
Abstract
Stenotrophomonas maltophilia are opportunistic, multi-drug-resistant Gram-negative pathogens increasingly prevalent in clinical settings. Quorum sensing (QS) systems play a crucial role in their pathogenesis, coordinating bacterial populations and enabling interactions within polymicrobial communities. While not the primary QS mechanism in S. maltophilia, these bacteria can respond to acyl-homoserine lactone (AHL)-type autoinducers. Some isolates exhibit AHL-quorum quenching activity, though the responsible components remain unidentified. Homology searches in S. maltophilia K279a revealed a protein with the locus tag SMLT_RS07305 (old locus tag Smlt1522), annotated as a putative penicillin acylase 2 precursor. Sequence and structural analyses classify this protein within the bacterial AHL-acylase group B, characterized by a heterodimeric structure consisting of α- and β-subunits connected by a spacer polypeptide. We experimentally confirmed the dual activity of Smlt1522 as an AHL-acylase and a penicillin acylase. This protein degrades AHLs with varying acyl chains and hydrolyses penicillin antibiotics in vitro, in vivo, and as an heterologously expressed product. Its physiological role includes the modulation of beta-lactam resistance, biofilm formation and bacterial fitness under specific conditions. Evolutionary analysis suggests structural and functional conservation, pointing to its potential role in the adaptation of S. maltophilia to diverse and competitive environments.
Similar content being viewed by others

Introduction
Bacteria possess the ability to swiftly adapt to changing environments by modulating complex social behaviours via a physiological mechanism known as quorum sensing (QS). This form of cellular communication enables bacteria to cooperate and compete within the same species or between different species. They achieve this by the production, release, and detection of extracellular signalling molecules known as autoinducers (AIs)1. Through the detection of these AIs, bacteria are capable of sensing and responding to changes in cell density by altering gene expression. Consequently, QS facilitates the synchronization of gene expression within bacterial populations, allowing for coordinated activities such as biofilm formation, virulence factor production, motility regulation and both the production of and resistance to antimicrobials2,3.
Among Gram-negative bacteria, the most important AIs used in QS communication are N-acyl-L-homoserine lactones (AHLs)4. The concept of targeting AHLs to degrade them externally, known as quorum quenching (QQ), is particularly appealing as a strategy to potentially modulate the virulence of certain pathogens by disrupting QS within competitive environments, like the rhizosphere or the lungs of cystic fibrosis patients5,6. QQ-based strategies have gained importance in various areas for combating bacterial biofilms, including healthcare, industry, aquaculture, and water treatment7. Furthermore, this approach aims to reduce pathogenicity without promoting antibiotic resistance8, which is essential for the design of new antimicrobials.
To date, three primary QQ enzymes to target AHL molecules have been described: AHL-acylases, which cleave the amide linkage between the acyl chain and homoserine lactone (HSL) ring, AHL lactonases, which break the ester bond within the HSL ring, and AHL oxidoreductases, which modify the acyl side chain’s third carbon9. The hydrolysis by acylases and lactonases typically results in AHLs losing their activity, highlighting these enzymes as potent QS inhibitors10. Most studied AHL-acylases belong to the N-terminal nucleophile (Ntn) hydrolase superfamily. These enzymes exhibit an αββα fold and possess a nucleophilic catalytic residue at the N-terminus11. They are synthesized as a single polypeptide chain comprising a signal peptide, an α-subunit, a spacer peptide, and a β-subunit, with the α- and β-subunits typically weighing around 20 and 60 kDa, respectively12,13,14. Due to the structural similarities between AHLs and beta-lactam antibiotics, which both contain an acyl side chain, a few bifunctional AHL-acylases have been identified. These enzymes not only disrupt intercellular communication but also mediate beta-lactam antibiotic resistance15,16,17. This dual functionality has also been observed in other enzymes from this superfamily, such as penicillin acylases, a few of which have been reported to exhibit AHL acylase activity18,19.
In the ubiquitous Gram-negative bacterium Stenotrophomonas maltophilia, no canonical AHL synthases have been identified20, although AHL-like activities have been documented within the Stenotrophomonas genus21. Nevertheless, S. maltophilia can respond to AHL signals via its unique LuxR-type receptor SmoR. This receptor specifically recognizes N-(3-Oxooctanoyl)-homoserine lactone (3OC8-HSL), increasing bacterial motility22. Furthermore, transcriptomic studies demonstrated that S. maltophilia regulates the expression of a set of genes when AHLs are added exogenously23. On the other hand, regarding QQ, certain strains of S. maltophilia isolated from the microbiota of aquatic organisms can degrade AHLs24,25. More recently, a novel AHL lactonase was identified in S. maltophilia with antivirulence activity against Pseudomonas aeruginosa26. These findings highlight the bacterium’s ability to detect and control AHL molecules in its environment and adjust its gene expression accordingly, underscoring the adaptive mechanisms that facilitate its survival and behaviour in diverse ecological niches23. Although the most studied QQ mechanisms in S. maltophilia involve disruption of AHL signalling, the endogenous molecular mechanisms responsible for the degradation of QS AIs are not fully understood, especially regarding AHL-acylases.
In this study, we identified a novel candidate gene (old locus tag Smlt1522 in the K279a genome) encoding a bifunctional acylase in S. maltophilia, based on structural analysis and its phylogenetic relationship with previously characterized acylases in other bacteria. The catalytic function of this protein was experimentally investigated by loss-of-function mutagenesis and overexpression of the coding gene. The protein, whether expressed homologously or heterologously, demonstrated the ability to degrade both AHLs and penicillin antibiotics in vitro and in vivo. Importantly, we also detected, for the first time, the production of AHL-like molecules in S. maltophilia, specifically in the mutant strain lacking this enzyme, further elucidating its role in bacterial physiology. The identification and characterization of QQ enzymes such as Smlt1522 provide deeper insights into bacterial survival strategies and present promising opportunities for therapeutic development. This bifunctional acylase may represent a group of bacterial enzymes that have evolved to retain broad substrate specificity, enabling precise adaptation to environmental challenges in competitive niches.
Results
Smlt1522 belongs to the bacterial N-acyl-L-homoserine lactones acylase group B
In the S. maltophilia K279a genome, the gene with locus tag SMLT_RS07305 (old locus tag Smlt1522) encodes a protein with 775 amino acids, which was annotated as a penicillin acylase family protein by automated computational analysis using protein homology. This protein is present and relatively conserved (80–100% identity) in all S. maltophilia strains whose sequenced genomes have been deposited in the NCBI, as well as in other species of the same genus. In many of these strains this protein is annotated as AHL acylase PvdQ, most likely due to its remote homology (41% similarity, 22% query coverage) to the well-studied AHL-hydrolysing enzyme PvdQ from P. aeruginosa27. To decipher the possible function and evolutionary origin of this protein in S. maltophilia, we first compared the Smlt1522 sequence with other AHL-acylases and potential homologues.
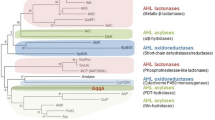
Protein Smlt1522 belongs to the Interpro family Penicillin/GL-7-ACA/AHL/aculeacin-A acylase (code IPR002692) representing self-cleaving precursor proteins of Ntn acylases. We conducted a phylogenetic analysis of bacterial Ntn hydrolase proteins based on their amino acid sequences and have found that they are categorized into three groups as proposed by Daly, J. W. et al.28 : the beta-lactam acylase group, and AHL-acylase groups A and B (Fig. 1a). Smlt1522 was clustered into the AHL-acylase group B, thereby becoming the second bifunctional AHL-acylase identified in this group alongside PfmA from Pseudoalteromonas flavipulchra29. Previously, other bifunctional acylases have been reported within AHL-acylase group A (AhlM, AuAHLA and MacQ) or within the beta-lactam acylase group (KcPGA). Members of the AHL-acylase group B included in this analysis are more divergent from each other, for example, Smlt1522 shares only 41% similarity with PfmA, over their entire sequences.
