
Abstract
Machine learning can be applied to identify key genes associated with osteoarthritis (OA). This study aimed to explore the differential expression of necroptosis-related genes (NRGs) during the progression of OA, identify key gene modules strongly linked to the onset of OA, and assess the role of CASP1 and its correlation with immune cell infiltration in OA. Gene expression profile data were obtained for OA and normal tissues: GSE55235 (10 OA and 10 normal synovial tissues) and GSE46750 (12 OA and 12 normal synovial tissues). Differential expression analysis identified 44 NRGs. Weighted gene co-expression network analysis revealed that the turquoise module, including 2037 genes, showed a strong correlation with OA. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes enrichment analyses showed that these genes were predominantly involved in regulating the JNK cascade, cellular response to oxidative stress, and Toll-like receptor signalling pathways. The support vector machine model exhibited the highest predictive performance (area under the curve of 0.883). Additionally, CASP1 expression in OA tissues was considerably elevated compared to normal tissues and was strongly associated with immune cell infiltration. These findings deepen our understanding of the pathophysiological foundation of OA and identify possible molecular targets for creating innovative OA therapies.
Similar content being viewed by others


Introduction
Osteoarthritis (OA), a progressive degenerative joint disorder with various causes, is characterised by clinical features including joint pain, stiffness, swelling, and varying degrees of inflammation. The pathogenesis of OA is currently unclear, and treatment is mainly focused on symptom relief, with no available methods to intervene in disease progression1,2; however, the mechanisms underlying necroptosis have garnered increasing attention. Necroptosis, which is strongly linked with the pathogenesis of OA, is a form of regulated cell death1,3. Research indicates that RIPK1 and RIPK3 are key regulatory factors of necroptosis. Their activation promotes chondrocyte necroptosis, which accelerates the development of OA. Inhibiting TRADD can effectively reduce chondrocyte necroptosis and combat OA by blocking the RIPK1–TAK1 signalling pathway and restoring autophagic function4. Thus, research on the necroptosis pathway not only reveals the pathogenic mechanism of OA but also provides potential targets for developing new therapeutic strategies.
Recently, the application of machine learning technologies in medical research has grown significantly, particularly for identifying key genes that contribute to disease onset and progression, including OA. By integrating bioinformatic analyses and machine learning methods, researchers can confirm biomarkers and immune infiltration characteristics associated with diseases, thereby gaining a deeper understanding of the underlying pathological mechanisms5,6,7,8. For example, studies based on bioinformatic analyses and machine learning have successfully identified aging-related biomarkers and immune-related genes associated with metabolic syndrome, providing insights into the diagnosis of OA9. Additionally, machine learning has been employed in the early identification of OA, whereby models have analysed large datasets to identify biomarkers in patients with OA, thereby improving diagnostic accuracy and efficiency10. These studies not only validate the application value of machine learning in screening key genes related to OA but also provided new strategies and goals for the early detection and treatment. However, research utilising machine learning methods to screen necroptosis-related genes (NRGs), that serve as key players in the onset and progression of OA, remains limited.
This study utilised machine learning techniques to comprehensively analyse the expression of NRGs in OA, identify key gene modules highly related to OA, and investigate the role of CASP1 in OA and its relationship with immune cell infiltration. By employing a series of advanced bioinformatics and machine learning techniques, this study not only highlights the importance of NRGs in the pathogenesis of OA but also reveals that the specific mode of expression of CASP1 is strongly linked with the immune response in OA. The results of this study are anticipated to provide a new perspective on the treatment and diagnosis of OA. Furthermore, we demonstrate the potential of using machine learning techniques to screen and evaluate disease-related genes.
Methods
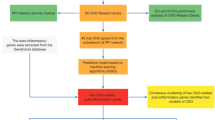
All methods were performed in strict accordance with the relevant guidelines and regulations to ensure ethical compliance. This research received formal authorisation and approval from the Ethics and Institutional Review Committee at Hainan Provincial People’s Hospital (Approval No: Med-Eth-Re [2023] 01). Furthermore, all patients provided written informed consent after being thoroughly briefed about the study prior to their participation. The flow diagram of our study design is shown in Supplementary Fig. 1.
