
Abstract
Ebsulfur and ebselen derivatives that were proven to be potent inhibitors against the main protease (MPro) of SARS-CoV-2 which is an essential enzyme for viral replication were chosen to study the quantitative structure–activity relationship (QSAR) analysis using a classical multiple linear regression (MLR) and a machine learning approach of random forest (RF) and artificial neural network (ANN) in order to find the relationship between molecular structural properties and biological inhibitory activities. With the statistical criteria, the R2 values of MLR, RF, and ANN models for the training set were 0.83, 0.82, and 0.92, respectively. The RMSE values of the test were considered for model evaluation, and the results were 0.27, 0.18, and 0.09 for MLR, RF, and ANN models, respectively. Therefore, the ANN model was the best-obtained model for predicting the MPro inhibitory activity of thirteen new synthetic ebselen analogs that haven’t tested the biological assay before. Notably, our predicted inhibitory activities against SARS-CoV-2 were then examined using enzyme-based assays and cytotoxicity tests, which found that compound P8 resulted in a good potential candidate for SARS-CoV-2 MPro inhibitory activity. Furthermore, the molecular dynamics simulations were performed to study the dynamic interaction of ligand and binding site; the results showed a binding pathway and mechanism of compound P8 with key residues surrounding the active site of SARS-CoV-2 MPro, which is useful for further development of ebselen derivatives.
Similar content being viewed by others


Introduction
The coronavirus family, formally known as Coronaviridae, is a large and diverse group of viruses that infect the respiratory tract with a wide range of hosts, including humans and other mammals1,2,3. This family causes respiratory illnesses with mild symptoms known as causing severe respiratory diseases such as severe acute respiratory syndrome (SARS), Middle East Respiratory Syndrome (MERS), and the ongoing global pandemic, SARS-CoV-2. The coronaviruses genome is a positive-sense single-stranded virus, approximately 26 to 32 kilobases in size4. In this study, one of the best-characterized drug targets is the main protease (MPro, also known as 3CLPro)5,6, which involves viral replication and a highly conservative pocket site of about 80% in clusters of the coronavirus family7.
The emergence of SARS-CoV-2 in Wuhan, China, in December 2019 was the beginning of a global health emergency that rapidly evolved into a pandemic. This newly emerging disease, for which there were no existing protections or medications, caused a surge in fatalities during the early phase of the pandemic8. In response, the World Health Organization (WHO) announced the official name of the disease as coronavirus disease 2019 (COVID-19), and the new coronavirus has been named severe acute respiratory syndrome 2 (SARS-CoV-2)9,10. Treatments for COVID-19 are categorized into several approaches, including vaccines such as Pfizer-BioNTech, Moderna, AstraZeneca, etc.11. In tandem with vaccination efforts, antiviral medications have played a crucial role in the therapeutic landscape for COVID-19. For example, Remdesivir, a broad-spectrum antiviral drug like Ebola, inhibits virus replication within host cells12. Another innovative antiviral medication is Molnupiravir, an oral drug designed to introduce errors during viral RNA replication, impeding the virus’s ability to proliferate13.
Previous studies evaluated the efficacy of ebselen derivatives against HIV14, HSV15, HCV16, and Zika virus infections17, and also SARS-CoV-218,19,20,21. Ebselen derivatives potent and effective inhibition of the main protease of SARS-CoV-2 via covalent inhibition via S-Se interaction22. Furthermore, it has computational techniques that were employed in many studies22,23,24 to confirm the inhibitory efficiency of ebselen and understand insight, such as molecular docking and molecular dynamic simulations (MDs) techniques. Moreover, a recent report presents a new ebselen and ebsulfur series that were synthesized and tested as inhibitors of SARS-CoV-2 MPro by fluorescence resonance energy transfer (FRET) technique by Sun, Le-Yun et al.25 which would attract some theoretical research on ligand-based drug design.
Computational approaches have emerged as powerful tools for drug discovery, offering a faster and more cost-effective alternative is the quantitative structure–activity relationship (QSAR) methodology26. It is useful in drug discovery to understand and predict the biological activity of compounds based on molecular structure. Drug discovery, based on extensive laboratory testing and experimentation, is time-consuming and expensive27,28,29,30. By integrating QSAR with machine learning algorithms to predict the biological activity of compounds based on their chemical structure. This allows for the rapid screening and prioritization of potential drug candidates31,32,33, enabling researchers to focus on molecules with higher probabilities of exhibiting the desired activity against SARS-CoV-2.
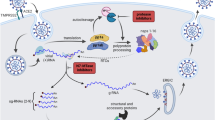
Thus, in this work, firstly, the application of QSAR with the three distinct methods, Genetic Function Approximation-Multiple Linear Regression (GFA-MLR), Random Forests (RF), and Artificial Neural Network (ANN), was employed to construct the relationship between structural properties and biological activity on ebselen and ebsulfur derivatives. The validation of our findings would be further confirmed by biological activity prediction on an external set compound. Insightly, the models further applied on SAR-CoV-2 inhibitory activity prediction of new ebselen and ebsulfur derivatives reported by Qing-Feng et al.34, we further investigated experimental inhibitory activity and toxicity drug evaluation, specifically focusing on drug-likeness and toxicity, grounded in the chemical structure of newly synthesized compounds. The successful process has led to the development of new effective inhibitor candidates. An overview of the study’s workflow shown in Fig. 1.

