
Abstract
Type 2 diabetes (T2D) is a crucial risk factor for both pancreatic cancer (PC) and kidney cancer (KC). However, effective common drugs for treating PC and/or KC patients who are also suffering from T2D are currently lacking, despite the probability of their co-occurrence. Taking disease-specific multiple drugs during the co-existence of multiple diseases may lead to adverse side effects or toxicity to the patients due to drug-drug interactions. This study aimed to identify T2D-, PC and KC-causing common genomic biomarkers (cGBs) highlighting their pathogenetic mechanisms to explore effective drugs as their common treatment. We analyzed transcriptomic profile datasets, applying weighted gene co-expression network analysis (WGCNA) and protein-protein interaction (PPI) network analysis approaches to identify T2D-, PC-, and KC-causing cGBs. We then disclosed common pathogenetic mechanisms through gene ontology (GO) terms, KEGG pathways, regulatory networks, and DNA methylation of these cGBs. Initially, we identified 78 common differentially expressed genes (cDEGs) that could distinguish T2D, PC, and KC samples from controls based on their transcriptomic profiles. From these, six top-ranked cDEGs (TOP2A, BIRC5, RRM2, ALB, MUC1, and E2F7) were selected as cGBs and considered targets for exploring common drug molecules for each of three diseases. Functional enrichment analyses, including GO terms, KEGG pathways, and regulatory network analyses involving transcription factors (TFs) and microRNAs, along with DNA methylation and immune infiltration studies, revealed critical common molecular mechanisms linked to PC, KC, and T2D. Finally, we identified six top-ranked drug molecules (NVP.BHG712, Irinotecan, Olaparib, Imatinib, RG-4733, and Linsitinib) as potential common treatments for PC, KC and T2D during their co-existence, supported by the literature reviews. Thus, this bioinformatics study provides valuable insights and resources for developing a genome-guided common treatment strategy for PC and/or KC patients who are also suffering from T2D.
Similar content being viewed by others

Introduction
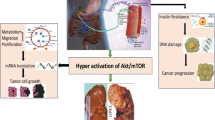
Type-2 diabetes (T2D) is a chronic metabolic disorder that is gradually increasing worldwide1. The International Diabetes Federation (IDF) estimated that there would be around 629 million adult diabetes patients worldwide by 20452. A population-based study reported that the prevalence of diabetes in all age groups is 2.8% in 2000 and is projected to increase to 4.4% by 20303. It is characterized by β-cell dysfunction, excessive glucose production from the liver and insulin resistance which impairs the ability of glucose to bind with insulin in blood4. T2D leads to serious health complications due to insulin resistance and hyperinsulinemia. It is also associated with obesity, cardiovascular disease, and cancers, including kidney cancer (KC)5,6 and pancreatic cancer (PC)7,8. Some studies showed that T2D stimulates about 80% of PC patients9 and 40% of KC patients10. Pancreatic cancer (PC) remains one of the most difficult cancers to diagnose and treat. In 2018, it was the 7th leading cause of cancer-related deaths worldwide, with approximately 466,000 deaths and a 5-year survival rate of just 10%11. Clear-cell renal cell carcinoma (ccRCC) is one of the most prevalent cancers worldwide. Kidney cancer (KC) includes various types, with renal cell carcinoma (RCC) being the most common. ccRCC, a subtype of RCC, accounts for approximately 70–80% of all kidney cancers12. It had the 17th highest cancer-related mortality in 2018 with 175,098 deaths worldwide13. In 2020, the death rate of KC patients was around 42%14. By 2030, pancreatic cancer (PC) is projected to become the second leading cause of cancer-related deaths15, while KC is expected to be the 10th most common cancer16. In PC, hyperinsulinemia associated with T2D elevates IGF-1 levels, which upregulates IGF-1R expression on pancreatic cells. This activation triggers cellular proliferation, suppresses apoptosis, and promotes genomic instability, fostering genetic mutations that may lead to cancer development17,18,19. Similarly, for KC, hyperinsulinemia stimulates IGF-1 production which developed renal cell proliferation and inhibits apoptosis19. Additionally, chronic inflammation and oxidative stress associated with T2D contribute to DNA damage, further increasing the risk of cancer development18,20. Thus, a schematic diagram about the link of PC and/or KC with T2D is given in Fig. 1.

A schematic diagram about the link of PC and/or KC with T2D
Mainly, PC can disrupt insulin regulation, leading to insulin resistance and hyperinsulinemia, which contribute to the development of T2D. Once T2D is established, chronic hyperinsulinemia and elevated IGF-1 levels promote cellular proliferation and inhibit apoptosis in various tissues, including the kidneys. Additionally, T2D is associated with chronic inflammation and oxidative stress, which cause DNA damage and genomic instability in renal cells. This environment fosters the development of KC, creating a cascade where pancreatic cancer leads to T2D, which in turn increases the risk of kidney cancer21. Doctors often prescribe disease-specific medications for patients with multiple conditions22, which can lead to polypharmacy and potential drug-drug interactions (DDIs) causing adverse effects or toxicity23,24,25. To mitigate this risk, it is preferable to prescribe a smaller number of common drugs that effectively address the multiple conditions. However, no study has yet proposed a common drug for patients with prostate cancer (PC) and/or kidney cancer (KC) who also suffer T2D. This bioinformatics-based study aims to: (i) identify common genomic biomarkers (cGBs) associated with T2D, PC, and KC, highlighting their shared pathogenetic mechanisms, and (ii) find cGBs-guided repurposable drugs for treating PC and/or KC in T2D patients.
Methodology
To explore repurposable drugs for treating PC and KC in patients with T2D, it is essential to identify common genomic biomarkers (cGBs) that can serve as drug targets. However, selecting top-ranked cGBs and potential therapeutic agents from numerous alternatives solely through wet-lab experiments is challenging due to the time, effort, and cost involved. To address these challenges, bioinformatics analysis plays a crucial role in streamlining and enhancing the drug discovery process. Transcriptomics profile analysis through bioinformatics tools is a popular approach to detect disease-causing or genomic biomarkers (GBs) as the targets of drug molecules26,27,28,29,30,31,32,33,34. The detailed methodology of this study is given in the following subsections 2.1–2.8.
Data source and descriptions
We considered gene-expression profiles for exploring T2D-, KC- and PC-causing cGBs, as well as meta-drugs to identify common drug molecules for these three diseases. It should be noted here that a group of drugs is considered as meta-drugs, which are already recommended for T2D, KC, or PC by individual studies.
Transcriptomics profiles collection (case/control)
There are several individual studies in the literature that explored multiple diseases causing cGBs from different independent transcriptomics datasets35,36,37,38. In this study, we used three independent transcriptomic datasets from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), GSE3689539 for KC, GSE1651540 for PC, and GSE7689641 for T2D. Where, GSE36895 includes 29 KC and 23 controls, GSE16515 includes 36 PC and 16 controls, and GSE76896 includes 55 T2D and 116 controls. Datasets were carefully selected based on their larger sample sizes in both case and control groups, ensuring statistical robustness and reliable results. Priority was given to datasets generated using the same platform, specifically the Affymetrix Human Genome U133 Plus 2.0 Array (GPL570), to maintain uniformity in data quality and experimental design. The inclusion of larger sample sizes helped reduce variability and enhance the power to detect true differences in gene expression between case and control groups, thereby supporting the identification of meaningful biomarkers and molecular mechanisms.
Collection of meta-drugs
A total of 110 KC-associated meta-drug agents (Table S1), 103 PC-associated meta-drug agents (Table S2), and 224 T2D-associated meta-drug agents (Tables S3) were collected to identify the potential common drug agents for each of T2D, KC, and PC. Specifically, the drug data were obtained from peer-reviewed published articles (Table S1, S2 & S3) and reliable online databases, including Drug Bank42, the National cancer institute43, and Drug.com44. Also, we individually selected the top-ranked 10 publicly available T2D -causing KGs (S4 Table), PC -causing KGs (S5 Table), and KC-causing KGs (S6Table) by the literature review to verify the performance of the proposed candidate drug agents via molecular docking analysis against the independent receptors.