The precursor protein Smlt1522, like others from the same family13,14, is composed of an N-terminal signal peptide (32 first amino acids) for secretion, and an α-subunit and a β-subunit separated by a spacer polypeptide. In Smlt1522, the spacer polypeptide has been predicted to start at amino acid A240 and ends in G253 (Supplementary Figs. 1 and 2). The AlphaFold prediction for the full sequence of Smlt1522 (Fig. 1b) yielded five models with pLDDT scores between 95.03 and 93.67, showing great confidence in both subunits, leaving the region predicted as the spacer polypeptide with lower score (Supplementary Fig. 2). A superposition of all models over the best-ranking one resulted in root-mean-square deviation (RMSD) values below 0.3 Å, indicating a high similarity between them. Superposing each subunit independently from structures 4HSR and 4YF9 over the predicted model (Supplementary Table 1) allowed us to predict the active site residues (Supplementary Tables 2 and Supplementary Figs. 1 and 3) including the catalytic serine S254 (Fig. 1b and Supplementary Fig. 1). Residue S254 defines one cleavage site for the spacer polypeptide and the N-terminal residue of the β-subunit (Ser1β) which can act as a nucleophile to start amide bond cleavage of different substrates14, typical of beta-lactam acylases.

Smlt1522 belongs to the bacterial N-acyl-L-homoserine lactone acylase group B (a) Phylogenetic analysis using the Neighbor-Joining method based on precursor protein sequences of Smlt1522 and other acylases from the Ntn-hydrolase superfamily identified experimentally. The phylogenetic tree was divided into three groups: Beta-lactam acylase group (green) and AHL acylase groups A and B (yellow). Known bifunctional acylases are indicated in red and Smlt1522 is highlighted in green. Ntn-hydrolases with reported AHL acylase activity (NCBI accession numbers): AhlM, from Streptomyces sp. M664 (AAT68473); SlPVA, from Streptomyces lavendulae ATCC 13,664 (AAU09670); AuAHLA, from Actinoplane utahensis NRRL 12,052 (WP_052163432); MacQ, from Acidovorax sp. strain MR-S7 (BAV56778); AiiD, from Ralstonia sp. XJ12B (AAO41113); QqaR, from Deinococcus radiodurans R1 (WP_010889514); HacA, from Pseudomonas syringae B728a (AAY37014); PvdQ, from Pseudomonas aeruginosa PAO1 (AAG05773); Aac, from Shewanella sp. strain MIB015 (BAF94155); HacB, from P. syringae B728a (AAY39885); AiiC, from Nostoc sp. strain PCC 7120 (BAB75623); Smlt1522, from S. maltophilia K279a (CAQ45057); QuiP, from P. aeruginosa PAO1 (AAG04421) and PfmA, from Pseudoalteromonas flavipulchra JG1 (ASS36259). Ntn-hydrolases with reported penicillin G acylase activity (NCBI accession numbers): KcPGA, from Kluyvera cryocrescens ATCC 21,285 (AAA25047); EcPGA, from Escherichia coli MG1655 (HBP1287286); PrPGA, from Providencia rettgeri (AAP86197); AcPGA, from Achromobacter sp. strain CCM 4824 (AAY25991); AfPGA, from Alcaligenes faecalis ATCC 19,018 (AAB71221); BmPGA, from Priestia megaterium ATCC 14,945 (AAB41343) and AvPGA, from Rhizobium viscosum ATCC 15,294 (AAA22077). (b) AlphaFold predicted structure of Smlt1522 without the signal peptide (residues from 33 to 775). α-subunit depicted in yellow, β-subunit in green, spacer polypeptide in orange and residue S254 (Ser1β) represented as spheres.

In vivo AHL-degrading activity in S. maltophilia is dependent on Smlt1522. In all bioassays shown, the blue halo in the agar plates indicates AHL detection by the biosensor strain A. tumefaciens KYC55. In each experiment, 20 µL of culture supernatants from the indicated strains were previously incubated with the specified AHLs for 24 h and spotted on the bioassay plate (indicated with an x). (a) Diameter of the AHL detection halos, comparing the wild-type S. maltophilia K279a strain with a smlt1522-deficient strain, and a sterile control which involved incubating the medium with AHLs without any microorganisms, thus no degradation was expected. This demonstrates that the wild-type strain can degrade AHLs, while the smlt1522 mutant completely loses this activity. Tukey’s multiple-comparison test (two-way ANOVA) was used to determine the significance of the data between groups (***, p < 0.001). (b) Molecular structure of the tested AHLs (N-octanoyl-HSL, C8-HSL; N-(3-Oxooctanoyl)-HSL, 3OC8-HSL; N-decanoyl-HSL, C10-HSL). (c) AHL-degrading activity in the presence of C8-HSL, showing that complementing the smlt1522 mutation achieves total degradation of the present AHLs. (d) Bioassays showing how E. coli DH5α acquires the capability to degrade AHLs when Smlt1522 is heterologously expressed.
S. maltophilia K279a can degrade AHLs through Smlt1522
The intrinsic AHL-acylase activity of S. maltophilia K279a was first evaluated using a bioassay based on the biosensor strain Agrobacterium tumefaciens KYC55, chosen for its high sensitivity and variability in detecting a broad range of exogenous AHLs. The lacZ indicator gene, which is regulated by the TraR-AHL complex, facilitates the detection of exogenous AHLs by forming a blue halo in the assay plates. This colour change is made possible by the hydrolysis of X-gal in the medium, which is catalysed by the enzyme β-galactosidase30. As illustrated in Fig. 2, S. maltophilia K279a can significantly degrade various AHLs (C8-HSL, 3OC8-HSL, C10-HSL) added exogenously in the medium at concentrations of 5 or 10 µM. In this assay, we also examined a mutant K279a strain in which the smlt1522 gene was deleted (K279aΔsmlt1522). This mutant strain completely lost the ability to degrade exogenously added AHLs, as evidenced by the absence of a reduction in the blue halo on the biosensor plates compared to the untreated group (Fig. 2). Encouraged by this result, we wanted to assess the impact of the lack of a protein with AHL degrading activity on AHL production by S. maltophilia K279a, although neither the synthesis of AHLs nor the presence of canonical AHL synthases has been demonstrated in this species so far. Of note, ethyl acetate extracts from concentrated culture supernatants resulted in the formation of a blue halo around the sample of the mutant in contrast to that of the wild-type strain (Supplementary Fig. 4), indicating an increased amount of AHL-like molecules secreted by the mutant. It is therefore attractive to speculate that the mutant can develop AHL-like activity, suggesting the existence of a non-canonical AHL synthase in S. maltophilia.
By complementing the deleted gene in the mutant strain K279aΔsmlt1522 with a broad host range replicative vector carrying the wild-type gene, the ability to degrade AHLs was restored (Fig. 2c). Moreover, it was observed that the complemented strains gained an increased ability to degrade AHLs to undetectable levels in the bioassay (Fig. 2c), probably due to the multicopy effect of the complementation plasmid and the promoter used. The AHL-acylase activity of Smlt1522 was also studied in vivo in a heterologous system. An E. coli DH5α strain carrying the complementation vector was shown to degrade chemically different AHLs to levels undetectable by the bioassay (Fig. 2d). This further demonstrates the AHL-acylase activity of Smlt1522 and its ability to degrade a variety of AHLs.