An overview of the study’s workflow, illustrating the key stages of developing the effective SARS-CoV-2 inhibitors which are composed of ligand based QSAR machine learning, enzyme-based assay on new designed ebselen, and structure based MD simulations.
Materials and methods
Data set
A data set of twenty-seven ebselen and ebsulfur derivatives and their MPro inhibitory activity (IC50) were synthesized by Sun et al.25. IC50 was then converted into pIC50 using Eq. (1), as shown in Fig. 2. The data set was divided by using the Kennard–Stone35,36 algorithm, a technique for selecting which data was suitable to be a training set or test set from the feature value distribution in the whole data set and calculated based on a distance metric between data points18. The training set was used to construct the QSAR model. While the test set was used for QSAR model validation.

The 2D structures and pIC50 values of ebselen and ebsulfur derivatives25. a is the IC50 (μM) and b is the pIC50. values.
All structures were built and minimized, and their molecular descriptors were then generated using the Materials Studio version 8.0 program37, which consisted of thirty-five molecular descriptors listed in Table S1. The molecular descriptors served as independent variables.
Descriptor selection and model construction
To find descriptors that are critical to significant SARS-CoV-2 MPro inhibitory activity by two algorithms are genetic functional algorithms (GFA); the GFA is a novel optimization technique that can be used to search for variables that are suitable for model construction38,39. The MLR model used the GFA to select important descriptors in Material Studio version 8.0. The condition to construct the GFA-MLR model in Material Studio version 8.0 with the population and maximum generation set at 100 and 500, respectively, and a mutation probability of 0.100.
Another one is Gini’s importance40,41, which is applied to select the crucial features of the RF and ANN models. The features with the highest Gini importance values indicate that compound structures significantly impact potency and bioactivity. Gini importance varies between 0 and 1, with 0 representing the lowest and best possible importance. A higher Gini importance indicates greater. Also, the RF and ANN models used Gini importance by Google Colab42. In addition, Fig. S1 displays the correlation matrix between two descriptors used to indicate the relationship between the descriptors in the model.
To find the greatest RF model, a hyperparameter for finding suitable conditions should be performed by varying four parameters: (1) Max feature is the maximum number of features considered for splitting a node. (2) Min_sample_leaf is the minimum number of data points allowed in a leaf node. (3) Min_samples_split is the minimum number of data points placed in a node before the node is split, and (4) The number of estimators is the number of trees in the forest. The optimized hyperparameter of the RF model is shown in Table 1(a).
The ANN is machine learning processing based on artificial neurons, which transform input data into output predictions via mathematical operations. It is a machine learning algorithm based on the structure and function of biological neurons and mimics human brain processing, which processes it using a non-linear activation function. The ANN model construction varied by two parameters: the number of nodes in the input layers, representing the number of descriptors, and the number of nodes and layers in the hidden layers43.
To improve the predictive performance and generalization capacity of the ANN model, we optimized the hyperparameters to obtain the optimal combination. Hyperparameter optimization for the ANN includes determining the number of hidden layers, the number of neurons, the maximum number of iterations (max_iter), the learning rate, and the batch size. A range of hyperparameters of the machine learning tools is varied to obtain the most robust and predictive non-linear models based on an n-fold cross-validation scheme using the Grid-Search CV of Scikit-learn44,45. The range of metrics for the grid search, where the five hyperparameters of the ANN model are examined, is presented in Table 1(b).
Model evaluation statistical terms
To investigate the degree of linear correlation between two descriptors by calculating the correlation coefficient (r)46,47. A correlation coefficient of 1.0 or − 1.0 indicates that two variables are highly correlated, while a coefficient of 0.0 shows no correlation, as shown in Eq. (2). When C(x,y) is covariance, which is the joint variance of two variables, x and y, the variance of a variable X (\({V}_{x}\)) and the variance of a variable Y (\({V}_{y}\)).
The variance inflation factor (VIF) indicates collinearity between descriptors in multiple regression models, indicating statistical significance48,49,50 as determined by Eq. (3).
The quality model was validated using statistical parameters, with R-Squared (R2) being a measure of the fit model’s quality, which should be greater than 0.6. When the predicted y values (\({y}_{pred}\)) and the mean values (\(\overline{y}\))
Root mean square error (RMSE) is a measure of prediction accuracy calculated as the square root of the average squared errors. A lower RMSE indicates better prediction quality, ideally closer to zero, as shown in Eq. (5).
Enzyme-based assay
The MPro activity and inhibition assay at 100 μM compound concentration was performed exactly as previously described51,52,53. Briefly, SARS-CoV-2 MPro with no tags at the termini was expressed and purified as described for SARS-CoV-1 MPro54. All assays were performed with BioTek Synergy H1 microplate reader using PBS containing 1 mM DTT and 1% DMSO as the reaction buffer. The fluorogenic substrate E(EDANS)TSAVLQSGFRK(DABCYL) (Biomatik) at 25 µM was used with 0.2 µM of MPro in the total reaction volume of 100 µL. The excitation and emission wavelengths employed were 340 and 490 nm, respectively. The percentage of the enzymatic activity was calculated from the initial rate of the reaction when the compound being tested was present relative to the initial rate of the reaction without the inhibitor. PF-07321332 at 100 nM was used as a positive control55. GraphPad Prism 856 (San Diego, California USA, https://www.graphpad.com) was used for graphing.