Identification of DEGs by weighted gene Co-expression network analysis (WGCNA)
At first differentially expressed genes (DEGs) between cases (T2D/PC/KC) and control groups were identified by using “CEMiTool”45 from three datasets: GSE36895 for KC, GSE16515 for PC and GSE76896 for T2D. The detail discussion can be found in our previous study about how CEMiTool provides DEGs between disease and control groups46. Then weighted gene co-expression network analysis (WGCNA)47 technique was used for further filtering of those DEGs by removing the clusters (modules) of less correlated DEGs. Module-trait relationships were determined by computing the Pearson correlation coefficient between module eigengenes (MEs) and traits. In this study, disease status was treated as a clinical trait, where each disease group—T2D, PC, and KC was encoded using a binary classification system. Specifically, a value of “1” was assigned to individuals in the disease group, while a value of “0” was assigned to the control/ non-disease group. For example, for T2D, “T2D = 1” represented patients diagnosed with the disease, and “T2D = 0” represented healthy controls. The same approach was applied to PC and KC. Modules with significant correlations (|r| ≥ 0.6, p-value < 0.001) were selected for further analysis. We detected differentially expressed gene-set (DEGs-set) by combining all genes from all significant modules for each of T2D, PC and KC, separately. Subsequently, we separated the up- and down-regulated DEGs by satisfying the criterion alog2FCi > 1 and alog2FCi < -1, respectively, where alog2FC values indicates average of log2 fold-change values and is computed as.
Here \(\:{z}_{gi}^{D}\) and \(\:{z}_{gj}^{C}\) are the responses/expressions for the gth gene with the ith disease and jth control samples, respectively.
Identification of common differentially expressed genes (cDEGs)
We identified shared up- and down-regulated DEGs separately that were common across three datasets (GSE36895 for KC, GSE16515 for PC, and GSE76896 for T2D). Then we combined shared up- and down-regulated DEG-sets to create a unified set of common differentially expressed genes (cDEGs) among T2D, KC, and PC.
Local genetic association among T2D, PC and KC through cDEGs
Although the average log2 fold change (aLog2FC) values were calculated for each of T2D, PC, and KC using independent datasets as per Eq. 1, these values were derived from the same set of cDEGs across T2D, PC, and KC. A cDEG is considered upregulated for two or more diseases if aLog2FC > 0 and downregulated if aLog2FC < 0. Assuming that a gene functions similarly across individuals, the genetic association between any two diseases, A and B, can be assessed using their aLog2FC values corresponding to the cDEGs through Pearson’s correlation coefficient, defined as:
where, \(\:{a}_{i}={\text{a}Log}_{2}{\text{F}\text{C}}_{i}\left(X\right)\) and \(\:{b}_{i}={\text{a}Log}_{2}{FC}_{i}\left(Y\right)\) are the aLog2FC values of the ith gene for the two diseases A and B, respectively; \(\:\stackrel{-}{a}\:\)and \(\:\stackrel{-}{b}\:\)are the means of \(\:{a}_{i}{\prime\:}s\) and \(\:{b}_{i}{\prime\:}s\), respectively.
Identification of three disease-causing common genomic biomarkers (cGBs)
To explore common genomic biomarkers (cGBs), an online database and analysis tool STRINGv11.5 (https://string-db.org/) was used to create the protein-protein interaction (PPI) network of cDEGs. The network was visualized using the Cytoscape3.10.2 software (https://manual.cytoscape.org/en/3.10.2/)48. The CytoHubba plugin in Cytoscape49 was employed to identify cGBs by applying six topological criteria: Closeness, Degree, EPC, MCC, MNC and DMNC. During the analysis, we identified genes that ranked highly across all six measures, as these represent nodes of critical importance to the network’s structure and function. Only genes that demonstrated significant scores in all six measures were considered key candidates. This integrative approach ensured the robustness of the selection process, as it minimized bias from any single metric and highlighted genes that are universally central to the network.
Verification of association of cGBs with cGBs and T2D, PC, and KC using independent datasets and databases
To verify the association of cGBs with T2D, PC and KC through the independent datasets and databases, we performed disease- cGBs interaction analysis and expression analysis cGBs with T2D, PC and KC as discussed in the following subsections 2.6.1–2.6.2.
Disease-cGBs interaction analysis
We considered GeneCodis 450 web tool based DisGeNET database51 to perform disease- cGBs enrichment analysis for exploring the association of cGBs with different diseases including differents T2D, KC and PC.
Expression analysis of cGBs with T2D, PC and KC based on independent datasets
The differential expression patterns of cGBs in T2D, PC, and KC were verified using box plot analysis with independent datasets from NCBI. We used the The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) databases through the GEPIA2 web tool (http://gepia2.cancer-pku.cn/)52 to confirm the differential expression of cGBs between KC/PC and control samples. For these analyses, the cutoff thresholds were set at a p-value of 0.01 and a log2FC of 1. For validating the differential expression of cGBs between T2D and control samples, we used the independent dataset GSE15932. Box plots were constructed to compare cGBs expression between T2D/KC/PC and control groups. Additionally, we developed a prediction model based on random forest (RF) using three independent expression profiles (GSE36895 for KC, GSE16515 for PC, and GSE76896 for T2D) from the NCBI database and evaluated the predictive performance using ROC curves generated with the R-package “ROCR” https://www.rdocumentation.org/packages/ROCR/versions/1.0-1153.
Disclosing common pathogenetic mechanisms of PC and KC with T2D
In order to disclosing common pathogenetic mechanisms of PC and KC with T2D, functional enrichment analysis with gene ontology (GO)-terms and Kyoto Encyclopedia of Genes and Genomes (KEGG)-pathways, regulatory network analysis with transcription factors (TFs) and microRNAs and DNA methylation analysis were performed as discussed in the following subsections 2.6.1–2.6.3.
Regulatory network analysis of cGBs
We conducted regulatory network analysis with transcription factors (TFs) and micro-RNAs (miRNA) to investigate the regulators of cGBs. In order to determine the primary TFs connected with cGBs, we analyzed the TFs-cGBs connection network with JASPAR database54. By examining the links between miRNA and cGBs using the TarBase55 databases, it was possible to identify the significant miRNAs that have an impact on cGBs at the post-transcriptional stage. NetworkAnalyst47 was used to replicate these interactions. The post-transcriptional regulators of cGBs were selected from top-ranked miRNAs. We used Cytoscape56 to visualize the networks of their interactions.
The cGBs -set enrichment analysis with GO-terms and KEGG-pathways
The Gene Ontology (GO) project is a bioinformatics tool that uses domain-specific ontologies to provide a complete source of functional data on gene products and descriptions of activities57. To investigate the Gene Ontology and KEGG pathway of cGBs, we considered GeneCodis 458 database, and a P-value of 0.03 was chosen as the threshold.
DNA methylation analysis of cGBs in PC and KC
DNA methylation of cGBs involves adding methyl groups to their DNA, influencing gene expression and regulation. Statistically, analyzing these methylation patterns helps to identify significant changes in gene activity associated with diseases, providing insights into gene regulation mechanisms and contributing to understanding disease processes. MethSurv web tools (https://biit.cs.ut.ee/methsurv/) with TCGA-KIRC methylation data were used to investigate DNA methylation, a complex epigenetic process that controls gene expression in both normal and malignant cells59. DNA methylation values (ranging from 0 to 1) were represented by β values, which were computed as M/ (M + U + 100) for every CpG site. The intensities of methylation and unmethylation are represented by M and U, respectively. We classified the methylation levels into two groups based on the difference in methylation β value between the cut-off point and higher (methylation β value above the cut-off point) in order to assess the impact on patient survival. One can use data quantiles or the means to get the grouping cut-off point.
The cGBs -guided drug repurposing
To explore cGBs-guided repurposable common drug molecule for T2D, PC and KC, we performed molecular docking and ADME/T analysis as discussed in the subsections 2.8.1–2.8.2.
Exploring candidate drugs by molecular Docking
To explore potential repurposable drug molecules, we performed molecular docking between six cGBs and their top two associated transcription factors (TFs) as target receptors and repurposable drug molecules using AutoDock Vina60. To identify cGBs-guided drug molecules, we gathered 434 candidate molecules from published articles and online databases related to T2D, PC, and KC, as detailed in Tables S1, S2 & S3. Receptor proteins’ 3D structures were obtained from Protein Data Bank61 and AlphaFold databases62. All 434 T2D, PC and KC-related meta-drug candidates’ 3D structures were taken from the PubChem database63. Following this, the binding affinity scores (in kcal/mol) between receptors and ligands (drug molecules) were determined through molecular docking. The receptor proteins were arranged in descending order based on the average binding affinity scores for each row, while the drug candidates were ranked according to the average scores in each column of the score matrix. This method allowed for the selection of the top-ranked drug molecules for further analysis.
ADME/T analysis
ADME/T analysis evaluates a drug candidate’s absorption, distribution, metabolism, excretion, and toxicity to predict its safety and efficacy. In drug repurposing, a compound may demonstrate strong binding to a new target during docking studies, indicating potential for a new therapeutic application. However, if it does not meet ADMET criteria—such as poor absorption, quick metabolism, or high toxicity—it is unlikely to succeed in its new role. By filtering out unsuitable candidates, ADMET analysis ensures that only those with favorable pharmacokinetic and safety profiles progress further in the repurposing process. We analyzed the drug-like properties and ADME/T (absorption, distribution, metabolism, excretion, and toxicity) profiles of the top six ranked drug compounds to better understand their structural features and chemical descriptors. The SCFBio web application64 was used to evaluate compliance with Lipinski’s rule. ADME/T parameters were then predicted using the online databases SwissADME65 and pkCSM66, utilizing the optimal structures of the drug compounds in SMILES format for the calculations.