Acquiring and refining data
For this study, we retrieved the gene expression profile statistics of OA and normal tissues from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/, accessed on 21 November 2023). Using ‘osteoarthritis’ as the search term, we obtained sequencing data for OA synovial tissues from two datasets, GSE55235 and GSE46750, along with their respective platform data, GPL96 and GPL10558. Specifically, GSE55235 included sequencing data from 10 OA and 10 normal synovial tissue samples, while GSE46750 provided sequencing data from 12 OA and 12 normal synovial tissue samples. NRGs were sourced from two key resources: 159 NRGs were identified from the necroptosis pathway (hsa04217) in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (https://www.kegg.jp/, accessed on 21 November 2023), and an additional 67 NRGs were curated from existing literature11. Finally, we integrated the intersections of these two gene sets, including the 204 NRGs, for subsequent analyses.
Differential analysis of NRGs
We utilised R software (version 4.3.1) to normalise the expression data matrix obtained from the GSE55235 dataset. Differential expression analysis was conducted on the normalized gene expression using the ‘limma’ R package to identify statistically significant differences between OA and normal synovial tissues. A boxplot was subsequently generated to visualize the differentially expressed NRGs between OA and normal synovial tissues, ensuring a clear depiction of the variations. An adjusted P value of < 0.05 and |log2FC| > 1 were used as the threshold for significance, and a heatmap was produced using the ‘pheatmap’ package.
Weighted gene co-expression network analysis (WGCNA)
The ‘WGCNA’ R package was employed to construct a weighted gene co-expression network using the OA tissue expression matrix. A sample tree was initially drawn to detect and exclude any potential outliers. The adjacency matrix was subsequently transformed into a topological overlap matrix (TOM), ensuring that the scale-free topology property was preserved with an R2 value set to 0.90. Modules were identified using the dynamic tree-cut algorithm, with parameters deepSplit set to 2 and minModuleSize set to 50. Finally, we conducted a correlation analysis between the identified modules and occurrence of OA, pinpointing the module that demonstrated the strongest correlation with the disease phenotype.
Enrichment analysis
Using bioinformatics and evolutionary genomics tools, we intersected the differentially expressed genes (DEGs) identified from the WGCNA results that showed a positive correlation with OA. To visually represent the overlap, a Venn diagram was constructed, enabling us to pinpoint the NRGs specifically associated with OA. This approach allowed for a clear identification of OA-related NRGs through the intersection of relevant gene sets. The gene set was then analysed using the ‘ClusterProfiler’ R package for Gene Ontology (GO) and KEGG enrichment analysis. GO enrichment analysis provided insights into the biological roles of core genes, whereas KEGG enrichment analysis identified core gene signalling pathways, with significance criteria set at P value < 0.05 and q value < 0.05.
Protein–protein interaction (PPI) networks of core genes
The gene sets identified through the Venn analysis were imported into the STRING database (https://string-db.org/, accessed on 15 January 2024) to construct the PPI network. The analysis was restricted to Homo sapiens to ensure species specificity. An interaction score threshold of > 0.7 was applied to select active interaction sources for constructing the PPI networks. A PPI enrichment p-value of < 0.05 was set to determine the statistical significance of the network. The resulting PPI network was further analyzed using Cytoscape software (version 3.9.1), where the degree plugin was utilized to rank genes based on their connectivity. The top 10 genes with the highest interaction scores were selected for the PPI network analysis.
Machine learning-based screening of OA-related NRGs
We merged the GSE55235 and GSE46750 datasets from the GEO database and applied three different machine learning algorithms, including support vector machine (SVM), random forest (RF), and extreme gradient boosting (XGB), to establish classifier models for OA and normal tissues. Using these methods, we produced residual box plots, residual reverse cumulative distribution plots, and gene importance distribution plots. Using the GEO data, ROC curves were plotted, and the AUC was computed to assess the predictive accuracy of the classifier models. We classified the accuracy levels as excellent (0.9 ≤ AUC < 1), good (0.8 ≤ AUC < 0.9), and non-informative (AUC = 0.5) according to the guidelines.
Correlation between CASP1 and immune cell infiltration in OA tissues
We used the single-sample gene set enrichment analysis (ssGSEA) method to appraise the level of immune cell infiltration in OA tissues. Box plots showed the differential expression of 29 immune-related gene sets between OA and normal tissues. The CIBERSORT algorithm assessed immune cell infiltration levels in OA and normal tissues. The xCell algorithm analysed the infiltration levels of 64 types of immune cells in OA tissues, with box plots displaying the differential expression levels of these cells.