Cytotoxicity testing
Cytotoxic (CC50) tests were evaluated according to the previous description36. Vero E6 cells were seeded and incubated overnight before the test. The compounds were prepared in DMSO for a final concentration of 500 µM. The compounds were twofold serially diluted to 8 concentrations before addition to Vero E6 cells. Cells were incubated for 48 h, and cytotoxicity was measured using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay Kit (MTS) (Promega, Madison, WI, USA) according to the manufacturer’s instructions and analyzed by spectrophotometry at 490 nm. The concentration required for 50% cell death (CC50) was determined by three independent experiments.
The efficacy study was conducted according to the guidelines of the Declaration of Helsinki and Chulalongkorn University Institutional Biosafety Committee (CU-IBC 003/2021). The Institutional Review Board of the Faculty of Medicine, Chulalongkorn University certified the protocol exemption (COE 017/2021, IRB No. 297/64). The SARS-CoV-2 B.1.617.2 (accession number ON381169) were propagated in Vero E6 cells with MEM supplemented with 1% fetal bovine serum, 100 I.U./ml penicillin, and 100 μg/ml streptomycin, 10 mM HEPES, NEAA, and sodium pyruvate at 37 °C humidified chamber under 5% CO2. Virus titers were determined as TCID50/ml in confluent cells in 96-well cell culture plates. All experiments with live SARS-CoV-2 MPro were performed in a certified biosafety level 3 facility of the research affair-Medical Research Center (MRC), Faculty of Medicine, Chulalongkorn University.
Seven ebselen analogs were tested against four strains of SARS-CoV-2 MPro. Briefly, Vero E6 cells at 5 × 104 cells per well were seeded into a 24-well plate and incubated overnight at 37 C under 5% CO2. Cells were infected with SARS-CoV-2 at 1000TCID50 for 1 h. After infection, cells were washed with phosphate buffer saline (PBS) and incubated with 1 ml of maintenance medium. The compounds were prepared at the indicated concentrations in 0.1% DMSO in the maintenance medium during and after infection. Cells were incubated at 37 °C for 72 h under 5% CO2 humidified chamber. Supernatants were collected for analysis of the viral infectivity by TCID50/ml (v2.1—20-01-2017_MB* by Marco Binder; adapted @ TWC. 5. 6, accessed on 16 May 2022). The compound was serially diluted to 6–8 different concentrations and was added to final concentrations into SARS-CoV-2-infected cells. Dimethyl sulfoxide at 0.1% was used as a vehicle, with no inhibition control. Cells were incubated for 72 h and supernatants were collected for subsequent TCID50/ml analysis43,57. Data were plotted and effective concentration EC50 values were calculated using nonlinear regression analysis.
Molecular dynamic simulations
This study used ligand-binding path sampling parallel cascade selection MD (LB-PaCS-MD)58, an extension of the original PaCS-MD59,60. PaCS-MD was developed to sample the transition paths of proteins between a set of endpoint structures, where multiple short-timescale MD simulations are repeated from reasonable structures to promote their conformational transitions from a reactant to a product61,62,63. In the case of LB-PaCS-MD, this technique repeats short timescale (about 100-ps) MD simulations from reasonable protein–ligand configurations, focusing on ligand-unbinding states. In this application, configurations are ranked based on the center-of-mass (COM) distance between the Se atom of each ligand and the sulfur atom in the active site (C145) of SARS-CoV-2 Mpro, termed dCOM. Top-ranked (five) snapshots from each cycle serve as initial structures for subsequent simulations. LB-PaCS-MD terminates automatically after 100 cycles, with 10 independent replications conducted by changing their initial velocities to ensure reliable results.
To generate the parameters and perform geometry optimization of the compound P8, the B3LYP/6-31 + G(d,p) method of calculations64 were applied to generate the electrostatic potential (ESP) charges using Gaussian 1665. Subsequently, the ligand-charged fitting was constructed by restricted ESP and topological parameters of the ligands (frcmod and prep files) using MCPB.py66 in AmberTools2167, together with the generalized Amber force field 2 (GAFF2)68. The 3D structure of ebselen covalently bound dimeric SARS-CoV-2 Mpro (PDB ID: 7BAK19) was utilized as the protein receptor. To construct the initial structure for the LB-PaCS-MD simulation, P8 was placed far from C145 located at the active site of SARS-CoV-2 Mpro on chain A, around 30 Å in a cubic box. The tLEaP module included in the AmberTools2167 was used to set up the complex by adding hydrogen atoms, TIP3P water molecules, and neutralized ions. This complex was converted to the GROMACS input file format to conduct the multiple MD simulations under NPT (T = 300 K and P = 1 bar) in each LB-PaCS-MD cycle using GROMACS (version 2019.6)69. The MD condition was used according to the previously described70,71.