Results
Identification of DEGs by weighted gene Co-expression network analysis (WGCNA)
Differentially expressed genes (DEGs) for T2D, PC, and KC were identified using the “CEMiTool” and “WGCNA” (Weighted correlation network analysis) approaches across three datasets: GSE9348, GSE121248, and GSE76896. Initially, the “CEMiTool” analysis identified 4,589 DEGs from GSE16515, 4,233 DEGs from GSE16515, and 7,548 DEGs from GSE76896. These DEGs were further refined using the “WGCNA” approach. For each filtered gene expression matrices, a soft threshold β value was chosen (14 for GSE76896, 11 for GSE16515 and 15 for GSE36895) based on a cutoff R2 value of 0.85 (Figure S1). Following that, some modules were found by hierarchical clustering with the minimal module size 30. To merge the modules, cut height of module eigengene was set 0.15 for GSE76896, 0.09 for GSE16515 and 0.13 for GSE36895 (Figure S2). To uncover the relationship between modules and clinical traits (KC/PC/T2D and non-KC/non-PC/non-T2D samples and control), We selected six modules from GSE76896, four modules from GSE16515 and six modules from GSE36895 based on the module-trait relationship (correlation greater than 0.6 or less than − 0.6 along with p-value < 4e-08) (Figure S3).
Identification of common differentially expressed genes (cDEGs)
We identified a total of 85 common differentially expressed genes (cDEGs), including 55 upregulated and 30 downregulated genes, across three comparisons: control vs. T2D, control vs. PC, and control vs. KC. These cDEGs were visualized using a Venn diagram, as shown in Fig. 2 (see also Table S7). The Venn diagrams were made using the Venn Web Tool at https://bioinformatics.psb.ugent.be/webtools/Venn/. DEGs-sets for each disease were pasted into the tool, and it generated the diagram showing overlaps and differences between the sets. The diagram was then customized and downloaded for further use.

Common DEGs and the trends of their aLog2FC values in T2D, PC and KC. (A) Upregulated cDEGs; (B) Downregulated cDEGs and, (C) The trends of aLog2FC vales of cDEGs in T2D, PC and KC.
Local genetic association among T2D, PC and KC through cDEGs
To understand the link of T2D with PC and KC, we computed pairwise local correlation coefficients using Eq. 2 for T2D, PC and KC based on the aLog2FC values of cDEGs (see Fig. 2C; Table 1 and Table S8). The correlation coefficient between each pair of the three diseases was ≥ 0.83 (see Table 1), indicating that T2D, PC and KC are locally associated with each other through the expressions of cDEGs.
Identification of three disease-causing common genomic biomarkers (cGBs)
The protein-protein interaction (PPI) network of cGBs was built that consist of 76 nodes and 459 edges. We select the top-ranked 6 cGBs (ALB, MUC1, TOP2A, BIRC5, RRM2 and E2F7) based on six topological methods with threshold Degree = 20, Closeness = 46.16, EPC = 14.35, MNC = 20, Betweenness = 245.0877 and Radiality = 3.37 in the PPI network. cGBs that ranked highly across all six measures were identified as key candidates, ensuring their critical importance to the network. This integrative approach minimized bias and highlighted genes central to the network’s structure and function. Here, six cGBs were upregulated and one cGBs was downregulated. Scores of six topological measures in six cGBs were shown in Table S9. And the graphical representation of PPI-network was displayed in Fig. 3.

Protein-protein interactions (PPIs) network analysis of cDEGs. Nodes in an Octagon shape with yellow color indicate the cGBs.
Verification of association of cGBs with cGBs and T2D, PC, and KC using independent datasets and databases
To verify the association of cGBs with T2D, PC and KC through the independent datasets and databases, we performed disease- cGBs interaction analysis and expression analysis cGBs with T2D, PC and KC as discussed in the following subsections 3.5.1–3.5.2.
Disease-cGBs interaction analysis
The disease- cGBs interaction analysis showed that the top-ranked 25 diseases (including T2D, PC and KC) these are significantly associated with cGBs (Table S10). The p-value < 0.03 was chosen as cutoff for the significance test.
Expression analysis of cGBs with T2D, PC and KC based on independent datasets
Then, we verified the differential expression patterns of cGBs in two independent databases (GTEx and TCGA) for PC and KC that combinedly contained 171 controls and 179 PC samples, and 523 controls and 100 KC samples through box plot analysis (Figure S4A.). We examined the differential expression patterns of cGBs between T2D and control samples using box plot analysis. This analysis was based on the gene expression profiles from the NCBI dataset with accession ID GSE15932, which includes 8 T2D and 8 control samples. We excluded the pancreatic cancer samples from our analysis (Figure S4B). We found that 5 cGBs are upregulated, and one is downregulated in these three diseases that support our findings. To assess the prediction performance of cGBs, we developed a Random Forest (RF) based prediction model using 60% samples as training data. The remaining 40% of the data was used as test data. We also considered 3 additional independent test datasets (GSE71989 for PC, GSE66272 for KC and GSE19420 for T2D) from NCBI database. For each of these three diseases, we constructed ROC curves for both test datasets (Figure S5) and calculated some performance scores (AUC, TPR, TNR, and Accuracy) (Table S11). The performance of the cGBs in both prediction models was strong, with an AUC greater than 0.98 and an accuracy (ACC) exceeding 0.83.
Disclosing common pathogenetic mechanisms of PC and KC with T2D
In order to disclosing common pathogenetic mechanisms of PC and KC, with T2D, functional enrichment analysis with gene ontology (GO)-terms and Kyoto Encyclopedia of Genes and Genomes (KEGG)-pathways, regulatory network analysis with transcription factors (TFs) and microRNAs (miRNAs) and DNA methylation analysis were performed as discussed in the following subsections 3.6.1–3.6.3.
The cGBs-set enrichment analysis with GO-terms and KEGG-pathways
We performed GO and KEGG pathway enrichment analysis for 6 cGBs to explore the shared pathogenetic processes of T2D, KC and PC by Enrichr. Table 2 gives the top-ranked five BPs, MFs, and KEGG pathways.
Regulatory network analysis of cGBs
We employed cGBs-TF-miRNA coregulatory network to detect the regulators of cGBs at transcriptional and post-transcriptional levels. We found the top-ranked three TFs (NFIC, GATA2 and KLF5) based on degree and betweenness with a threshold degree of 3 and betweenness of 44.38, and two miRNAs (hsa-mir-16-5p and hsa-mir-103a-3p) based on degree and betweenness with a threshold degree of 5 and betweenness of 632.36 (Fig. 4).

cGBs-TF-miRNA coregulatory networks. Here KGs were marked as red color with octagon shapes, TFs were marked as blue color with ellipse and the miRNAs were marked as green color with demand shape. Key TF and miRNAs are in the in the right-hand side of this figure.
DNA methylation of cGBs in KC and PC
DNA methylation is an epigenetic mechanism that regulates gene expression by recruiting repressive proteins or inhibiting transcription factor binding to DNA61. We examined the DNA methylation status at CpG sites for all cGBs (ALB, BIRC5, E2F7, MUC1, TOP2A, and RRM2) using MethSurv. Our analysis revealed that ALB, BIRC5, and E2F7 were hypomethylated, while MUC1, TOP2A, and RRM2 were hypermethylated, with all cGBs showing significant CpG sites (p-value ≤ 0.01) (Table 3).
The cGBs -guided drug repurposing
To identify cGBs-guided repurposable drug molecules for T2D, PC, and KC, we conducted molecular docking and ADME/T analyses as detailed in subsections 3.7.1–3.7.2.
Exploring candidate drugs by molecular Docking
To identify cGBs-guided repurposable drug molecules, we performed molecular docking between cGBs-mediated receptors and candidate drug molecules. We obtained the 3D structures of six receptors (TOP2A, RRM2, ALB, MUC1, GATA2, and KLF5) from the Protein Data Bank (PDB) using the following PDB codes 1zxm, 3olj, 2Z5V, 2acm, 5o9b, and 2ebt, respectively. The remaining targets (BIRC5, NFIC, and E2F7) were sourced from the AlphaFold Protein Structure Database with UniProt IDs Q5RAH9, P08651, and Q96AV8. Through molecular docking analysis, we calculated the binding affinity scores (BAS) between the receptors and candidate drugs. We ranked the receptors and drug agents based on the row and column averages of the BAS matrix (Table S12). Out of 434 drugs, the top six potential candidates (NVP.BHG712, Irinotecan, Olaparib, Imatinib, RG-4733, and Linsitinib) were selected as multi-targeted drugs, all showing significant BAS values of < -7.0 kcal/mol with all target proteins (Fig. 5A).