Purified recombinant Smlt1522 heterologously expressed in E. coli shows penicillin-degrading activity
After several attempts to produce the Smlt1522 protein heterologously, we were successful in introducing an E. coli codon-optimized DNA fragment that expressed the protein precursor (without the N-terminal signal peptide) of the putative AHL-acylase into E. coli Origami™ B (DE3). Although the production yield was low under these conditions, it was possible to purify a sufficient amount of the recombinant protein for activity assays (Supplementary Fig. 5). The Immobilized-metal affinity chromatography (IMAC) showed five eluted peaks, and protein fractions with the highest purity content, corresponding to elution peaks 3, 4 and 5, were pooled, dialyzed, and concentrated separately for further analysis (Supplementary Fig. 5a). A major protein band of approximately 80 kDa was present in these protein peaks along with several other smaller protein bands, with a higher proportion of the 80 kDa protein band in peaks 4 and 5 compared to peak 3. This suggests that the precursor polypeptide was not fully processed into the two subunits in E. coli using this expression system (expected molecular weight of 23 kDa for α-subunit, 57 kDa for β-subunit), and contamination or nonspecific proteolysis was detected. The retention of the spacer polypeptide in the recombinant version of the protein was also verified by peptide mass fingerprinting (Supplementary Fig. 5b). After chromatography the protein purity was sufficient for enzymatic activity characterization. An initial screening of the activity of the three elution peaks against two penicillins was performed. The penicillin-degrading activity of the protein peaks against piperacillin was detected in all peaks, with the highest activity in peak 3 (Supplementary Fig. 6). Additionally, remarkable activity was detected in all peaks in contact with ampicillin. The higher activity of protein peak 3 against piperacillin of the ureidopenicillin class could be explained by the presence of more processed subunits as seen by SDS-PAGE analysis (Supplementary Fig. 5a). The susceptibility assay to determine the activity of the elution peaks was carried out using both P. aeruginosa and Staphylococcus aureus as indicator bacteria and ampicillin or piperacillin as target antibiotics. From this point forward, all subsequent assays were performed with the protein purified from this elution peak containing the highest enzymatic activity against both tested antibiotics.
Smlt1522 has dual activity capable of degrading diverse AHLs and penicillin antibiotics
To confirm whether Smlt1522 can degrade both AHLs and penicillins, we conducted in vitro degradation assays for a diverse set of AHLs and beta-lactam antibiotics using the purified recombinant variant of the protein. Dose-dependent degradation by the enzyme was observed for the penicillin antibiotics ampicillin and piperacillin and for the AHLs C8-HSL, 3OC8-HSL and C10-HSL (Fig. 3a and Supplementary Fig. 7). The enzyme’s ability to degrade other beta-lactam antibiotics, including carbapenems and cephalosporins, was also investigated, but no activity was detected for non-penicillin beta-lactams (Supplementary Fig. 7b). In this experiment, using S. aureus as indicator strain, partial degradation of ticarcillin was detected. Interestingly, the observed activity against piperacillin was inhibited by the beta-lactamase inhibitor tazobactam. Tazobactam, a structural analog of penicillins, irreversibly binds to beta-lactamases and can also interact with penicillin-binding proteins (PBPs)31. Our findings propose a novel role for tazobactam in inhibiting penicillin acylases, warranting further investigation.

Dual enzymatic activity of purified recombinant Smlt1522. (a) Antibiograms and AHL bioassays to detect the presence of penicillins and AHLs after incubation with increasing concentrations of the enzyme Smlt1522. Five microliters of each indicated solution were applied to each disk. It was observed that higher concentrations of Smlt1522 lead to a greater loss of antibiotic activity, indicating degradation, and a lower amount of AHLs, also indicating their degradation by the enzyme. P. aeruginosa PAO1 and S. aureus Newman were used as indicator strains in the antibiograms. (b,c) GC-MS metabolite analyses of C10-HSL and piperacillin degradation products of Smlt1522 activity. The mass spectra corresponding to the untreated molecules can be seen in Supplementary Fig. 8. (d) Conceptual diagram of the deacylation reactions catalysed by Smlt1522 for N-acylhomoserine lactones (AHLs) and beta-lactam antibiotics. The diagrams illustrate the fundamental structures of C10-HSL and piperacillin as well as the respective reaction mechanisms for their degradation.
To conclusively demonstrate the nature of molecular cleavage by Smlt1522, we utilized gas chromatography-mass spectrometry (GC-MS) to monitor the generation of homoserine lactone from the deacylation of C10-HSL and (R)-2-(4-Ethyl-2,3-dioxopiperazine-1-carboxamido)-2-phenylacetic acid lactone from the deacylation of piperacillin. The corresponding compounds were identified with GC retention times of 13.32 min and 21.17 min. Mass spectrometric analysis of these GC fractions revealed [M-H] ions at m/z values of 101 and 316 (Fig. 3b, c), corresponding to the molecular weights of homoserine lactone and (R)-2-(4-Ethyl-2,3-dioxopiperazine-1-carboxamido)-2-phenylacetic acid, respectively. These results definitively confirmed that Smlt1522 has bifunctional deacylating activity and can efficiently degrade both AHLs and beta-lactam antibiotics (Fig. 3d).
Knowing that Smlt1522 displays a beta-lactam degrading activity, we investigated the antibiotic resistance mediating ability of Smlt1522 using the MIC method. The MICs for S. maltophilia K279a against a range of beta-lactams were determined and compared with those of the mutant strain K279aΔsmlt1522 and its complemented variant (Table 1). No differences in MIC values of more than 2-fold were observed, most likely due to the high intrinsic resistance of S. maltophilia K279a to this antibiotic family by the presence of the genes for two major beta-lactamases, the metallo-beta-lactamase BlaL1 and the Ambler class A beta-lactamase BlaL232. However, it is important to highlight the increased susceptibility of the mutant to piperacillin and a modest two-fold increase in resistance to some of these antibiotics in the strain carrying multiple copies of the gene smlt1522 on a plasmid for complementation (Table 1). The susceptibility profile to beta-lactams was also determined in an E. coli DH5α strain carrying the complementation plasmid, which expresses Smlt1522 heterologously. In this case, a four-fold increase in resistance to both ampicillin and piperacillin (penicillin antibiotics) was observed compared to the control strain carrying the empty vector (Table 1), indicating that Smlt1522 can confer beta-lactam antibiotic resistance to a more susceptible host strain by hydrolysing penicillin antibiotics.
Potential physiological role of Smlt1522 in S. maltophilia
The mutant strain K279aΔsmlt1522 showed somewhat different phenotypes compared to its wild-type variant for QS-regulated activities such as growth kinetics, biofilm formation, and cell motility (Fig. 4). When these strains were grown in a rich medium such as LB, no growth defect was detected (Fig. 4a). In the minimal medium MMGC, however, the mutant strain showed a more extended lag phase (Fig. 4b). Remarkably, the mutant only showed this deviating growth pattern when the cells were derived from overnight cultures grown in the same minimal medium. When the inoculum was derived from cultures grown in LB, no delayed lag phase was observed (Supplementary Fig. 9), suggesting that the lack of the Smlt1522 protein has negative effects on metabolic fitness and the preparation of cells for the onset of replication in a nutrient-deficient medium. In this minimal medium, the mutant cells have an increased capacity to form biofilms under static growth conditions (Fig. 4d). Swarming was evaluated on BM2 agar medium because we have observed that the iron-limited minimal medium MMGC hinders this motility in S. maltophilia. Under these conditions, limited swarming motility in BM2 medium could be observed for the smlt1522 deletion strain (Fig. 4e). Growing in BM2 medium, a delayed lag phase in growth was also observed for the mutant strain, however there were no differences in biofilm formation (Supplementary Fig. 9). In all experiments the complemented mutant strain carrying plasmid pBBR1-BAD-smlt1522 showed phenotypes quite similar to the wild-type strain.

Smlt1522 plays a role in QS-related phenotypes in S. maltophilia grown in minimal media. (a,b) Growth profiles under different growth conditions. Bacterial cells were grown in LB (a) or the minimal medium MMGC (b), in microtiter plates at 30 °C and with continuous shaking. Optical density at 550 nm (OD 550 nm) was measured every 15 min for 24 h. To prepare the initial inoculum, the cells were grown overnight in the same assay medium. (c,d) Biofilm formation on polystyrene surface quantified by crystal violet staining. Cells were grown statically in a microtiter plate in LB (c) or MMCG (d) at 30 °C. Biofilm formation was normalized by cell growth and reported as relative biofilm formation. Tukey’s multiple-comparison test (one-way ANOVA) was used to determine the significance of the data between groups (ns, non-significant; **, p < 0.01; ***, p < 0.001). (e) Bacterial motility on BM2 swarming agar plates after incubation at 30 °C for four days. Swarming experiments were done in triplicate and representative images are shown. All assays include the wild-type strain S. maltophilia K279a, its isogenic mutant strain K279aΔsmlt1522 and the complemented strain carrying plasmid pBBR1-BAD-smlt1522.