All 10 LB-PaCS-MD trajectories were used to calculate the free-energy profile (kBT) as a function of the distances of S(C145)–Se(P8) and Nε(H41)–N(P8), which were then plotted as a two-dimensional free energy landscape (2D-FEL). The complex sampled from the Global Minimum State (GMS) was evaluated for binding interaction energy using the LigandScout 4.4.6 program, following standard protocol72,73. The 3D and 2D interactions of the complex at GMS were visualized using Visual Molecular Dynamics (VMD) version 1.9.474,75 and BIOVIA Discovery Studio Visualizer76.
Results
Classical QSAR
The Kennard–Stone algorithm was applied to divide the data set into twenty-one training sets and six test sets. The MLR model was crafted using a selection of 5 descriptors of the training data set determined through the GFA algorithm, as shown in Eq. (6), and the definition of descriptors was explained in Table S2. This was a predicted pIC50 value, which shows residue values less than 1. The model validation parameters of Eq. (6): R2 of the training set = 0.69, RMSE of the training set = 0.16, and RMSE of the test set = 0.56, indicating that the model was predictively accurate and acceptable (Fig. 3).

A scatter plot of the predicted pIC50 for each model (Blue circles = Training set, Red triangles = Test set) (A) MLR, (B) RF, and (C) ANN.
After that, investigate the correlation matrix of descriptors and the VIF presented in Table S3. The correlation matrix between the two descriptors is less than 0.7. Further confirmed, the VIF values for each descriptor were less than 10, indicating that the five descriptors were not multicollinear and could not lead to problems in model interpretation and stability.
QSAR-ML
The RF and ANN models were developed utilizing the Gini importance method (Fig. S2), with emphasis on key descriptors such as shadow length along the Y-axis, AlogP98, shadow area fraction in the YZ plane, and principal moment of inertia along the Y-axis. Detailed definitions for each descriptor can be found in Table S2. To ascertain the significance of these descriptors, VIF helps identify multicollinearity among predictors by measuring how much the variance of an estimated regression coefficient increases if your predictors are correlated.
The best RF model was constructed by conditions consisting of a max depth of 10, a max feature of 4, a min_sample_leaf of 2, and a min_samples_split of 2. The number of estimators is 30. The results have the acceptable statistical parameters: R2 of the training set = 0.82; RMSE of the training set = 0.14; RMSE of the test set = 0.18. (Fig. 3) For the development of better predictive models according to Fig. S2. This method obtained four descriptors from the same RF model by selecting descriptors based on Gini importance. The good performance of the ANN architecture was 4-(5-5-5)-1, which represents the number in the first position as one input layer of four neurons, which is the number of descriptors selected by the Gini importance method. The number in the second position is three hidden layers with each with five neurons, and the number in the last is one output layer with inhibitory layers. The artificial neural network (ANN) model in Fig. 3 illustrated robust and stable performance with the notable statistical parameters: R2 of 0.89 for the training set, RMSE of 0.10 for the training set, and RMSE of 0.05 for the test set. Like the MLR model, the correlation analysis revealed that the four descriptors did not exhibit high correlations in RF and ANN models, as shown in Table S4. Compared to the MLR and RF models, the ANN shows much higher prediction accuracy in Table S5.
External validation
To evaluate the predictive ability and robustness of the ANN model developed in the previous step, an external validation was conducted using ebselen data reported from Amporndanai et al.19. Three compounds from this work were selected, including MR6-17-1, D_MR6-18-4, and D_MR6-26-2. The analysis revealed an RMSE of 0.35, which demonstrates good predictive performance (Fig. 4).

The data of the external compounds and their predicted and experimental pIC50 values predicted by ANN model SARS-CoV-2.
Inhibitory activity of the new synthetic ebselen analogs prediction
The structures of thirteen new ebselen analogs were synthesized by Qing-Feng et al.34. The external set used in this work came from a collaboration with Osaka University. The molecular descriptors for the new ebselen analogs calculated by Material Studio version 8.0 were displayed in Table S7. The pIC50 values of each new ebselen analog were then predicted using the ANN model, and it was found that the pIC50 values (Table S8) were all within the range of the data set shown in Fig. 5.

The new synthetic ebselen structures and the predicted pIC50 values by ANN model (Blue circles = Training set, Red triangles = Test set, Grey squares = New designed set).
Enzyme based assay
The ebselen analogs were initially tested for their inhibitory activities against SARS-CoV-2 MPro. At 100 μM concentration, the compounds P1, P3, P4, P5, P7, P8, and P12 showed modest inhibitory activity in Fig. 6A. The most potent inhibitor is P8 that caused reduction of enzymatic activity to 64.5%. Therefore, these ebselen analogs could serve as starting points for further modification to improve inhibitory potency.

(A) and (B) Effect of the new synthetic ebselen analogs in Vero E6 cells viability. (C) Inhibitory activity of ebselen analogs (100 μM) against SARS-CoV-2 MPro.