Image of drug-target binding affinity matrices (A) X-axis indicates the top-ordered 50 drug agents (out of 434) and Y-axis indicates ordered proposed receptor proteins. (B) the top 6 proposed and published drugs (out of 434) in the Y-axis with a red tint. In the X-axis, blue color indicated the 10 independent receptors of T2D, red color indicated the 10 independent receptors of PC, black color indicated the 10 independent receptors of KC.
To verify their binding performance against other independent receptors, we considered top-ranked six key-genes for each of T2D, PC and KC separately, by the literature review. We observed that the proposed drug molecules significantly bind (BAS<-7.0 kcal/mol) with most of the independent receptors (Fig. 5B & Table S13). Therefore, the proposed cGBs-guided these top-ranked six lead drugs could be promising as the candidate drug molecules for the treatment of KC and PC with T2D. To provide more information about the proposed drugs and targets, top-ranked three drug-target complexes highlighting their 3-dimension (3D) view and interacting residues were given in Table S14.
ADME /T analysis
Among the detected six drugs by molecular docking analysis, six suggested (NVP.BHG712, Irinotecan, Olaparib, Imatinib, RG-4733 and Linsitinib) were satisfied at least four parameters of Lipinski’s rule of five, indicating their drug-like characteristics (Table S15). To assess the efficacy and indemnity of the suggested drugs, many parameters can be used to analyze their toxicity and ADME (absorption, distribution, metabolism and excretion) analyses (Table 4). All drug molecules exhibited high gastrointestinal absorption (HIA ≥ 68%), strong P-glycoprotein inhibition (P-gpI), and almost all were classified as non-toxic based on AMES, LD50, and LC50 tests, with LC50 values > 1.0 log mM. None effectively crossed the blood-brain barrier (LogBB < -1), but this limitation is not critical for non-CNS therapies. All candidates efficiently inhibited CYP3A4, supporting their metabolic suitability. Overall, these drugs demonstrate strong absorption, safety, and drug-like profiles, making them promising candidates for oral consumption. Among these six drugs, their effectiveness and potential were evaluated based on ADME/T properties, toxicity profiles, and drug-likeness characteristics, highlighting distinct advantages and limitations for each. Imatinib emerged as a highly effective candidate due to its excellent gastrointestinal absorption (HIA: 93.84%), strong Caco-2 permeability (1.09), low toxicity (LC50: 2.08 log mM), and efficient CYP3A4 inhibition, demonstrating its therapeutic promise. Similarly, NVP-BHG712 showed exceptional absorption (HIA: 95.71%), moderate permeability (0.51), low toxicity (LC50: 1.73 log mM), and favorable metabolic properties, reinforcing its safety and suitability. Olaparib also displayed strong absorption (HIA: 91.93%), excellent Caco-2 permeability (1.08), low toxicity (LC50: 1.97 log mM), and efficient metabolism, further highlighting its potential as a drug candidate. RG-4733, while having a relatively lower HIA (68.00%) and negative Caco-2 permeability (-0.59), demonstrated minimal toxicity with the highest LC50 value (2.83 log mM), ensuring a strong safety profile. In contrast, Irinotecan showed mixed results, with the highest absorption score (HIA: 99.88%) and moderate permeability (0.648), but its higher toxicity (LC50: 0.79 log mM) presents a challenge for its therapeutic application. Linsitinib, although exhibiting strong absorption (HIA: 93.26%) and the highest permeability (Caco-2: 1.188), was hindered by significant toxicity (negative LC50: -1.10 log mM), limiting its suitability as a safe drug candidate. Overall, Imatinib, NVP-BHG712, Olaparib, and RG-4733 displayed a favorable balance of absorption, safety, and metabolism, making them promising candidates for further exploration, while Irinotecan and Linsitinib may require optimization to improve their profiles.
Discussion
Population-based studies have shown that type-2 diabetes (T2D) is linked to kidney cancer (KC)67 and pancreatic cancer (PC)68. In this study, we aimed to explore the genetic connection between these diseases by identifying 78 common differentially expressed genes (cDEGs) that distinguish T2D, PC, and KC samples from control groups through transcriptomic analysis (see Figs. 1 and 2). We then identified the top 6 cDEGs (ALB, BIRC5, E2F7, MUC1, TOP2A, and RRM2) as candidate genetic biomarkers (cGBs) by constructing a protein-protein interaction (PPI) network for the cDEGs (Fig. 3), aimed at finding common drug molecules. The association of cGBs with T2D, PC, and KC was further validated through literature review, disease-cGB interaction analysis, and expression analysis of cGBs in these diseases. Additionally, functional enrichment analysis using Gene Ontology (GO) terms and KEGG pathways, regulatory network analysis involving transcription factors (TFs) and microRNAs, and DNA methylation analysis based on independent databases were also conducted. A graphical summary of this study is provided in Figure S6. The association of cGBs-with T2D, PC and KC also supported by some previous individual studies including ALB69,70,71, TOP2A72,73,74,75, BIRC576,77,78,79, MUC180,81,82, RRM283,84,85 and E2F786,87,88, as displayed in Fig. 6A. We investigated the shared pathogenetic mechanisms of KC, PC, and T2D by analyzing cGBs and identified some key enriched biological processes, molecular functions, and KEGG pathways include DNA damage response, cell cycle regulation, apoptosis, DNA binding, pyridoxal phosphate binding, N-methyltransferase activity, protein kinase C binding, copper ion binding, pyrimidine metabolism, glutathione metabolism, p53 signaling, drug metabolism, and cancer pathways (Table 2).

Verification of proposed cGBs and common drug molecules for T2D, PC and KC through literature review. (A) Verification of cGBs and (B) Verification of drug molecules, ellipse in green represent FDA-approved drugs, yellow represent investigational drugs, and red represent non-FDA approved drugs.
Among them, BIRC5 is a key inhibitor of apoptosis, involved in regulating the cell cycle and inhibiting programmed cell death89. This gene is essential for cell cycle regulation and mitotic cytokinesis, both of which are vital for tumor progression in KC and PC76. It’s overexpression is a well-established marker for poor prognosis in various cancers, including KC and PC. In T2D, its association with the regulation of apoptotic processes and positive regulation of cell cycle might reflect the dysregulation of cell survival and proliferation in insulin-resistant tissues79. The presence of BIRC5 in the p53 signaling pathway further connects its role in apoptosis regulation, highlighting its potential as both a prognostic marker and therapeutic target for tumors and diabetic complications, particularly kidney dysfunction90. MUC1 is a membrane-associated glycoprotein involved in several cellular functions, such as adhesion, signaling, and protecting against oxidative stress. It plays an essential role in regulating apoptosis and responding to DNA damage, which is vital for maintaining cell integrity91. In the context of T2D, MUC1 has been implicated in insulin signaling and glucose homeostasis, and its dysregulation may contribute to insulin resistance and dysfunction in pancreatic beta-cells82. In KC and PC, MUC1 aids in cancer cell migration and invasion, facilitating metastasis80,81. Its participation in the glutathione metabolism pathway also highlights its role in mitigating oxidative damage, a common feature in both diabetes and cancer92. Albumin (ALB), primarily synthesized in the liver, is a key protein that helps maintain osmotic pressure and facilitates the transport of various molecules in the bloodstream. In T2D, ALB is closely associated with kidney dysfunction, with its presence in urine (albuminuria) serving as an early indicator of diabetic nephropathy. The involvement of albumin in regulating apoptotic processes and DNA damage response pathways suggests it could serve as a biomarker for renal cell injury in diabetes-related kidney disease69. In KC, albumin plays a significant role in the glutathione metabolism and drug metabolism pathways, underscoring its role in cellular defense mechanisms and response to chemotherapy, especially in the context of oxidative stress71. Research on PC further emphasizes ALB’s involvement in regulating cell survival and proliferation, pointing to its potential as a therapeutic target for both diabetes complications and cancer treatment72. E2F7 is a member of the E2F family of transcription factors and plays a role in controlling the G1/S transition of the cell cycle, a key checkpoint in cell division. It is involved in the DNA damage response and has been implicated in maintaining genomic stability. In T2D, the aberrant regulation of cell cycle progression could contribute to the pathological changes observed in tissues like the kidney88. In KC, E2F7’s regulatory role in the G1/S transition and cell cycle processes suggests its involvement in tumor proliferation and resistance to apoptosis, which is critical for cancer progression87. The connection to positive regulation of mitotic cytokinesis in PC underscores its potential contribution to uncontrolled cellular division and tumor metastasis86. TOP2A is an enzyme involved in DNA replication and mitotic chromosome segregation, essential for cell division. Its overexpression is frequently associated with rapid cell proliferation and is a known marker of cell cycle regulation in cancer cells. In T2D, it may influence the regulation of apoptotic processes and contribute to tissue remodeling and fibrosis in the kidney. In KC, it is often upregulated in tumor tissues, where it enhances DNA damage repair and cell survival, promoting cancer progression73,74. The overexpression of TOP2A in pancreatic cancer (PC) has been linked to increased metastasis and poorer patient survival outcomes93. Its association with p53 signaling pathways further suggests its role in modulating responses to DNA damage, making it a valuable target for therapies aimed at both cancer treatment and preventing diabetic nephropathy94. RRM2 is a critical enzyme for the synthesis of deoxyribonucleotides, essential for DNA replication and repair. In the context of T2D, it’s regulation of the pyrimidine metabolism pathway could influence DNA replication in tissues affected by chronic hyperglycemia, such as in diabetic nephropathy85. In KC and PC, it is overexpressed in many tumor types, suggesting its role in sustaining the high proliferative rate of cancer cells83,84. Its involvement in glutathione metabolism and the p53 signaling pathway underscores its potential role in cellular stress responses, especially in cancers that depend on rapid cell division and survival under oxidative stress conditions. The enzyme’s regulation in drug metabolism pathways also points to its potential involvement in drug resistance mechanisms95. These findings demonstrate that the cGBs identified in this study—ALB, BIRC5, E2F7, MUC1, TOP2A, and RRM2—are intricately connected to key biological processes that are dysregulated in T2D, KC, and PC. Their involvement in cell cycle regulation, apoptosis, DNA damage response, and metabolism pathways highlight their potential as biomarkers and therapeutic targets for these diseases.