Sequence variability and selective pressure detected on gene smlt1522 among S. maltophilia strains
Due to the suspected role of Smlt1522 in adaptation to the environment, we finally decided to investigate what evolutionary forces have acted on its encoding gene. The complete nucleotide sequences coding for our AHL-acylase were obtained from 82 available genomes of S. maltophilia strains collected from different places and sources and codon aligned. Based on these sequences, this gene was found to have an overall pairwise nucleotide sequence identity of 92.1% (amino acid identity of 94.4%), with a number of polymorphic nucleotide sites in the codon alignment of 1,000. Along the alignment, a high variability was found at both nucleotide and amino acid level, affecting both subunits of the protein (Fig. 5). Interestingly, the sequence comprising the spacer polypeptide accumulates numerous sites of variability. We used the codon alignment to evaluate recombination and positive selection acting on this gene. Five significant recombination breakpoints were identified which were used to split the alignment for positive selection analysis. Only one potentially positively selected codon position (L362; p < 0.01) was identified in the β-subunit region of the protein by the SLAC method. This site, which is subject to diversifying selection, was located on the protein surface without interfering with the catalytic site. On the contrary, highly significant purifying selection was detected at 299 sites distributed throughout the entire sequence, suggesting that despite the observed sequence variability, most of the amino acids are under strong structural constraints to maintain protein function.

Diversity of the coding region of Smlt1522 among S. maltophilia strains. (a) Variability profile generated from Smlt1522 precursor protein sequence alignment from residue A32 to the end. Shanon Entropy was employed in this case. Higher values represent higher variability in that particular position in the alignment. The α and β subunits are indicated, as well as the spacer polypeptide (SP). (b) Line graph reporting dn–ds values estimated for each codon position of the Smlt1522 coding region. The rhombus indicates a position under positive selection. Amino acid and codon positions are represented according to the K279a sequence. The amino acids positions correspond to the sequence of protein Smlt1522 with Uniprot entry B2FIC3.
Discussion
Here we report a novel enzyme in S. maltophilia belonging to the Ntn hydrolase superfamily with bifunctional QQ and penicillin-degrading activity. Ntn hydrolase proteins are produced as inactive precursor polypeptides that undergo self-cleavage for activation, resulting in a mature hetero-dimeric enzyme consisting of an α- and β-subunit14,33. The S. maltophilia protein Smlt1522 retains the structural fold characteristic of this family, based on its closest homologs with resolved 3D structure, i.e. MacQ from Acidovorax sp.14 and a cephalosporin acylase from Pseudomonas sp.34. Based on these structural similarities, the active site, the spacer polypeptide, and the nucleophilic catalytic serine residue (Ser1β) could be identified in the AlphaFold-generated model. Recombinant expression of Smlt1522 in E. coli shown in this study produced mostly the uncleaved protein, as SDS-PAGE analysis revealed a major band at around 80 kDa and the spacer polypeptide was detected by mass spectrometry in the purified protein. Even so, the enzymatic activity of this protein towards both AHLs and penicillin substrates was not affected. Although the substrate binding cavity is shielded by the spacer polypeptide13, subsequent studies with MacQ and PvdQ revealed that self-cleaving maturation is not essential for enzymatic activation14,35. In addition to incomplete processing into subunits, the recombinant production of Smlt1522 poses further challenges, such as a possible conformational heterogeneity evidenced by the elution of multiple protein peaks with distinct activities. An heterogeneous enzyme sample was also observed during the purification of PvdQ, because the spacer peptide remained attached to the α-subunit35. On the other hand, in some cases the elution of His-tagged proteins in multiple elution peaks with varying activities has been attributed to the differential interactions of protein subpopulations within a complex mixture of protein conformers36.
We have demonstrated both in vivo and in vitro the ability of the protein Smlt1522 to degrade both AHL with different side chains and penicillin antibiotics. Comparative analyses revealed that such enzymes have structural features that enable broad substrate specificity, which may have evolved from ancestral QQ enzymes that diversified under selective pressures12,34,37,38. The dual activity of Smlt1522 reflects a similar bifunctionality observed in other related bacterial enzymes15,16,17,18,28,29,39. This duality suggests a convergent evolutionary adaptation that allows these enzymes to respond to various environmental stresses such as antibiotic exposure and QS interference. While AHL degradation disrupts QS-mediated behaviours in polymicrobial communities, penicillin acylase activity provides resistance to beta-lactam antibiotics, a critical survival trait in competitive or hostile environments40,41. S. maltophilia are environmental organisms that inhabit moist environments and are mainly associated with the plant rhizosphere42, a highly competitive niche. As an opportunistic human pathogen, the presence of such an enzyme does not pose an additional risk for the development of resistance to beta-lactam antibiotics as penicillin acylases are not usually considered a primary resistance mechanism. However, the activity of the protein Smlt1522 could shape the susceptibility profile of less resistant strains to beta-lactams, and, on the other hand, it could help to clean the environment of these molecules, thereby preventing the development of resistance in microbial communities43. Moreover, the unexpected detection of AHL-like activity in the mutant strain suggests autoregulatory interplay between QQ and potential QS pathways, indicating a more complex ecological role for this enzyme. Given that S. maltophilia senses exogenous AHLs via the LuxR solo SmoR to regulate behaviours like swarming motility22, Smlt1522 might modulate such processes by influencing the availability of environmental AHLs as seen for some strains24. In any case, the mutant strain lacking Smlt1522 is a valuable tool to identify the gene(s) encoding the observed AHL-like activity in future studies.
QQ serves as an adaptive bacterial strategy to interfere with QS systems, disrupting intercellular communication that regulates collective behaviours such as biofilm formation, motility, and virulence factor production44. The data of this study allow us to draw the conclusion that the physiological role of Smlt1522 may extend beyond substrate degradation in S. maltophilia. Deletion mutant lacking Smlt1522 showed delayed growth kinetics, enhanced biofilm formation, and impaired swarming motility, especially under nutrient-limited conditions and when using specific minimal media. The growth defects in the mutant strain suggest that Smlt1522 may contribute to bacterial fitness by fine-tuning metabolic and behavioural responses through the control of the accumulation of AHL-like molecules and/or participation in the metabolism of other compounds. It has already been shown that, when exposed to AHL molecules, S. maltophilia can regulate the expression of genes related to oxidation-reduction and energetic processes, transcription and amino acid and nitrogen metabolism23. In other organisms, AHL-acylases and related enzymes have been reported to perform additional energy-providing or energy-consuming processes. For example, it has been suggested that degradation of AHLs in marine environments, in addition to its role in QQ, could also serve the purpose of using these signals as an alternative energy source45. On the other hand, PvdQ plays an additional role in pyoverdine biosynthesis in P. aeruginosa by removing an incorporated fatty acid that is not present in mature pyoverdine46. Mutants deficient in PvdQ and related enzymes in P. aeruginosa showed defective biofilm formation and swarming motility phenotypes47,48. The reduced swarming motility of the pvdQ mutant was influenced by differences in the production of rhamnolipids and iron deficiency in the medium, but not by the accumulation of AHLs48. Our findings also highlight the need for further investigation into the role of iron in QQ-related processes in S. maltophilia. Iron serves as a key signalling element playing a crucial role in biofilm formation, the production of extracellular virulence factors, responses to oxidative stress, QS via diffusible signal factors, and siderophore synthesis in S. maltophilia49.
The evolutionary conservation of Smlt1522, despite sequence variability, highlights its importance in the adaptive strategies of S. maltophilia. The strong purifying selection detected across its sequence suggests that its bifunctional activity and structural integrity are critical for its role in environmental adaptation. Our findings contribute to a broader understanding of QQ mechanisms and their evolutionary relationships with antibiotic resistance. The suggested evolutionary link between AHL acylases and beta-lactam acylases, as well as the overlapping functions of antibiotics and known quorum-sensing molecules39,50, aligns with the structural and functional similarities observed in the protein Smlt1522 and related enzymes. This dual role in QS interference and antibiotic resistance highlights the ecological and clinical importance of enzymes like Smlt1522, offering valuable insights into bacterial survival strategies and identifying potential targets for antimicrobial development.