Toxicity and efficacy testing
The compounds P1, P3, P4, P5, P7, P8, and P12 were tested for cytotoxicity in Vero E6 cells in Fig. 6B. Cytotoxicity was not observed in P1, P3, P4, P5, P7, and P8 in any tested concentrations to 100 µM; therefore, we concluded that the compounds ‘cytotoxicity was higher than 100 µM. However, the P12 showed a cytotoxic effect at higher concentrations, calculated to 51.58 ± 5.90 µM. Moreover, the efficacy was tested against SARS-CoV-2 in the BSL-3 facility. The P3 compound showed 1–1.5 log TCID50 reduction from the initial concentration, and the inhibition was consistent through the higher concentrations. The P4, P5, P7, P8, and P12 showed 1–2 log TCID50 reduction from the initial concentration, but the inhibitions were reduced in higher concentrations in Fig. 6C. We speculated that the finding could correlate to the solubility issue as crystals were found in those respective tested concentrations. Finally, the P1 compound showed fluctuating SARS-CoV-2 titers, suggesting the inconsistent solubility of the compound.
P8 binding pathway towards the catalytic dyad region
To sample the plausible binding pathway and configuration of P8 towards the active site of SARS-CoV-2 MPro, LB-PaCS-MD simulation was conducted for 10 individual replications (#1–10) using the same initial coordinates but with varying initial velocities (Fig. 7A). The 2D free-energy profile (2D-FEL) for each replication, derived from LB-PaCS-MD trajectories based on the Markov State Model (MSM), shows the relative free energy (kBT) of P8 across its conformational space. Analyzing the representative trajectory (#1), P8 reveals a global minimum state (GMS, Fig. 7B), indicating its search for an optimal conformation facilitating binding at the active site of SARS-CoV-2. The binding process of the P8 observed in this study was similar to that of ebselen, as recently reported77. The P8 rearranged its conformation by orienting the Se of the benzoselenazole moiety toward the S of C145 (Fig. 7C), resulting in a binding interaction energy of − 16.15 kcal/mol (Fig. 7D). We found that chalcogen-bonding interaction between S atoms in P8 and C145, and also a π-donor hydrogen interaction with N142, could induce and stabilize the binding mode of P8. Additionally, the influence of the naphthalene ring at R1 of the benzoselenazole ring, introduced from the QSAR-ML study, could maintain ligand binding through interactions with M49 and M165 via alkyl-π interaction, as well as van der Waals interactions with residues within sub-pockets S2 and S4 of the SARS-CoV-2 MPro active site (Fig. 7C). Our findings are congruent with previous studies that confirmed the existence of the naphthalene moiety in compound CDD-1733 leads to a full occupation of the sub-pocket S278, aligning with raised hydrophobicity. Moreover, the P2 of α-Ketoamide inhibitors and Nirmatrelvir fit well into the sub-pocket S2, contributing to hydrophobic interactions with M49, M165, and D18779,80. This sub-pocket S2 could also accommodate the benzene ring of flavonoid and the bicycloproline moieties of boceprevir and telaprevir through hydrophobic interactions81,82,83.

(A) The P8 binding pathway towards the catalytic dyad region of SARS-CoV-2 MPro is elucidated using LB-PaCS-MD with 10 independent runs (#1–10), each individually set by varying their initial velocities. (B, C) 2D-FEL of the representative trajectory is chosen to visualize the binding pathway and the metastable stage at GMS (×). (D) The binding pattern and interaction of P8 in 3D and 2D are illustrated.
Conclusions
In this study, The QSAR provides a significant understanding of the properties of compounds that significantly inhibit SARS-CoV-2 MPro activity (pIC50) by using several algorithms, including MLR, RF, and ANN. When comparing all models together, the statistical parameters of the ANN model had the highest R2, which was 0.89, and the lowest RMSE of the test set was 0.05, which indicates that the performance of this model was accurate. Consequently, the ANN model was used to predict inhibitory SARS-CoV-2 MPro activity of the thirteen new synthetics ebselen analogs and then examined the enzyme base activity and toxicity testing found that the compound P8 was notable inhibitory SARS-CoV-2 MPro activity and passed the enzyme base activity examination and non-toxicity. The LB-PaCS-MD study was conducted for 10 individual replications, analyzing the representative trajectory of P8 that demonstrates a global minimum state with a binding interaction energy of − 16.15 kcal/mol. It can effectively bind to the active site of SARS-CoV-2, while P2 in α-Ketoamide inhibitors plays a role in hydrophobic interactions with M49, M165, and D187 residues in pocket S2.
Data availability
All other data are available either in the main text or as supplementary materials. The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Change history
17 June 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-04533-z
References
Pal, M. et al. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): an update. Cureus 12(3), e7423 (2020).
Pillaiyar, T., Meenakshisundaram, S. & Manickam, M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today 25(4), 668–688 (2020).
Steiner, S. et al. SARS-CoV-2 biology and host interactions. Nat. Rev. Microbiol. 22(4), 206–225 (2024).
Saha, S. et al. Complete genome sequence of a novel coronavirus (SARS-CoV-2) isolate from Bangladesh. Microbiol. Resour. Announc. 9(24), e00568-20. https://doi.org/10.1128/mra.00568-20 (2020).
Ullrich, S. & Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 30(17), 127377 (2020).
Begum, M. M. et al. Virological characteristics correlating with SARS-CoV-2 spike protein fusogenicity. Front. Virol. 4, 1353661 (2024).
Chen, Y., Liu, Q. & Guo, D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 92(4), 418–423 (2020).
Jeon, S. et al. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 64(7), e00819-20. https://doi.org/10.1128/aac.00819-20 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395(10223), 497–506 (2020).