The Random Forest (RF) prediction model using cGBs effectively separated the three diseases (T2D, PC, and KC) from the control groups, with an AUC greater than 0.91 and an accuracy (ACC) over 0.83. This highlights the significance of cGBs in classifying these diseases (Figure S5, Table S11). Box-plot analysis revealed that five cGBs were significantly upregulated and one was downregulated in T2D, PC, and KC compared to the control groups, supporting our findings (Figure S4). Additionally, three transcription factors (NFIC, GATA2, and KLF5) and two miRNAs (hsa-mir-16-5p and hsa-mir-103a-3p) were identified as key regulators and post-regulatory elements connected with the cGBs (Fig. 4). NFIC enhances binding accessibility in all malignancies and is significantly overexpressed in cells of KC96.In a study, GATA2, NFIC were identified as the best transcriptional regulatory signatures in PC patients97. The progression of KC toward a more aggressive stage is influenced by GATA2 proteins98. Another study reported that GATA2 is an important risk factor for T2D99. According to recent study, KLF5 is linked to the earliest stages and development of KC100. In low-grade primary pancreatic tumors and pre-neoplastic lesions, KLF5, a regulator of PC differentiation, is expressed, maintaining epithelial gene expression and promoting glandular epithelial organization in xenografts101. KLF5 is linked to renal issue related to T2D102.
We used molecular docking analysis to find possible medication candidates for the therapy of T2D, PC and KC (Fig. 5A, Table S12) and found six of the top-ranked drug agents (NVP.BHG712, Irinotecan, Olaparib, Imatinib, RG-4733 and Linsitinib) out of 434 that show strong binding affinities with the target proteins (cGBs and their TFs). Subsequently, we compared the effectiveness of these six chemical molecules against 10 independent receptors separately for each of T2D, PC and KC (published by others) that supported our results (Fig. 5B, Table S13). The literature review provided further support for the potential effectiveness of our suggested drugs as treatments for T2D, PC and KC individually. NVP.BHG71226,103,104,105, Irinotecan106,107,108, Olaparib109,110,111, Imatinib111,112,113, RG-4733114,115,116 and Linsitinib117,118,119, as displayed in (Fig. 6B). All of our proposed drugs were supported as common candidate molecules for T2D, PC, and KC based on individual studies of each disease. Among the identified candidate drugs, The FDA has approved irinotecan and Olaparib with Drug Bank (DB) accession codes (DB00762 and DB09074) for the treatment of PC106. It has been suggested that combining Olaparib with some drugs, such as irinotecan, is essential for the treatment of PC109. The kinase inhibitor NVP-BHG712 affects blood flow, oxygen shortage, and the development of cancer103. Additionally, it specifically inhibits EphB4 kinase, a potential therapeutic target for T2D and insulin resistance104,105. NVP-BHG712 has been suggested as a possible PC treatment26. According to the Drug Bank database, RG-4733 is an experimental medication being investigated for cancer (accesssion number DB11870). This new gamma secretase inhibitor, an essential part of the enzyme complex that cleaves and activates Notch, is being investigated as an anti-cancer drugs114. FDA-approved cancer drug imatinib (DB00619) has been associated with renal failure in 14% of patients with 800 mg of KC, particularly those who have had a nephrectomy in the past112. imatinib increases the anti-tumor efficaciousness of gemcitabine in drug-resistant PC xenografts113. PC development and metastasis are affected in different ways by the experimental cancer medication linsitinib (DB accession number DB06075), which inhibits insulin-like growth factor receptor-1 (IGF-1R)26,117. To validate the drug molecules computationally, we conducted ADME/T analysis and assessed their drug-likeness. This evaluation confirmed the effectiveness of the proposed drug molecules. Each identified drug molecule complied with at least four of Lipinski’s Rule of Five, demonstrating their suitability as drug candidates (Table S15). The six drug molecules that were chosen for analysis showed favorable ADME/T profiles (Table 4), high HIA percentages ranging from 68 to 99.88%, sufficient water solubility, and no carcinogenic characteristics. Among the screened drugs, NVP-BHG712, Olaparib, Imatinib, and RG-4733 stand out for their favorable drug-likeness profiles, good absorption potential, and promising molecular characteristics, making them well-suited for clinical use in treating T2D, PC, and KC. These drugs show strong pharmacokinetic properties, indicating better bioavailability and efficacy. In contrast, Irinotecan and Linsitinib are less effective due to suboptimal drug-likeness profiles, including lower absorption and permeability, though they may still be considered for combination therapies based on clinical needs. Thus, the findings of this study might be vital resources for diagnosis and therapies PC and KC patients who are also suffering from T2D.However, a limitation of this study is that the proposed T2D-, PC- and KC-causing cGBs and candidate therapeutic agents are not validated yet to the patients who are suffering from these three diseases. However, a limitation of this study is that T2D-, PC- and KC- causing cGBs and candidate therapeutic agents are not validated yet to the patients who are suffering from these three diseases.
Conclusion
Initially, we identified 78 common differentially expressed genes (cDEGs) associated with PC and KC in the context of T2D. Expression analysis of these cDEGs revealed a genetic connection between PC, KC, and T2D. We then pinpointed six top-ranked cDEGs (ALB, BIRC5, E2F7, MUC1, TOP2A, and RRM2) as common genomic biomarkers (cGBs) for these diseases. The differential expression patterns of these cGBs were validated using independent datasets from NCBI, TCGA, and GTEx databases for T2D, PC, and KC.We explored shared pathogenetic mechanisms of PC, KC, and T2D through cGBs-set enrichment analysis, including biological processes, molecular functions, cellular components, and KEGG pathways. We also conducted regulatory network analysis involving cGBs, transcription factors (TFs), proteins, and miRNAs, as well as DNA methylation analysis. Ultimately, we identified six top-ranked candidate drug molecules (NVP-BHG712, Olaparib, Imatinib, RG-4733, Irinotecan and Linsitinib) as potential treatments for PC and/or KC with T2D. Both the proposed genomic biomarkers and drug molecules were supported by literature reviews of individual studies on T2D, PC, and KC. This study’s findings provide valuable resources for developing effective common drugs for treating PC and/or KC with T2D.
Data availability
The datasets analyzed in this study were downloaded from NCBI database with the following links that are freely availablehttps://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE36895https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=gse16515https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE76896https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE71989https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE66272https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE19420https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE15932.
References
Zimmet, P., Alberti, K. G. M. M. & Shaw, J. L20 Diabetes Epidemic. 414. (2001).
Ogurtsova, K. et al. IDF diabetes atlas: global estimates of undiagnosed diabetes in adults for 2021. Diabetes Res. Clin. Pract. 183 https://doi.org/10.1016/j.diabres.2021.109118 (2022).
Zhang, P. H. et al. Increased risk of Cancer in patients with type 2 diabetes mellitus: A retrospective cohort study in China. BMC Public. Health. 12 (1). https://doi.org/10.1186/1471-2458-12-567 (2012).