Methods
Bacterial strains and general growth conditions
All bacterial strains and plasmids used in this work are listed in Table 2. S. maltophilia K279a served as the wild-type and reference strain51, and Agrobacterium tumefaciens KYC55 functioned as the AHL-reporter strain30. Pseudomonas aeruginosa PAO1 and Staphylococcus aureus Newman (Table 2) were used as indicator strains in susceptibility tests.
Unless otherwise specified, all strains were routinely cultured at 37 °C in Lysogeny Broth (LB) medium (10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl, pH 7.5) on a rotary shaker at 200 rpm, supplemented with the appropriate antibiotics as needed. Antibiotic susceptibility assays in liquid and in agar plate were done in Mueller-Hinton broth (MHB). For other phenotypic assays, two minimal media were used throughout this work. Modified BM2-glucose minimal medium52 was composed of 62 mM potassium phosphate buffer, pH 7.0, 2 mM MgSO4, 10 µM FeSO4, 0.5% casamino acids and 0.4% glucose. An iron-limited minimal medium enriched with a carbon source and casamino acids (MMGC) was prepared according to Xavier et al.53 with minor modifications. It contained a phosphate buffered solution (2.4 g Na2HPO4, 3 g KH2PO4, pH 6.7 and 0.5 g NaCl for 1 L) with 1 mM MgCl2, 0.1 mM CaCl2, 0.5% casamino acids and 0.2% glucose. A. tumefaciens KYC55 was cultured at 28 °C in BM2 minimal medium supplemented with tetracycline (5 µg/mL), gentamicin (100 µg/mL), and spectinomycin (100 µg/mL). In general, bacterial growth was measured in a GENESYS 30 spectrophotometer at 550 nm (OD550nm).
Chemical reagents
Synthetic QS signals C8-HSL (N-octanoyl-HSL), 3OC8-HSL (N-(3-Oxooctanoyl)-HSL) and C10-HSL (N-decanoyl-HSL) were purchased from Sigma-Aldrich (Darmstadt, Germany). Stock solutions were prepared at 20 mg/mL in methanol and stored at -20°C until needed. Beta-lactam antibiotics for susceptibility testing and enzymatic assays were obtained from Sigma-Aldrich, except ticarcillin and ticarcillin-clavulanate mixture which were supplied by Apollo Scientific Ltd (Cheshire, United Kingdom). Antibiotic stock solutions were prepared according to CLSI guidelines54. X-gal (5-bromo-4-chloro-3-indolyl β-D-galactopyranoside) was purchased from Sigma-Aldrich and stock solution dissolved in N’N’-dimethylformamide.
Smlt1522 protein production in Escherichia coli and purification
The E. coli codon-optimized genes encoding Smlt1522, along with hexahistidine tagged sequences at both ends, were devised and integrated into the pET22b vector (Novagen, Madison, WI, USA) via NdeI and HindIII restriction enzymes. To remove the predicted signal peptide for cytoplasmic expression, the cloning strategy included residue A32 to the end of the protein sequence (Uniprot entry B2FIC3). The resulting plasmid was procured from GeneArt (ThermoFisher, Waltham, MA, USA) and transformed into E. coli Origami™ B (DE3) (Novagen) for optimization of recombinant protein expression. To produce an N- and C-terminally His-tagged Smlt1522 protein, transformed Origami™ B (DE3) cells were grown in LB with 100 µg/mL ampicillin, 12.5 µg/mL tetracycline and 166 µg/mL kanamycin, and protein expression was induced with 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Cells were harvested after 20 h of incubation at 16 ºC by centrifugation at 5000x g for 15 min at 4 ºC and the pellet was stored at -80 ºC until use.
For protein purification, cells were resuspended in 50 mM sodium phosphate, pH 7.5, 300 mM NaCl and 10% glycerol and disrupted with Emulsiflex C5 (Avestin, Ottawa, Canada) and a one-minute sonication step at 10% amplitude in a Branson digital sonifier (Emerson, St Louis, MO, USA). The lysate was centrifuged at 15,000xg for 45 min at 4 ºC, and the supernatant, once filtered, was subjected to immobilized metal affinity chromatography (IMAC) on an ÄKTA Pure FPLC system from Cytiva (Marlborough, MA; USA) using the HisTrap HP column 5 mL from Cytiva. Elution was performed with an increasing linear gradient of imidazole over 20 column volumes in 50 mM sodium phosphate, pH 7.5, 300 mM NaCl and 10% glycerol. The fractions corresponding to the eluted peaks were dialyzed in the storage buffer containing 50 mM Tris-HCl pH 7.6, 150 mM NaCL, 10% Glycerol, concentrated to 0.5 mg/mL using 3 kDa MWCO Vivaspin centrifugal filtration devices and stored at 4 ºC. For protein detection, samples were analysed by SDS-PAGE in TGX Stain-free acrylamide gels visualized in Chemidoc Touch Imaging System (Bio-Rad, Hercules, CA, USA). Specific detection of histidine-tagged proteins was done by Western blot with monoclonal anti-his Clontech antibodies. Protein identification was done by peptide mass fingerprinting by the proteomics (U30) Unit of CIBER in Bioengineering, Biomaterials & Nanomedicine CIBER-BBN, Spain (https://www.nanbiosis.es/platform-units/).
Construction of K279a smlt1522 markerless deletion mutant and complementation
A markerless smlt1522 mutant of S. maltophilia K279a was generated using the pGPI-SceI/pDAI-SceI-SacB system55,56, which was initially developed for multidrug-resistant Burkholderia species. The deletion plasmid, pΔsmt1522-US’DS’, was created from the mobilizable suicide vector pGPI-SceI-XCm containing the flanking regions of the target gene from the K279a genome (NC_010943). This plasmid was maintained in E. coli SY327 (refer to Tables 2 and 3 for plasmid construction and primer details). It was then transferred to S. maltophilia K279a by triparental mating using E. coli DH5α/pRK2013 as the helper strain and E. coli SY327 as the donor strain. Selection of K279a co-integrates was performed at 37 ºC on LB agar plates supplemented with norfloxacin (5 µg/mL) to counterselect against E. coli, and chloramphenicol (60 µg/mL) to select for K279a transconjugants. The integration of the suicide plasmid was confirmed by streaking single colonies and observing the development of a yellow colour when sprayed with 0.45 M pyrocatechol. Subsequently, pDAI-SceI-SacB was introduced into the co-integrates via electroporation, and selection was performed on LB agar plates containing tetracycline (60 µg/mL). The removal of the pGPI-SceI-XCm plasmid was verified by the absence of colour change upon pyrocatechol treatment and susceptibility to chloramphenicol (60 µg/mL). Mutations were confirmed through PCR and DNA Sanger sequencing. The markerless mutant was ultimately established by removing the pDAI-SceI-SacB plasmid using sucrose counterselection. The deleted smlt1522 gene was complemented by cloning the complete CDS (including the N-terminal signal peptide) into the broad-host range plasmid pBBR1-BAD (Table 2) using primers Smlt1522_Comp_Fw and Smlt1522_Comp_Rv (Table 3).
Extraction of AHLs
AHLs were extracted from 24-h stationary-phase culture supernatants with the ethyl acetate method57. Cultures of 150 mL were centrifuged, and the remaining supernatants were filtered using a Kitasatos flask and a vacuum pump. The supernatants were acidified with drops of 32% HCl to pH 6 to reverse lactone-ring hydrolysis caused by the increased pH during liquid cultures. The organic phase was separated twice with 1:1 ethyl acetate supplemented with 0.1% glacial acetic acid. The organic phase in ethyl acetate was dehydrated with anhydrous magnesium sulphate and evaporated on a rotary evaporator to obtain a volume of approximately 5 mL. The volume was recovered, and the extract was lyophilized entirely using SpeedVac. The lyophilized residues were resuspended in 1 mL of methanol, resulting in organic extracts concentrated 150 times from the original supernatants.
