotes bladder cancer progression. Clin. Transl Med. 12, e738. https://doi.org/10.1002/ctm2.738 (2022).
Du, L., Li, J., Tian, Y. & Feng, R. Pleckstrin-2-promoted PPM1B degradation plays an important role in transforming growth factor-beta-induced breast cancer cell invasion and metastasis. Cancer Sci. 114, 2429–2444. https://doi.org/10.1111/cas.15791 (2023).
Wang, H. et al. DCAF4L2 promotes colorectal cancer invasion and metastasis via mediating degradation of NFkappab negative regulator PPM1B. Am. J. Transl Res. 8, 405–418 (2016).
Cho, H. J. et al. Protein phosphatase 1B dephosphorylates rho guanine nucleotide dissociation inhibitor 1 and suppresses cancer cell migration and invasion. Cancer Lett. 417, 141–151. https://doi.org/10.1016/j.canlet.2018.01.002 (2018).
Cheng, A., Kaldis, P. & Solomon, M. J. Dephosphorylation of human cyclin-dependent kinases by protein phosphatase type 2 C alpha and beta 2 isoforms. J. Biol. Chem. 275, 34744–34749. https://doi.org/10.1074/jbc.M006210200 (2000).
Sun, W. et al. PPM1A and PPM1B act as IKKbeta phosphatases to terminate TNFalpha-induced IKKbeta-NF-kappaB activation. Cell. Signal. 21, 95–102. https://doi.org/10.1016/j.cellsig.2008.09.012 (2009).
Prajapati, S., Verma, U., Yamamoto, Y., Kwak, Y. T. & Gaynor, R. B. Protein phosphatase 2Cbeta association with the IkappaB kinase complex is involved in regulating NF-kappaB activity. J. Biol. Chem. 279, 1739–1746. https://doi.org/10.1074/jbc.M306273200 (2004).
Wang, H. et al. DCAF4L2 promotes colorectal cancer invasion and metastasis via mediating degradation of NFκb negative regulator PPM1B. Am. J. Translational Res. 8, 405–418 (2016).
Wang, S. et al. TRIM67 activates p53 to suppress Colorectal Cancer initiation and progression. Cancer Res. 79, 4086–4098. https://doi.org/10.1158/0008-5472.Can-18-3614 (2019).
Guo, P. et al. TRIM31 is upregulated in hepatocellular carcinoma and promotes disease progression by inducing ubiquitination of TSC1-TSC2 complex. Oncogene 37, 478–488. https://doi.org/10.1038/onc.2017.349 (2018).
Tan, P. et al. TRIM59 promotes breast cancer motility by suppressing p62-selective autophagic degradation of PDCD10. PLoS Biol. 16, e3000051. https://doi.org/10.1371/journal.pbio.3000051 (2018).
Montori-Grau, M. et al. GNIP1 E3 ubiquitin ligase is a novel player in regulating glycogen metabolism in skeletal muscle. Metab. Clin. Exp. 83, 177–187. https://doi.org/10.1016/j.metabol.2018.02.005 (2018).
Mei, P. et al. E3 ligase TRIM25 ubiquitinates RIP3 to inhibit TNF induced cell necrosis. Cell Death Differ. 28, 2888–2899. https://doi.org/10.1038/s41418-021-00790-3 (2021).
Yang, M. et al. SINAT E3 Ligases Control the light-mediated Stability of the Brassinosteroid-activated transcription factor BES1 in Arabidopsis. Dev. Cell. 41, 47–58. https://doi.org/10.1016/j.devcel.2017.03.014 (2017). .e44.
Ying, H. et al. TRIM59 promotes tumor growth in hepatocellular carcinoma and regulates the cell cycle by degradation of protein phosphatase 1B. Cancer Lett. 473, 13–24. https://doi.org/10.1016/j.canlet.2019.12.030 (2020).
Liu, Y. et al. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 11, 348. https://doi.org/10.1038/s41467-019-14190-2 (2020).
Zhou, S. et al. TRIM25 regulates oxaliplatin resistance in colorectal cancer by promoting EZH2 stability. Cell. Death Dis. 12, 463. https://doi.org/10.1038/s41419-021-03734-4 (2021).
Wang, Q. et al. Vimentin affects colorectal cancer proliferation, invasion, and migration via regulated by activator protein 1. J. Cell. Physiol. 236, 7591–7604. https://doi.org/10.1002/jcp.30402 (2021).
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 82273361), the Joint Funds for the Innovation of Science and Technology, Fujian Province (No. 2020Y9017), the Natural Science Foundation of Fujian Province (No. 2022J02032, No. 2023J06032), the Fujian Provincial Health Department Innovation Project (No. 2020CXA032, 2022CXA024), Fujian Provincial Finance Project (Nos. BPB-YJX2021, BPB-ZGW2021) and the Open Project of Fujian Provincial Key Laboratory of Tumor Microbiology (No. FMUGIC-202101).
Author information
Authors and Affiliations
Contributions
G.Z. and J.Y. designed this experiment. H.C. and S.L. conducted experiments. H.C. and S.L. analyzed data. G.Z. and H.C. explained the experimental results. C.L. and P.L. prepared these figures. G.Z. and H.C. wrote this manuscript. J.Y. revised the manuscript. All authors participated in reading and discussing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All animal studies were approved by the Fujian Medical University’s laboratory animal ethics committee and Chinese government guidelines for animal experiments. All methods used in animal experiments were carried out in accordance with relevant guidelines, and handled according to standard use protocols and animal welfare regulations, and we also confirmed that all animal methods were reported in accordance with ARRIVE guidelines (http://arriveguidelines.org) for the reporting of animal experiments.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, Gw., Chen, H., Liu, Sy. et al. PPM1B degradation mediated by TRIM25 ubiquitination modulates cell cycle and promotes gastric cancer growth. Sci Rep 15, 6160 (2025). https://doi.org/10.1038/s41598-025-89519-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-89519-7