Leahy, J. L. Pathogenesis of type 2 diabetes mellitus. Arch. Med. Res. 36, 197–209. https://doi.org/10.1016/j.arcmed.2005.01.003 (2005).
Psutka, S. P. et al. Diabetes mellitus is independently associated with an increased risk of mortality in patients with clear cell renal cell carcinoma. J. Urol. 192, 1620–1627. https://doi.org/10.1016/j.juro.2014.06.014 (2014).
Cheng, X. & Hou, Y. Importance of metabolic and immune profile as a prognostic Indicator in patients with diabetic clear cell renal cell carcinoma. Front. Oncol. 13, 1–13. https://doi.org/10.3389/fonc.2023.1280618 (2023).
Eibl, G. et al. Diabetes mellitus and obesity as risk factors for pancreatic Cancer. J. Acad. Nutr. Diet. 118, 555–567. https://doi.org/10.1016/j.jand.2017.07.005 (2018).
Gallo, M. et al. Early prediction of pancreatic Cancer from New-Onset diabetes: an associazione Italiana oncologia medica (AIOM)/Associazione Medici diabetologi (AMD)/Società Italiana endocrinologia (SIE)/Società Italiana Farmacologia (SIF) multidisciplinary consensus Pos. ESMO Open. 6 https://doi.org/10.1016/j.esmoop.2021.100155 (2021).
C., P. P. T. L. N. P.; M.S., R.; K., X.; A., A. Pancreatic Cancer triggers diabetes through TGF-β-Mediated selective depletion of islet β-Cells. Life Sci. Alliance 3. (2020).
, A. V. et al. Pre-Existing type 2 diabetes mellitus is an independent risk factor for mortality and progression in patients with renal cell carcinoma. Med. (United States). 93, nopagination (2014).
Overview. A.C.S.P.C. Key Statistics for Pancreatic Cancer.
Barth, D. A. et al. Current concepts of Non-Coding RNAs in the pathogenesis of Non-Clear cell renal cell carcinoma. Cancers (Basel). 11 https://doi.org/10.3390/cancers11101580 (2019).
Sung, W. W., Ko, P. Y., Chen, W. J., Wang, S. C. & Chen, S. L. Trends in the kidney Cancer Mortality-to-Incidence ratios according to health care expenditures of 56 countries. Sci. Rep. 11 https://doi.org/10.1038/s41598-020-79367-y (2021).
Bahadoram, S. et al. Renal cell carcinoma: an overview of the epidemiology, diagnosis, and treatment. G Ital. Di Nefrol. 39, 32–47 (2022).
Rahib, L. et al. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States (Cancer Research 74 (2913-21)). Cancer Res. 2014, 74, 4006. (2014).
Cancer Facts & Fig. 2024.
Tracz, A. F., Szczylik, C., Porta, C. & Czarnecka, A. M. Insulin-like growth Factor-1 signaling in renal cell carcinoma. BMC Cancer. 16 https://doi.org/10.1186/s12885-016-2437-4 (2016).
Giovannucci, E., Harlan, D. M., Archer, M. C. & Bergenstal, R. M. Diabetes and Cancer: A Consensus Report. 60, 207–221, (2010). https://doi.org/10.3322/caac.20078.Available
Pollak, M. Insulin and Insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer. 8, 915–928. https://doi.org/10.1038/nrc2536 (2008).
Coussens, L. M. & Werb, Z. Inflammation and Cancer. Nature 420, 860–867. https://doi.org/10.1038/nature01322 (2002).
Chan, M. T. et al. Effects of insulin on human pancreatic Cancer progression modeled in vitro. BMC Cancer. 14, 1–12. https://doi.org/10.1186/1471-2407-14-814 (2014).
Lucke, T. et al. Disease-Specific medication, Patient-Reported diagnoses and their relation to COPD severity for common comorbidities in COPD. PA698, (2017). https://doi.org/10.1183/1393003.congress-2017.pa698
Zhang, P., Wang, F., Hu, J. & Sorrentino, R. Label propagation prediction of Drug-Drug interactions based on clinical side effects. Sci. Rep. 5 https://doi.org/10.1038/srep12339 (2015).
Sommer, J., Seeling, A. & Rupprecht, H. Adverse drug events in patients with chronic kidney disease associated with multiple drug interactions and polypharmacy. Drugs Aging. 37, 359–372. https://doi.org/10.1007/s40266-020-00747-0 (2020).
Merel, S. E. & Paauw, D. S. Common drug side effects and drug-Drug interactions in elderly adults in primary care. J. Am. Geriatr. Soc. 65, 1578–1585. https://doi.org/10.1111/jgs.14870 (2017).
Hossen, M. B. et al. Robust identification of common genomic biomarkers from multiple gene expression profiles for the prognosis, diagnosis, and therapies of pancreatic Cancer. Comput. Biol. Med. 152, 106411. https://doi.org/10.1016/j.compbiomed.2022.106411 (2023).
Chang, H. T. Biomarker discovery using Dry-Lab technologies and High-Throughput screening. Biomark. Med. 10, 559–561. https://doi.org/10.2217/bmm-2016-0111 (2016).
Alam, M. S. et al. Integrated bioinformatics and statistical approaches to explore molecular biomarkers for breast Cancer diagnosis, prognosis and therapies. PLoS One. 17, e0268967. https://doi.org/10.1371/journal.pone.0268967 (2022).
Mosharaf, M. P., Reza, M. S., Gov, E., Mahumud, R. A. & Mollah, M. N. H. Disclosing Potential Key Genes, Therapeutic Targets and Agents for Non-Small Cell Lung Cancer: Evidence from Integrative Bioinformatics Analysis. Vaccines 10, 771, (2022). https://doi.org/10.3390/vaccines10050771
Reza, M. S. et al. Metadata analysis to explore hub of the hub-Genes highlighting their functions, pathways and regulators for cervical Cancer diagnosis and therapies. Discov Oncol. 13 https://doi.org/10.1007/s12672-022-00546-6 (2022).
Islam, M. A. et al. Exploring core genes by comparative transcriptomics analysis for early diagnosis, prognosis, and therapies of colorectal Cancer. Cancers (Basel). 15 https://doi.org/10.3390/cancers15051369 (2023).
Horaira, M. A. et al. Bioinformatics screening of Colorectal-Cancer causing molecular signatures through gene expression profiles to discover therapeutic targets and candidate agents. BMC Med. Genomics. 16, 1–24. https://doi.org/10.1186/s12920-023-01488-w (2023).
Ajadee, A. et al. Screening of differential gene expression patterns through survival analysis for diagnosis, prognosis and therapies of clear cell renal cell carcinoma. PLoS One. 19, e0310843 (2024).
Ahmmed, R. et al. H. Bioinformatics analysis to disclose shared molecular mechanisms between Type-2 diabetes and Clear-Cell Renal-Cell carcinoma, and therapeutic indications. Sci. Rep. 14 https://doi.org/10.1038/s41598-024-69302-w (2024).
Zhang, J. et al. Identification of key genes associated with the process of hepatitis B inflammation and Cancer transformation by integrated bioinformatics analysis. Front. Genet. 12 https://doi.org/10.3389/fgene.2021.654517 (2021).
Liu, G. M., Zeng, H. D., Zhang, C. Y. & Xu, J. W. Key genes associated with diabetes mellitus and hepatocellular carcinoma. Pathol. Res. Pract. 215 https://doi.org/10.1016/j.prp.2019.152510 (2019).
Zou, J. et al. Identification of key genes associated with polycystic ovary syndrome (PCOS) and ovarian Cancer using an integrated bioinformatics analysis. J. Ovarian Res. 15 https://doi.org/10.1186/s13048-022-00962-w (2022).
Jian, L. & Yang, G. Identification of key genes involved in diabetic peripheral neuropathy progression and associated with pancreatic Cancer. Diabetes Metab. Syndr. Obes. 13, 463–476. https://doi.org/10.2147/DMSO.S235011 (2020).
Peña-Llopis, S. et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 44, 751–759. https://doi.org/10.1038/ng.2323 (2012).
Ellsworth, K. A. et al. Contribution of FKBP5 genetic variation to gemcitabine treatment and survival in pancreatic adenocarcinoma. PLoS One. 8, e70216. https://doi.org/10.1371/journal.pone.0070216 (2013).
Khamis, A. et al. Laser capture microdissection of human pancreatic Islets reveals novel EQTLs associated with type 2 diabetes. Mol. Metab. 24, 98–107. https://doi.org/10.1016/j.molmet.2019.03.004 (2019). EhehaltF.Marselli, L.; Distler, M.; Richter, D.; et al.
DRUGBANK. online Advanced Kidney Cancer-DRUGBANK Online.