Bioassay for AHL activity detection
The bioassay for AHL activity detection was done as described previously22. Briefly, cells from an overnight liquid culture of the biosensor strain A. tumefaciens KYC55 were centrifuged and resuspended in 1 mL of fresh BM2. The cells were then incorporated into sterile, tempered BM2 supplemented with 0.8% noble agar (BD Difco) and 40 µg/mL X-gal, achieving a final OD550 of 0.05. This medium was poured into Petri dishes and allowed to solidify in the dark. The samples were spotted onto the agar and subsequently incubated for 24 h at 28 °C. AHL activity was detected by the appearance of blue spots on the agar.
In vivo AHL-degrading activity
The capability of strains to degrade exogenous AHLs in vivo was assessed. The strains were incubated for 24 h at 37 ºC in LB in the presence of different synthetic AHLs, namely C8-HSL (5 µM), 3OC8-HSL (5 µM), and C10-HSL (10 µM). After this period, the cultures were centrifuged, and the supernatants filtered. Twenty microliters of these supernatants were applied to biosensor plates to detect the presence of AHLs.
Analyses of AHLs and beta-lactam antibiotics degradation in vitro
Different molecules of AHLs or beta-lactam antibiotics were individually mixed with the purified recombinant Smlt1522 protein at different concentrations up to 4 µM. The mixtures were prepared in protein storage buffer (50 mM Tris-HCl pH 7.6, 150 mM NaCl, 10% Glycerol) and incubated at 37 °C for 12 h with agitation. After incubation, 5 µL of each reaction were embedded onto individual filter paper disks which were placed accordingly on (i) BM2 plates containing the biosensor strain A. tumefaciens KYC55 to assess the presence of AHLs; or (ii) MHB plates seeded with confluent cultures of P. aeruginosa PAO1 or S. aureus Newman to evaluate the presence of beta-lactam antibiotics by inhibiting bacterial growth around the disks. The plates were incubated for 20 h at (i) 28 ºC or (ii) 37 ºC, and the diameter of the halos indicated (i) the presence of AHLs or (ii) the presence of beta-lactam antibiotics.
Metabolite analyses of C10-HSL and Piperacillin degradation
The purified Smlt1522 protein (100 µg) was mixed with 7.4 M C10-HSL or 3.7 M piperacillin in its protein storage buffer and incubated at 37 °C for 12 h. After incubation, the digestion mixtures were extracted with equal volumes of ethyl acetate three times; thereafter, the combined organic phase was evaporated to dryness in the SpeedVac. The redissolved samples in methanol were analysed in an Agilent GC-MS system equipped with an automatic PAL GC XT injector and an HP-5MS UI 30 M chromatographic column (0.24 mm internal diameter, 0.25 μm film thickness). Helium was used as the carrier gas at a flow rate of 6.2 mL/min. The column temperature profile initially started at 60 °C for 1 min, increased to 320 °C at a rate of 10 °C/min, and was finally held at 320 °C for 5 min. These analyses were carried out by the technical staff of the Organic Chemistry Section at the University of Barcelona, Spain (https://dqio.ub.edu/en/organic/services/).
Antimicrobial susceptibility testing
The minimum inhibitory concentration (MIC) of a broad panel of beta-lactam antibiotics was determined using the broth microdilution method following CLSI guidelines54. MICs were determined in sterile 96-well plates by preparing two-fold serial dilutions of the corresponding antibiotic in cation-adjusted MHB (CAMHB). Initially, two-fold serial dilutions of antibiotics were made in 100 µL of CAMHB in the microtiter plate wells. Overnight cultures of test strains in CAMHB were then diluted in the same media to achieve an initial inoculum of 5 × 105 cells/mL in the assay plate. Subsequently, each well containing the corresponding two-fold serial antibiotic dilution was supplemented with 100 µL of the cell suspension, resulting in a final volume of 200 µL/well. MIC plates were read after 24 h of incubation at 37 ºC without shaking. The MIC was determined as the lowest concentration at which no bacterial growth occurred, assessed through visual inspection and confirmed by adding 30 µL of 0.01% resazurin to each well to determine cell viability.
Bacterial phenotypic assays
To assess the growth of the mutant strain in comparison to its parent strain, fresh overnight cultures were diluted to an OD550nm of 0.05 using the appropriate media. Subsequently, 200 µL of each diluted culture was transferred into individual wells of a standard 96-well microtiter plate with 4–8 technical replicates. The plates were incubated for 24 h in a Multiskan FC microplate photometer (Thermo Fisher Scientific) at 30 °C with continuous shaking to support bacterial growth, while the OD550nm was recorded every 15 min.
Biofilm formation under static conditions was assessed as described previously58 using LB, MMGC or BM2 as growth medium. Four technical replicates were done per strain and condition. To calculate the relative biofilm formation value, the bacterial growth (OD550nm) of each well was first measured in the microtiter plate assay. The crystal violet absorbance value was then expressed as a ratio to this growth measurement, specifically as OD550nm (crystal violet) / OD550nm (growth). Swarming assay was done using BM2 minimal medium containing 0.5 % (w/v) BD Difco Noble agar22.
Protein sequence and structural analysis
Protein sequences were first retrieved and analysed using UniProt (http://www.uniprot.org/) and InterPro (https://www.ebi.ac.uk/interpro/) databases, and BLAST, PSI-BLAST and CDD within NCBI (http://ncbi.nlm.nih.gov/). Signal peptide prediction was done with SignalP 5.0 (https://services.healthtech.dtu.dk/services/SignalP-5.0/). DeepMind’s AlphaFold59 version 2.3.2, locally installed and running in a Docker container, was used to model the Smlt1522 protein. AlphaFold was obtained from https://github.com/deepmind/alphafold. The script provided by the developers was used to download all required databases (software and databases were downloaded on July 3rd, 2024). The AlphaFold script was run with default parameters using the “monomer_ptm” model, full databases and multiple sequence alignments constructed using all templates available at the download date.
For structural comparisons, homologous protein structures were obtained from the RCSB PDB database (https://www.rcsb.org/). Crystal structure data of MacQ from Acidovorax sp. (4YF9) and the class III cephalosporin acylase Glutaryl-7-ACA acylase from Pseudomonas sp. (4HSR) were used. Alignment of Smlt1522 with these representative proteins was constructed by superposing with Dali-lite v.560 each subunit independently (4HSR_A and 4YF9_A for Alpha and 4HSR_B and 4YF9_C for Beta) over the Smlt1522 predicted AlphaFold model. Linker sequences were aligned versus PFAM61 profile PF01804 (Penicillin amidase). Jalview62 was used to construct the final alignment. ProFit v3.3 (http://www.bioinf.org.uk/software/profit/) software was used to compute RMSDs and Pymol v 2.4.2 (https://pymol.org/) to generate structure representations.
Sequence diversity and phylogenetic analysis
Phylogenetic analyses were done using the MEGA11 software version 11.0.1363. Orthologs of the smlt1522 nucleotide sequence in all sequenced S. maltophilia genomes available at NCBI were obtained using tBLASTn. A total of 82 complete sequences from different S. maltophilia strains were retrieved and codon aligned using the ClustalW tool in MEGA11. The multiple sequence alignment was modified manually to minimize errors. The Protein Variability Server (http://imed.med.ucm.es/PVS/) was employed to calculate Shannon Entropy values for each residue position based on translated codon alignment. Analysis on recombination and evolutionary selective pressure acting on sites based on heuristic and maximum-likelihood approaches were carried out using programs GARD64 and SLAC65, respectively, both available through the Datamonkey web server (http://www.datamonkey.org/). The GARD predicted recombination breakpoint sites were used to partition the alignment prior to SLAC analysis. The dn and ds per site were calculated with SNAP (Synonymous Non-synonymous Analysis Program) at the HIV database website (www.hiv.lanl.gov).