Ahn, D.-G. et al. Current status of epidemiology, diagnosis, therapeutics, and vaccines for novel coronavirus disease 2019 (COVID-19). J. Microbiol. Biotechnol. 30, 313–324 (2020).
Bernal, J. L. et al. Effectiveness of the Pfizer-BioNTech and Oxford-AstraZeneca vaccines on covid-19 related symptoms, hospital admissions, and mortality in older adults in England: test negative case-control study. BMJ 373, n1088 (2021).
Malin, J. J. et al. Remdesivir against COVID-19 and other viral diseases. Clin. Microbiol. Rev. 34(1), e00162-20. https://doi.org/10.1128/cmr.00162-20 (2020).
Pourkarim, F., Pourtaghi-Anvarian, S. & Rezaee, H. Molnupiravir: A new candidate for COVID-19 treatment. Pharmacol. Res. Perspect. 10(1), e00909 (2022).
Thenin-Houssier, S. et al. Ebselen, a small-molecule capsid inhibitor of HIV-1 replication. Antimicrob. Agents Chemother. 60(4), 2195–2208 (2016).
Sartori, G. et al. Antiviral action of diphenyl diselenide on herpes simplex virus 2 infection in female BALB/c mice. J. Cell. Biochem. 117(7), 1638–1648 (2016).
Mukherjee, S. et al. Ebselen inhibits hepatitis C virus NS3 helicase binding to nucleic acid and prevents viral replication. ACS Chem. Biol. 9(10), 2393–2403 (2014).
Simanjuntak, Y. et al. Ebselen alleviates testicular pathology in mice with Zika virus infection and prevents its sexual transmission. PLoS Pathog. 14(2), e1006854 (2018).
Haritha, C., Sharun, K. & Jose, B. Ebselen, a new candidate therapeutic against SARS-CoV-2. Int. J. Surg. (London, England) 84, 53 (2020).
Amporndanai, K. et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 12(1), 3061 (2021).
Ma, C. et al. Ebselen, disulfiram, carmofur, PX-12, tideglusib, and shikonin are nonspecific promiscuous SARS-CoV-2 main protease inhibitors. ACS Pharmacol. Transl. Sci. 3(6), 1265–1277 (2020).
Menéndez, C. A. et al. Molecular characterization of ebselen binding activity to SARS-CoV-2 main protease. Sci. Adv. 6(37), eabd0345 (2020).
Parise, A. et al. The Se–S bond formation in the covalent inhibition mechanism of SARS-CoV-2 main protease by Ebselen-like inhibitors: a computational study. Int. J. Mol. Sci. 22(18), 9792 (2021).
Rudrapal, M. et al. Phytocompounds as potential inhibitors of SARS-CoV-2 Mpro and PLpro through computational studies. Saudi J. Biol. Sci. 29(5), 3456–3465 (2022).
Behera, S. K. et al. Drug repurposing for identification of potential inhibitors against SARS-CoV-2 spike receptor-binding domain: An in silico approach. Indian J. Med. Res. 153(1–2), 132 (2021).
Sun, L.-Y. et al. Ebsulfur and Ebselen as highly potent scaffolds for the development of potential SARS-CoV-2 antivirals. Bioorg. Chem. 112, 104889 (2021).
Achary, P. G. Applications of quantitative structure-activity relationships (QSAR) based virtual screening in drug design: A review. Mini Rev. Med. Chem. 20(14), 1375–1388 (2020).
Ishola, A. A. et al. QSAR modeling and Pharmacoinformatics of SARS coronavirus 3C-like protease inhibitors. Comput. Biol. Med. 134, 104483 (2021).
Gini, G. QSAR methods. In silico methods for predicting drug toxicity, 1–20 (2016).
Isarankura-Na-Ayudhya, C. et al. A practical overview of quantitative structure-activity relationship (2009).
Tropsha, A. et al. Integrating QSAR modelling and deep learning in drug discovery: the emergence of deep QSAR. Nat. Rev. Drug Discov. 23(2), 141–155 (2024).
Zhao, L. et al. Advancing computer-aided drug discovery (CADD) by big data and data-driven machine learning modeling. Drug Discov. Today 25(9), 1624–1638 (2020).
Paul, D. et al. Artificial intelligence in drug discovery and development. Drug Discov. Today 26(1), 80 (2021).
Ignacz, G. & Szekely, G. Deep learning meets quantitative structure–activity relationship (QSAR) for leveraging structure-based prediction of solute rejection in organic solvent nanofiltration. J. Membr. Sci. 646, 120268 (2022).
Xu-Xu, Q. F. et al. Synthesis of benzoisoselenazolones via Rh (III)-catalyzed direct annulative selenation by using elemental selenium. Chem. A Eur. J. 27(71), 17952–17959 (2021).
Svitliishyi, M. et al. Application Kennard–Stone algorithm for QSAR studies. Sci. Collect. 144, 534–539 (2023).
Saptoro, A., Tadé, M. O. & Vuthaluru, H. A modified Kennard–Stone algorithm for optimal division of data for developing artificial neural network models. Chem. Prod. Process Model. https://doi.org/10.1515/1934-2659.1645 (2012).