NIH NIH The Cancer Genome Atlas Program - NCI. Cancer Genome Atlas Progr.
Drugs.com Drugs. Used to Treat Renal Cell Carcinoma-Drugs.Com. Drugs used to treat Ren. Cell Carcinoma.
Russo, P. S. T. et al. CEMiTool: A bioconductor package for performing comprehensive modular Co-Expression analyses. BMC Bioinform. 19 https://doi.org/10.1186/s12859-018-2053-1 (2018).
Sarker, A., Aziz, M. A., Hossen, M. B., Mollah, M. M. H. & Al-Amin; Mollah, M. N. H. Discovery of key molecular signatures for diagnosis and therapies of glioblastoma by combining supervised and unsupervised learning approaches. Sci. Rep. 14, 27545. https://doi.org/10.1038/s41598-024-79391-2 (2024).
Horvath, S. & Langfelder, P. W. G. C. N. A. An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Christmas, R. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Chin, C. H. et al. Identifying hub objects and Sub-Networks from complex interactome. BMC Syst. Biol. 8, 1–7. https://doi.org/10.1186/1752-0509-8-S4-S11 (2014).
Nogales-Cadenas, R. et al. Interpreting gene lists through enrichment analysis and integration of diverse biological information. Nucleic Acids Res. 37 https://doi.org/10.1093/nar/gkp416 (2009).
Piñero, J. et al. The disgenet knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 48 https://doi.org/10.1093/nar/gkz1021 (2020).
Tang, Z., Kang, B., Li, C., Chen, T. & Zhang, Z. GEPIA2: an enhanced web server for Large-Scale expression profiling and interactive analysis. Nucleic Acids Res. 47, W556–W560. https://doi.org/10.1093/nar/gkz430 (2019).
Sing, T., Sander, O., Beerenwinkel, N. & Lengauer, T. R. O. C. R. Visualizing classifier performance in R. Bioinformatics 21, 3940–3941. https://doi.org/10.1093/bioinformatics/bti623 (2005).
Khan, A. et al. JASPAR 2018: update of the Open-Access database of transcription factor binding profiles and its web framework. 46, 260–266, (2018). https://doi.org/10.1093/nar/gkx1126
Sethupathy, P., Corda, B. & Hatzigeorgiou, A. G. TarBase: A comprehensive database of experimentally supported animal MicroRNA targets. Rna 12, 192–197. https://doi.org/10.1261/rna.2239606 (2006).
Paul, S. et al. 1;1; 2 Daniel Ramage, 2; Nada Amin, 2; Benno Schwikowski, 1, 5 and Trey Ideker2, 3, 4, 5; 山本隆久; 豊田直平; 深瀬吉邦; 大森敏行 Cytoscape: A Software Environment for Integrated Models. Genome Res. 13, 426, (1971). https://doi.org/10.1101/gr.1239303.metabolite
Dwight3, J. T. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25 (2000).
Garcia-Moreno, A. et al. Functional enrichment analysis of regulatory elements. Biomedicines 10 https://doi.org/10.3390/biomedicines10030590 (2022).
Modhukur, V. et al. MethSurv: A web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics 10, 277–288. https://doi.org/10.2217/epi-2017-0118 (2018).
Trott, O., Olson, A. J., AutoDock & Vina Improving the speed and accuracy of Docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, NA–NA. https://doi.org/10.1002/jcc.21334 (2009).
Berman, H. M. et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 58 https://doi.org/10.1107/S0907444902003451 (2002).
Varadi, M. et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-Sequence space with High-Accuracy models. Nucleic Acids Res. 50, D439–D444. https://doi.org/10.1093/nar/gkab1061 (2022).
Kim, S. et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 47, D1102–D1109. https://doi.org/10.1093/nar/gky1033 (2019).
Lipinski, C. A. Lead- and Drug-like compounds: the Rule-of-Five revolution. Drug Discov Today Technol. 1, 337–341. https://doi.org/10.1016/j.ddtec.2004.11.007 (2004).
Daina, A., Michielin, O. & Zoete, V. S. A. D. M. E. A free web tool to evaluate pharmacokinetics, Drug-Likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7 https://doi.org/10.1038/srep42717 (2017).
Pires; Blundell, T. L., Ascher, D. B. & PkCSM Predicting Small-Molecule Pharmacokinetic Properties Using Graph-Based Signatures (Theory- How to Enterpret PkCSM Result). pKCSM 5. (2015).
Graff, R. E. et al. Type 2 diabetes in relation to the risk of renal cell carcinoma among men and women in two large prospective cohort studies. Diabetes Care. 41, 1432–1437. https://doi.org/10.2337/dc17-2518 (2018).
Cho, J., Scragg, R. & Petrov, M. S. Postpancreatitis diabetes confers higher risk for pancreatic Cancer than type 2 diabetes: results from a nationwide Cancer registry. Diabetes Care. 43, 2106–2112. https://doi.org/10.2337/dc20-0207 (2020).
Wang, H. D., Zhong, Z. H., Weng, Y. J., Wei, Z. Z. & Zhang, Y. Q. Degraded sericin significantly regulates blood glucose levels and improves impaired liver function in T2d rats by reducing oxidative stress. Biomolecules 11 https://doi.org/10.3390/biom11081255 (2021).
Fan, Z. et al. The crp/albumin ratio predicts survival and monitors chemotherapeutic effectiveness in patients with advanced pancreatic Cancer. Cancer Manag Res. 11, 8781–8788. https://doi.org/10.2147/CMAR.S211363 (2019).
Wang, J. et al. Bioinformatics and functional analyses of key genes and pathways in human clear cell renal cell carcinoma. Oncol. Lett. 15, 9133–9141. https://doi.org/10.3892/ol.2018.8473 (2018).
Pei, Y., Yin, X. & Liu, X. min; qiang TOP2A Induces Malignant Character of Pancreatic Cancer through Activating β-Catenin Signaling Pathway. Biochim. Biophys. Acta - Mol. Basis Dis. 1864, 197–207, (2018). https://doi.org/10.1016/j.bbadis.2017.10.019
Yuan, L. et al. Identification of key genes and pathways in human clear cell renal cell carcinoma (CcRCC) by Co-Expression analysis. 14, (2018). https://doi.org/10.7150/ijbs.23574
Ye, M. et al. A TOP2A-Derived Cancer panel drives Cancer progression in papillary renal cell carcinoma. Oncol. Lett. 16, 4169–4178. https://doi.org/10.3892/ol.2018.9179 (2018).
Vastrad, B. & Vastrad, C. Bioinformatics Analysis of Potential Key Genes and Mechanisms in Type 2 Diabetes Mellitus. bioRxiv 2021.03.28.437386. (2021).
Duffy, M. J., O’Donovan, N., Brennan, D. J., Gallagher, W. M. & Ryan, B. M. Survivin: A promising tumor biomarker. Cancer Lett. 249, 49–60. https://doi.org/10.1016/j.canlet.2006.12.020 (2007).
Melisko, M. A. Multigene assay to predict recurrence of Tamoxifen-Treated, Node-Negative breast Cancer. Women’s Oncol. Rev. 5, 45–47. https://doi.org/10.1080/14733400500093379 (2005).
Wang, H. W., Sharp, T. V., Koumi, A., Koentges, G. & Boshoff, C. Characterization of an Anti-Apoptotic glycoprotein encoded by Kaposi’s Sarcoma-Associated herpesvirus which resembles a spliced variant of human survivin. EMBO J. 21, 2602–2615. https://doi.org/10.1093/emboj/21.11.2602 (2002).
Wu, X. et al. Survivin is required for Beta-Cell mass expansion in the pancreatic Duct-Ligated mouse model. PLoS One. 7, 1–11. https://doi.org/10.1371/journal.pone.0041976 (2012).
Hinoda, Y. et al. Increased expression of MUC1 in advanced pancreatic Cancer. J. Gastroenterol. 38, 1162–1166. https://doi.org/10.1007/s00535-003-1224-6 (2003).
Bourdon, E. et al. Ren. Carcinomas 1–14. (2024).
Strawbridge, R. J. & Rona, J. Strawbridge Stockholm 2008. (2008).
Bhutia, Y. D. et al. Differential processing of Let-7a precursors influences RRM2 expression and chemosensitivity in pancreatic cancer: role of LIN-28 and SET oncoprotein. PLoS One. 8 https://doi.org/10.1371/journal.pone.0053436 (2013).
Szarkowska, J. et al. Original Article RRM2 gene expression depends on BAF180 subunit of SWISNF chromatin remodeling complex and correlates with abundance of tumor infiltrating lymphocytes in CcRCC. Am. J. Cancer Res. 11, 5965–5978 (2021).
S0888754322001525.