Statistical analysis
A one- or two-way ANOVA with Tukey’s multiple comparison test was used to determine the significance of the data between groups. All statistical analyses were performed by GraphPad Prism software (ver. 5.0; GraphPad Inc, San Diego, USA), with a p-value of less than 0.05 considered statistically significant.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Whiteley, M., Diggle, S. P. & Greenberg, E. P. Progress in and promise of bacterial quorum sensing research. Nature 551, 313–320 (2017).
Mayer, C., Borges, A., Flament-Simon, S. C. & Simões, M. Quorum sensing architecture network in Escherichia coli virulence and pathogenesis. FEMS Microbiol. Rev. 47, fuad031 (2023).
Rutherford, S. T. & Bassler, B. L. Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb Perspect. Med. 2, (2012).
Papenfort, K. & Bassler, B. L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 14, 576–588 (2016).
Park, B., Iwase, T. & Liu, G. Y. Intranasal application of S. epidermidis prevents colonization by methicillin-resistant Staphylococcus aureus in mice. PLoS ONE. 6, e25880 (2011).
Utari, P. D., Setroikromo, R., Melgert, B. N. & Quax, W. J. PvdQ quorum quenching acylase attenuates Pseudomonas aeruginosa virulence in a mouse model of pulmonary infection. Front. Cell. Infect. Microbiol. 8, 119 (2018).
Bzdrenga, J. et al. Biotechnological applications of quorum quenching enzymes. Chem. Biol. Interact. 267, 104–115 (2017).
Chen, F., Gao, Y., Chen, X., Yu, Z. & Li, X. Quorum quenching enzymes and their application in degrading signal molecules to block quorum sensing-dependent infection. Int. J. Mol. Sci. 14, 17477–17500 (2013).
Sikdar, R. & Elias, M. Quorum quenching enzymes and their effects on virulence, biofilm, and microbiomes: a review of recent advances. Expert Rev. Anti Infect. Ther. 18, 1221–1233 (2020).
Rehman, Z. U. & Leiknes, T. Quorum-Quenching Bacteria isolated from red sea sediments reduce biofilm formation by Pseudomonas aeruginosa. Front. Microbiol. 9, 1354 (2018).
Oinonen, C. & Rouvinen, J. Structural comparison of Ntn-hydrolases. Protein Sci. 9, 2329–2337 (2000).
Bokhove, M., Nadal Jimenez, P., Quax, W. J. & Dijkstra, B. W. The quorum-quenching N-acyl Homoserine lactone acylase PvdQ is an Ntn-hydrolase with an unusual substrate-binding pocket. Proc. Natl. Acad. Sci. U. S. A. 107, 686–691 (2010).
Kim, J. K. et al. Crystal structures of glutaryl 7-aminocephalosporanic acid acylase: insight into autoproteolytic activation. Biochemistry 42, 4084–4093 (2003).
Yasutake, Y. et al. Bifunctional quorum-quenching and antibiotic-acylase MacQ forms a 170-kDa capsule-shaped molecule containing spacer polypeptides. Sci. Rep. 7, 8946 (2017).
Kusada, H., Tamaki, H., Kamagata, Y., Hanada, S. & Kimura, N. A. Novel Quorum-Quenching N-Acylhomoserine Lactone Acylase from Acidovorax sp. Strain MR-S7 Mediates Antibiotic Resistance. Appl. Environ. Microbiol. 83, e00080–e00017 (2017).
Park, S. Y. et al. Identification of extracellular N-acylhomoserine lactone acylase from a Streptomyces sp. and its application to quorum quenching. Appl. Environ. Microbiol. 71, 2632–2641 (2005).
Serrano-Aguirre, L. et al. Novel bifunctional acylase from Actinoplanes utahensis: A versatile enzyme to synthesize antimicrobial compounds and use in quorum quenching processes. Antibiotics. 10, 922 (2021).
Mukherji, R., Varshney, N. K., Panigrahi, P., Suresh, C. G. & Prabhune, A. A new role for penicillin acylases: degradation of acyl Homoserine lactone quorum sensing signals by Kluyvera citrophila penicillin G acylase. Enzyme Microb. Technol. 56, 1–7 (2014).
Sunder, A. V. et al. Penicillin V acylases from gram-negative bacteria degrade N-acylhomoserine lactones and attenuate virulence in Pseudomonas aeruginosa. Appl. Microbiol. Biotechnol. 101, 2383–2395 (2017).
Huedo, P., Coves, X., Daura, X., Gibert, I. & Yero, D. Quorum sensing signaling and quenching in the Multidrug-Resistant pathogen Stenotrophomonas maltophilia. Front. Cell. Infect. Microbiol. 8, 122 (2018).
Hu, H. et al. Role of N-acyl-homoserine lactone (AHL) based quorum sensing on biofilm formation on packing media in wastewater treatment process. RSC Adv. 6, 11128–11139 (2016).
Martínez, P. et al. Stenotrophomonas maltophilia responds to exogenous AHL signals through the LuxR solo SmoR (Smlt1839). Front. Cell. Infect. Microbiol. 5, 41 (2015).
Coves, X. et al. A Stenotrophomonas maltophilia TetR-Like transcriptional regulator involved in fatty acid metabolism is controlled by quorum sensing signals. Appl. Environ. Microbiol. 89, e0063523 (2023).
Reina, J. C., Torres, M. & Llamas, I. Stenotrophomonas maltophilia AHL-Degrading strains isolated from marine invertebrate microbiota attenuate the virulence of Pectobacterium carotovorum and Vibrio coralliilyticus. Mar. Biotechnol. (NY). 21, 276–290 (2019).
Torabi Delshad, S., Soltanian, S., Sharifiyazdi, H., Haghkhah, M. & Bossier, P. Identification of N-acyl Homoserine lactone-degrading bacteria isolated from rainbow trout (Oncorhynchus mykiss). J. Appl. Microbiol. 125, 356–369 (2018).
Curcic, J. et al. A novel thermostable YtnP lactonase from Stenotrophomonas maltophilia inhibits Pseudomonas aeruginosa virulence in vitro and in vivo. Int. J. Biol. Macromol. 264, 130421 (2024).
Huang, J. J., Han, J. I., Zhang, L. H. & Leadbetter, J. R. Utilization of acyl-homoserine lactone quorum signals for growth by a soil pseudomonad and Pseudomonas aeruginosa PAO1. Appl. Environ. Microbiol. 69, 5941–5949 (2003).
Daly, J. W., Keely, S. J. & Gahan, C. G. M. Functional and phylogenetic diversity of BSH and PVA enzymes. Microorganisms 9, 732 (2021).
Liu, N. et al. PfmA, a novel quorum-quenching N-acylhomoserine lactone acylase from Pseudoalteromonas flavipulchra. Microbiology. 163, 1389–1398 (2017).
Zhu, J., Chai, Y., Zhong, Z., Li, S. & Winans, S. C. Agrobacterium bioassay strain for ultrasensitive detection of N-acylhomoserine lactone-type quorum-sensing molecules: detection of autoinducers in Mesorhizobium huakuii. Appl. Environ. Microbiol. 69, 6949–6953 (2003).
Urban, C., Go, E., Mariano, N. & Rahal, J. J. Interaction of sulbactam, clavulanic acid and Tazobactam with penicillin-binding proteins of imipenem-resistant and -susceptible Acinetobacter baumannii. FEMS Microbiol. Lett. 125, 193–197 (1995).
Sánchez, M. B. Antibiotic resistance in the opportunistic pathogen Stenotrophomonas maltophilia. Front. Microbiol. 6, 658 (2015).
Brannigan, J. A. et al. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature 378, 416–419 (1995).
Golden, E. et al. Structure of a class III engineered cephalosporin acylase: comparisons with class I acylase and implications for differences in substrate specificity and catalytic activity. Biochem. J. 451, 217–226 (2013).
Sio, C. F. et al. Quorum quenching by an N-acyl-homoserine lactone acylase from Pseudomonas aeruginosa PAO1. Infect. Immun. 74, 1673–1682 (2006).