Sharma, S., Kumar, P. & Chandra, R. Applications of BIOVIA materials studio, LAMMPS, and GROMACS in various fields of science and engineering. Molecular dynamics simulation of nanocomposites using BIOVIA materials studio 329–341 (Lammps and Gromacs, 2019).
Hasegawa, K. & Funatsu, K. Partial least squares modeling and genetic algorithm optimization in quantitative structure-activity relationships. SAR QSAR Environ. Res. 11(3–4), 189–209 (2000).
Sohail, A. Genetic algorithms in the fields of artificial intelligence and data sciences. Ann. Data Sci. 10(4), 1007–1018 (2023).
Menze, B. H. et al. A comparison of random forest and its Gini importance with standard chemometric methods for the feature selection and classification of spectral data. BMC Bioinform. 10(1), 1–16 (2009).
Nembrini, S., König, I. R. & Wright, M. N. The revival of the Gini importance?. Bioinformatics 34(21), 3711–3718 (2018).
Bisong, E. & Bisong, E. Building machine learning and deep learning models on google cloud platform: a comprehensive guide for beginners. Google Collaboratory 59–64 (2019).
Bullen, C. K., Davis, S. L. & Looney, M. M. Quantification of infectious SARS-CoV-2 by the 50% tissue culture infectious dose endpoint dilution assay. In SARS-CoV-2: Methods and Protocols 131–146 (Springer, 2022).
Garreta, R. & Moncecchi, G. Learning Scikit-learn: Machine Learning in Python Vol. 2013 (Packt Publishing, 2013).
Hao, J. & Ho, T. K. Machine learning made easy: a review of scikit-learn package in python programming language. J. Educ. Behav. Stat. 44(3), 348–361 (2019).
Cohen, I. et al. Pearson correlation coefficient. In Noise Reduction in Speech Processing 1–4 (2009).
De, P. et al. Prediction reliability of QSAR models: an overview of various validation tools. Arch. Toxicol. 96(5), 1279–1295 (2022).
Ghamali, M. et al. QSAR analysis of the toxicity of phenols and thiophenols using MLR and ANN. J. Taibah Univ. Sci. 11(1), 1–10 (2017).
Alin, A. Multicollinearity. Wiley Interdiscip. Rev. Comput. Stat. 2(3), 370–374 (2010).
Daoud, J. I. Multicollinearity and regression analysis. In Journal of Physics: Conference Series (IOP Publishing, 2017).
Deetanya, P. et al. Interaction of 8-anilinonaphthalene-1-sulfonate with SARS-CoV-2 main protease and its application as a fluorescent probe for inhibitor identification. Comput. Struct. Biotechnol. J. 19, 3364–3371 (2021).
Nutho, B. et al. Discovery of C-12 dithiocarbamate andrographolide analogues as inhibitors of SARS-CoV-2 main protease: In vitro and in silico studies. Comput. Struct. Biotechnol. J. 20, 2784–2797 (2022).
Sanachai, K. et al. Identification of repurposing therapeutics toward SARS-CoV-2 main protease by virtual screening. PLoS One 17(6), e0269563 (2022).
Xue, X. et al. Production of authentic SARS-CoV Mpro with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J. Mol. Biol. 366(3), 965–975 (2007).
Owen, D. R. et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 374(6575), 1586–1593 (2021).
Swift, M. L. GraphPad prism, data analysis, and scientific graphing. J. Chem. Inf. Comput. Sci. 37(2), 411–412 (1997).
Lei, C. et al. On the calculation of TCID 50 for quantitation of virus infectivity. Virol. Sin. 36, 141–144 (2021).
Aida, H., Shigeta, Y. & Harada, R. Ligand binding path sampling based on parallel cascade selection molecular dynamics: LB-PaCS-MD. Materials 15(4), 1490 (2022).
Harada, R. & Kitao, A. Parallel cascade selection molecular dynamics (PaCS-MD) to generate conformational transition pathway. J. Chem. Phys. 139(3), 035103 (2013).
Ikizawa, S. et al. PaCS-Toolkit: optimized software utilities for parallel cascade selection molecular dynamics (PaCS-MD) simulations and subsequent analyses. J. Phys. Chem. B 128(15), 3631–3642 (2024).
Baba, T. et al. On the induced- fit mechanism of substrate-enzyme binding structures of nylon-oligomer hydrolase. J. Comput. Chem. 35(16), 1240–1247 (2014).
Kitao, A. et al. Parallel cascade selection molecular dynamics for efficient conformational sampling and free energy calculation of proteins. In Proceedings of the International Conference of Computational Methods in Sciences and Engineering 2016 (ICCMSE-2016), 1790 (2016).
Fujita, J. et al. Identification of the key interactions in structural transition pathway of FtsZ from Staphylococcus aureus. J. Struct. Biol. 198(2), 65–73 (2017).
Zhao, Y. & Truhlar, D. G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 41(2), 157–167 (2008).
Frisch, M. J. et al. Gaussian 16 Rev. C.02 (2016).
Li, P. & Merz, K. M. Jr. MCPB.py: A Python based metal center parameter builder. J. Chem. Inf. Model. 56(4), 599–604 (2016).
Case, D. et al. AmberTools21 (University of California, 2021).