Luo, L., Zhang, G., Wu, T. & Wu, G. Prognostic value of E2F transcription factor expression in pancreatic adenocarcinoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 27, e933443. https://doi.org/10.12659/MSM.933443 (2021).
Healthcare Engineering, J. O. & Retracted Comprehensive Analysis of the Expression and Prognosis for E2Fs in Human Clear Cell Renal Cell Carcinoma. J. Healthc. Eng. 2022, 9807439, (2022). https://doi.org/10.1155/2022/9807439
Ma, Q. et al. Long Non-Coding RNA screening and identification of potential biomarkers for type 2 diabetes. J. Clin. Lab. Anal. 36, 1–10. https://doi.org/10.1002/jcla.24280 (2022).
Mita, A. C., Mita, M. M., Nawrocki, S. T., Giles, F. J. & Survivin Key regulator of mitosis and apoptosis and novel target for Cancer therapeutics. Clin. Cancer Res. 14, 5000–5005. https://doi.org/10.1158/1078-0432.CCR-08-0746 (2008).
Al-Yahya, S. et al. Post-Transcriptional regulation of BIRC5/Survivin expression and induction of apoptosis in breast Cancer cells by tristetraprolin. RNA Biol. 21, 1–15. https://doi.org/10.1080/15476286.2023.2286101 (2024).
Chen, W. et al. MUC1: structure, function, and clinic application in epithelial cancers. Int. J. Mol. Sci. 22 https://doi.org/10.3390/ijms22126567 (2021).
Nath, S. & Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in Cancer progression. Trends Mol. Med. 20, 332–342. https://doi.org/10.1016/j.molmed.2014.02.007 (2014).
Kisling, S. G. et al. Implications of Prognosis-Associated genes in pancreatic tumor metastasis: lessons from global studies in bioinformatics. Cancer Metastasis Rev. 40, 721–738. https://doi.org/10.1007/s10555-021-09991-1 (2021).
Youns, M., Abdel, H. & Hegazy, W. The natural flavonoid Fisetin inhibits cellular proliferation of hepatic, colorectal, and pancreatic Cancer cells through modulation of multiple signaling pathways. PLoS One. 12, e0169335. https://doi.org/10.1371/journal.pone.0169335 (2017).
Wang, H., Guo, M., Wei, H. & Chen, Y. Targeting P53 pathways: mechanisms, structures and advances in therapy. Signal. Transduct. Target. Ther. 1–35. https://doi.org/10.1038/s41392-023-01347-1 (2023).
Wu, Y. Chromatin Accessibility Shapes Tumor Architecture and Microenvironment in Clear Cell Renal Cell Carcinoma.
Ullah, M. A., Sarkar, B. & Akter, F. Prediction of biomarker signatures and therapeutic agents from blood sample against pancreatic ductal adenocarcinoma (PDAC): A Network-Based study. Inf. Med. Unlocked. 19, 100346. https://doi.org/10.1016/j.imu.2020.100346 (2020).
Peters, I. et al. Decreased MRNA expression of GATA1 and GATA2 is associated with tumor aggressiveness and poor outcome in clear cell renal cell carcinoma. Target. Oncol. 10, 267–275. https://doi.org/10.1007/s11523-014-0335-8 (2015).
Muiya, N. P. et al. A study of the role of GATA2 gene polymorphism in coronary artery disease risk traits. Gene 544, 152–158. https://doi.org/10.1016/j.gene.2014.04.064 (2014).
Fu, R. J. et al. DNMT1-Maintained hypermethylation of Krüppel-like factor 5 involves in the progression of clear cell renal cell carcinoma. Cell. Death Dis. 8, e2952. https://doi.org/10.1038/cddis.2017.323 (2017).
Diaferia, G. R. et al. Dissection of transcriptional and Cis‐Regulatory control of differentiation in human pancreatic Cancer. EMBO J. 35, 595–617. https://doi.org/10.15252/embj.201592404 (2016).
Kyriazis, I. D. et al. KLF5 is induced by FOXO1 and causes oxidative stress and diabetic cardiomyopathy. Circ. Res. 128, 335–357. https://doi.org/10.1161/CIRCRESAHA.120.316738 (2021).
Neuber, C. et al. The Pyrazolo[3,4-d]Pyrimidine-Based kinase inhibitor NVP-BHG712: effects of regioisomers on tumor growth, perfusion, and hypoxia in EphB4-Positive A375 melanoma xenografts. Molecules 25 https://doi.org/10.3390/molecules25215115 (2020).
P., G. M. B., C., H. E. B., J., S. & M., B. P., I. The small molecule specific EphB4 kinase inhibitor NVP-BHG712 inhibits VEGF driven angiogenesis. Angiogenesis 13, 259–267 (2010).
Liu, X. et al. Insulin induces insulin receptor degradation in the liver through EphB4. Nat. Metab. 4, 1202–1213. https://doi.org/10.1038/s42255-022-00634-5 (2022).
Wishart, D. S. et al. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 36 https://doi.org/10.1093/nar/gkm958 (2008).
Chui, C. Y., Moes, D. J. A. R., Koolen, S. L. W., Swen, J. J. & Gelderblom, H. Pharmacokinetic profile of Irinotecan in patients with chronic kidney disease: two cases and literature review. Br. J. Clin. Pharmacol. 89, 2920–2925. https://doi.org/10.1111/bcp.15833 (2023).
Capello, M. et al. CES2 expression in pancreatic adenocarcinoma is predictive of response to Irinotecan and is associated with type 2 diabetes. JCO Precis Oncol. 426–436. https://doi.org/10.1200/po.19.00330 (2020).
Yarchoan, M. et al. Olaparib in combination with Irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic Cancer. Oncotarget 8 https://doi.org/10.18632/oncotarget.17237 (2017).
Wang, Z., Gao, J., Ohno, Y., Liu, H. & Xu, C. Rosiglitazone ameliorates senescence and promotes apoptosis in ovarian Cancer induced by Olaparib. Cancer Chemother. Pharmacol. 85, 273–284. https://doi.org/10.1007/s00280-019-04025-8 (2020).
Oncologist, T. Kidney Cancer Research Summit Abstracts. Oncologist 2023, 28, S1–S1, (2023). https://doi.org/10.1093/oncolo/oyad216.001
Vuky, J. et al. Phase II trial of Imatinib (Gleevec®) in patients with metastatic renal cell carcinoma. Invest. New. Drugs. 24, 85–88. https://doi.org/10.1007/s10637-005-4543-z (2006).
Shi, Y. H., Xu, Q. C., Zhu, Y. Q., Liu, Z. & De Imatinib facilitates gemcitabine sensitivity by targeting epigenetically activated PDGFC signaling in pancreatic Cancer. Mol. Ther. 31, 503–516. https://doi.org/10.1016/j.ymthe.2022.11.004 (2023). ZhaoG.Y.Liu, Q.; Wang, X.Y.; Wang, J.Q.; Xu, X.; Su, Q.; et al.
Venkatesh, V. et al. Target. Notch Signal. Pathw. Cancer Stem Cells doi:https://doi.org/10.21037/sci.2018.02.02. (2018).
S030573721400125X.
Cho, E. S., Kang, H. E., Kim, N. H. & Yook, J. I. Therapeutic implications of Cancer Epithelial-Mesenchymal transition (EMT). Arch. Pharm. Res. 42, 14–24. https://doi.org/10.1007/s12272-018-01108-7 (2019).
Zheng, Y. et al. Insulin-like growth factor 1-Induced enolase 2 deacetylation by HDAC3 promotes metastasis of pancreatic Cancer. Signal. Transduct. Target. Ther. 5 https://doi.org/10.1038/s41392-020-0146-6 (2020).
Fassnacht, M. et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: A Double-Blind, randomised, phase 3 study. Lancet Oncol. 16, 426–435. https://doi.org/10.1016/S1470-2045(15)70081-1 (2015).
Xu, K. et al. Construction of five Disulfidptosis-Related LncRNA signature for predicting prognosis and immune activity in kidney renal clear cell carcinoma. (2023).
Author information
Authors and Affiliations
Contributions
Conceptualization: A.A. and M.N.H.M. Data curation and processing: A.A., S.M. and T.N. Transcriptomic data analysis: A.A and A.S. Molecular docking analysis: A.A. and S.M. Methodology: A.A., T.N. and M.N.H.M, Validation & Visualization: R.A and S.M., Writing – original draft: A.A. Writing - review & editing: R.A., A.S. & M.N.H.M. Project administration & Supervision: M.N.H.M. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ajadee, A., Mahmud, S., Sarkar, A. et al. Screening of common genomic biomarkers to explore common drugs for the treatment of pancreatic and kidney cancers with type-2 diabetes through bioinformatics analysis. Sci Rep 15, 7363 (2025). https://doi.org/10.1038/s41598-025-91875-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-91875-3