Carratalá, J. V. et al. Selecting subpopulations of High-Quality protein conformers among conformational mixtures of Recombinant bovine MMP-9 solubilized from inclusion bodies. Int. J. Mol. Sci. 22, 3020 (2021).
Mayer, J. et al. Crystal structures and protein engineering of three different penicillin G acylases from Gram-positive bacteria with different thermostability. Appl. Microbiol. Biotechnol. 103, 7537–7552 (2019).
Quiroga, I., Hernández-González, J. A. & Bautista-Rodríguez, E. Benítez-Rojas, A. C. Exploring the structurally conserved regions and functional significance in bacterial N-Terminal nucleophile (Ntn) Amide-Hydrolases. Int. J. Mol. Sci. 25, 6850 (2024).
Philem, P. D. et al. Structural and enzymatic analysis of a dimeric Cholylglycine hydrolase like acylase active on N-acyl Homoserine lactones. Biochimie 177, 108–116 (2020).
Cui, S. & Kim, E. Quorum sensing and antibiotic resistance in polymicrobial infections. Commun. Integr. Biol. 17, 2415598 (2024).
Park, S. J., Park, S. Y., Ryu, C. M., Park, S. H. & Lee, J. K. The role of AiiA, a quorum-quenching enzyme from Bacillus thuringiensis, on the rhizosphere competence. J. Microbiol. Biotechnol. 18, 1518–1521 (2008).
Brooke, J. S. Advances in the microbiology of Stenotrophomonas maltophilia. Clin. Microbiol. Rev. 34, e0003019 (2021).
Crofts, T. S. et al. Shared strategies for β-lactam catabolism in the soil Microbiome. Nat. Chem. Biol. 14, 556–564 (2018).
Gonzales, M. et al. Disrupting quorum sensing as a strategy to inhibit bacterial virulence in human, animal, and plant pathogens. Pathog. Dis. 82, ftae009 (2024).
Romero, M., Martin-Cuadrado, A. B. & Otero, A. Determination of whether quorum quenching is a common activity in marine Bacteria by analysis of cultivable Bacteria and metagenomic sequences. Appl. Environ. Microbiol. 78, 6345–6348 (2012).
Drake, E. J. & Gulick, A. M. Structural characterization and high-throughput screening of inhibitors of PvdQ, an NTN hydrolase involved in Pyoverdine synthesis. ACS Chem. Biol. 6, 1277–1286 (2011).
de Celis, M. et al. Acylase enzymes disrupting quorum sensing alter the transcriptome and phenotype of Pseudomonas aeruginosa, and the composition of bacterial biofilms from wastewater treatment plants. Sci. Total Environ. 799, 149401 (2021).
Jimenez, P. N. et al. Role of PvdQ in Pseudomonas aeruginosa virulence under iron-limiting conditions. Microbiology. 156, 49–59 (2010).
Kalidasan, V., Joseph, N., Kumar, S., Awang Hamat, R. & Neela, V. K. Iron and virulence in Stenotrophomonas Maltophilia: all we know so Far. Front. Cell. Infect. Microbiol. 8, 401 (2018).
Aminov, R. I. The role of antibiotics and antibiotic resistance in nature. Environ. Microbiol. 11, 2970–2988 (2009).
Avison, M. B., von Heldreich, C. J., Higgins, C. S., Bennett, P. M. & Walsh, T. R. A TEM-2beta-lactamase encoded on an active Tn1-like transposon in the genome of a clinical isolate of Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 46, 879–884 (2000).
Overhage, J., Lewenza, S., Marr, A. K. & Hancock, R. E. W. Identification of genes involved in swarming motility using a Pseudomonas aeruginosa PAO1 mini-Tn5-lux mutant library. J. Bacteriol. 189, 2164–2169 (2007).
Xavier, J. B., Kim, W. & Foster, K. R. A molecular mechanism that stabilizes cooperative secretions in Pseudomonas aeruginosa. Mol. Microbiol. 79, 166–179 (2011).
CLSI. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved standard. 10th edition. M07 A10. Clinical and Laboratory Standards Institute, Wayne, PA. (2015).
Aubert, D. F., Hamad, M. A. & Valvano, M. A. A markerless deletion method for genetic manipulation of Burkholderia cenocepacia and other multidrug-resistant gram-negative bacteria. Methods Mol. Biol. 1197, 311–327 (2014).
Hamad, M. A., Skeldon, A. M. & Valvano, M. A. Construction of aminoglycoside-sensitive Burkholderia cenocepacia strains for use in studies of intracellular bacteria with the gentamicin protection assay. Appl. Environ. Microbiol. 76, 3170–3176 (2010).
Schaefer, A. L., Hanzelka, B. L., Parsek, M. R. & Greenberg, E. P. Detection, purification, and structural Elucidation of the acylhomoserine lactone inducer of Vibrio fischeri luminescence and other related molecules. Methods Enzymol. 305, 288–301 (2000).
Coves, X. et al. The mla system and its role in maintaining outer membrane barrier function in Stenotrophomonas maltophilia. Front. Cell. Infect. Microbiol. 14, 1346565 (2024).
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596, 583–589 (2021).
Holm, L. Benchmarking fold detection by DaliLite V.5. Bioinformatics 35, 5326–5327 (2019).
Mistry, J. et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M. & Barton, G. J. Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Kosakovsky Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H. & Frost, S. D. W. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 23, 1891–1901 (2006).
Kosakovsky Pond, S. L. & Frost, S. D. W. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22, 1208–1222 (2005).
Huedo, P. et al. Two different Rpf clusters distributed among a population of Stenotrophomonas maltophilia clinical strains display differential diffusible signal factor production and virulence regulation. J. Bacteriol. 196, 2431–2442 (2014).
Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166, 557–580 (1983).
Miller, V. L. & Mekalanos, J. J. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170, 2575–2583 (1988).
Yero, D. et al. The Pseudomonas aeruginosa substrate-binding protein Ttg2D functions as a general glycerophospholipid transporter across the periplasm. Commun. Biol. 4, 448 (2021).
Baba, T., Bae, T., Schneewind, O., Takeuchi, F. & Hiramatsu, K. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of Staphylococcal genomes: polymorphism and evolution of two major pathogenicity Islands. J. Bacteriol. 190, 300–310 (2008).
Figurski, D. H. & Helinski, D. R. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. U S A. 76, 1648–1652 (1979).
Flannagan, R. S., Linn, T. & Valvano, M. A. A system for the construction of targeted unmarked gene deletions in the genus Burkholderia. Environ. Microbiol. 10, 1652–1660 (2008).
Funding
This work was funded by the Spanish MICINN PID2019-111364RB-I00 and PID2023-150290OB-I00. Authors also thank Catalan AGAUR (2021 SGR00092 and SGR00068). Protein production, purification and identification were performed by the ICTS “NANBIOSIS”, more specifically by the Protein Production Platform (PPP-U1) and by the proteomics (U30) Unit of CIBER in Bioengineering, Biomaterials & Nanomedicine (CIBER-BBN) (Platform units - Nanbiosis).
Author information
Authors and Affiliations
Contributions
MB, OCS, XC, AGN, ACG, MMM, NFM and DY performed experiments, analysed data and contributed to the writing of the manuscript. MB, OCS and DY wrote the original draft and conducted the final editing and review of the manuscript. NFM, XD, IG and DY supervised research and revised the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bravo, M., Conchillo-Solé, Ò., Coves, X. et al. An acyl-homoserine lactone acylase found in Stenotrophomonas maltophilia exhibits both quorum quenching activity and the ability to degrade penicillin antibiotics. Sci Rep 15, 8557 (2025). https://doi.org/10.1038/s41598-025-92749-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-92749-4
Keywords
This article is cited by
-
Quorum sensing in bacteria: insights into communication and inhibition strategies—a review
Archives of Microbiology (2026)
-
Quorum quenching pathoblockers from bacteria: an alternative approach for bacterial infection management
Folia Microbiologica (2025)