Vassetti, D., Pagliai, M. & Procacci, P. Assessment of GAFF2 and OPLS-AA general force fields in combination with the water models TIP3P, SPCE, and OPC3 for the solvation free energy of druglike organic molecules. J. Chem. Theory Comput. 15(3), 1983–1995 (2019).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015).
Hengphasatporn, K. et al. Promising SARS-CoV-2 main protease inhibitor ligand-binding modes evaluated using LB-PaCS-MD/FMO. Sci. Rep. 12(1), 17984 (2022).
Munei, Y. et al. Determination of the association between mesotrione sensitivity and conformational change of 4-hydroxyphenylpyruvate dioxygenase via free-energy analyses. J. Agric. Food Chem. 71(24), 9528–9537 (2023).
Wolber, G. & Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 45(1), 160–169 (2005).
Pojtanadithee, P. et al. Identification of promising sulfonamide chalcones as inhibitors of SARS-CoV-2 3CLpro through structure-based virtual screening and experimental approaches. J. Chem. Inf. Model. 63(16), 5244–5258 (2023).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14(1), 33–38 (1996).
Mackoy, T. et al. viewSq, a Visual Molecular Dynamics (VMD) module for calculating, analyzing, and visualizing X-ray and neutron structure factors from atomistic simulations. Comput. Phys. Commun. 264, 107881 (2021).
BIOVIA. Dassault Systèmes, Discovery Studio Visualizer (Dassault Systèmes, 2021).
Toopradab, B. et al. Machine learning-based QSAR and LB-PaCS-MD guided design of SARS-CoV-2 main protease inhibitors. Bioorg. Med. Chem. Lett. 110, 129852 (2024).
Jimmidi, R. et al. DNA-encoded chemical libraries yield non-covalent and non-peptidic SARS-CoV-2 main protease inhibitors. Commun. Chem. 6(1), 164 (2023).
He, J. et al. Potential of coronavirus 3C-like protease inhibitors for the development of new anti-SARS-CoV-2 drugs: Insights from structures of protease and inhibitors. Int. J. Antimicrob. Agents 56(2), 106055 (2020).
Hu, Y. et al. Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. ACS Central Sci. 9(8), 1658–1669 (2023).
Qiao, J. et al. SARS-CoV-2 M<sup>pro</sup> inhibitors with antiviral activity in a transgenic mouse model. Science 371(6536), 1374–1378 (2021).
Khamto, N. et al. Inhibitory activity of flavonoid scaffolds on SARS-CoV-2 3CLpro: Insights from the computational and experimental investigations. J. Chem. Inf. Model. 64, 874–891 (2024).
Hengphasatporn, K. et al. Halogenated baicalein as a promising antiviral agent toward SARS-CoV-2 main protease. J. Chem. Inf. Model. 62(6), 1498–1509 (2022).
Acknowledgements
This project is funded by the National Research Council of Thailand (NRCT, grant number N42A650231 for T.R. and N34A670082 for K.W.) and the Second Century Fund (C2F), Chulalongkorn University, and the National Research Council of Thailand (NRCT, grant number N42A650231). P.M. and TR would like to thank the Sci-super plus program of Chulalongkorn University and the Shanghai International Funding (G2023013060L). Y.S. acknowledges financial support from various sources, including the Ministry of Education, Culture, Sports, Science and Technology (MEXT) in Japan through the Grant-in-aid for innovative research “Biometal Sciences” (grant no: 22H04800) and the Japan Science and Technology Agency (JST) through the CREST program “Precise arrangement toward functionality” (grant no. JPMJCR20B3). Additional funding was provided by the Multidisciplinary Cooperative Research Program in CCS at the University of Tsukuba, the Quantum Information Life Science project at Tsukuba University, and the Toyota Riken Scholar Program 2024.
Author information
Authors and Affiliations
Contributions
T.R. and P.M. carried out the conceptualization of this work, while data curation and formal analysis were performed by P.M. Funding acquisition and the development of methodology were contributed by S.S., Y.N., Q.X., P.W., K.S., P.D., K.W., A.S., S.A., L.D., R.H., and K.H. Resources were managed by M.M. Validation, and project administration was done by P.M and T.R. Visualization was led by S.S. and K.H., and the original draft was prepared by S.S. and K.H. P.M. T.R handled the review and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained a typographical error in Equation 1, where the symbol log was missing and in Equation 4, where the symbol “1-” was missing.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sinsulpsiri, S., Nishii, Y., Xu-Xu, QF. et al. Unveiling the antiviral inhibitory activity of ebselen and ebsulfur derivatives on SARS-CoV-2 using machine learning-based QSAR, LB-PaCS-MD, and experimental assay. Sci Rep 15, 6956 (2025). https://doi.org/10.1038/s41598-025-91235-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-91235-1
Keywords
This article is cited by
-
Broad-spectrum antiviral activity of ebselen
Scientific Reports (2025)
-
AI-driven techniques for detection and mitigation of SARS-CoV-2 spread: a review, taxonomy, and trends
Clinical and Experimental Medicine (2025)
-
Unlocking urban soil secrets: machine learning and spectrometry in Berlin’s heavy metal pollution study considering spatial data
Environmental Monitoring and Assessment (2025)