Enrichment analysis of CASP1 higher and lower expression groups
The downloaded expression data matrix was normalised using R software. We used the ‘limma’ package for differential expression analysis of the normalised data, with criteria of |log2FC| > 1 and an adjusted P < 0.05 to verify DEGs. Volcano and heat maps were generated using the ‘ggplot2’ and ‘pheatmap’ packages. GO and KEGG enrichment analyses were performed on the gene sets using the ‘ClusterProfiler’ R package, which highlighted the signalling pathways and biological functions of the gene sets.
Validation of CASP1
We collected normal knee (n = 3) and OA (n = 3) synovial tissues from patients undergoing arthroscopic surgery for acute trauma and knee arthroplasty, respectively. Whole RNA was extracted using Beyozol (Beyotime Bio, Inc.), and cDNA was synthesised using the BeyoRT™ II cDNA Synthesis Kit. Quantitative real-time PCR (qRT-PCR) was then performed using the BeyoFast™ SYBR Green One-Step Kit on an Applied Biosystems Real-Time PCR System, and gene expression was quantified relative to GAPDH using the 2−ΔΔCt method.
In the western blot (WB) analysis, proteins were separated using sodium dodecyl sulphate–polyacrylamide gel electrophoresis (Bio-Rad) and transferred onto polyvinylidene fluoride membranes (Millipore). The membranes were then treated with a CASP1-specific primary antibody (Abcam) and subsequently with an HRP-conjugated secondary antibody (Santa Cruz Biotechnology). Signal detection was performed using enhanced chemiluminescence reagents (Thermo Fisher Scientific), and protein band intensity was measured with ImageJ software (version 1.53e), with GAPDH serving as the internal control.
Statistical analysis
All statistical analyses were carried out using R software for comprehensive data evaluation. Power calculations for sample size determination were also performed in R, with a power level set at 0.80 to ensure an adequate sample size for detecting the expected effect.
Results
Differentially expressed NRGs
After preprocessing the raw data, 44 differentially expressed NRGs were identified in the dataset GSE55235, as depicted in box plots (Fig. 1a,b). Among these, 22 were upregulated and 22 were downregulated.

Analysis of differential expression. (a) Heatmap of differentially expressed NRGs in osteoarthritis (OA) tissues compared to that in normal tissues based on sequencing data. (b) Box plot of differentially expressed necroptosis genes in OA tissues compared to that in normal tissues. *P < 0.05, **P < 0.01, ***P < 0.001 compared to normal tissues.
WGCNA
We applied WGCNA to analyse the sequencing data from the GSE55235 OA tissue dataset, with the goal of identifying crucial gene modules related to the molecular characteristics of OA (Fig. 2a,b). Seven modules were discovered in the GSE55235 dataset using average linkage hierarchical clustering and a soft-threshold power approach (Fig. 2c). The module that most positively correlated with OA in GSE55235 was the turquoise module (r = 0.97, P = 5e-12) (Fig. 2d), which contained 2037 genes.

Key modules associated with OA using weighted gene co-expression network analysis (WGCNA). (a,b) Scatter plots illustrating the fitting index versus power value in the GSE55235 dataset. (c) Dendrogram of all genes in the GSE55235 dataset, clustered based on different measures (1 – topological overlap matrix [TOM]). Each branch in the dendrogram corresponds to an individual gene, with colour indicating a co-expression module. (d) Heatmap illustrating the correlation between clustered gene modules and OA in the GSE55235 dataset, including correlation coefficients and adjusted P values for each module.
GO and KEGG enrichment analysis for differentially expressed genes
The overlap between the 44 differentially expressed NRGs and the 2037 genes positively associated with OA, as identified using WGCNA, yielded 15 core NRGs related to OA (Fig. 3a). According to the GO enrichment analysis, these genes were primarily linked to biological functions, including the regulation of the JNK cascade, regulation of cytokine secretion, response of cells to oxidative stress, modulation of the apoptotic signalling pathway, and regulation of the inflammatory response (Fig. 3b). According to KEGG enrichment analysis, these genes were primarily involved in specific pathways such as the Toll-like receptor signalling pathway, FoxO signalling pathway, PI3K–Akt signalling pathway, NOD-like receptor signalling pathway, and necroptosis (Fig. 3c).

Selection of core gene sets related to OA. (a) A Venn diagram illustrating the intersection of genes with differential expression and WGCNA of positively correlated module genes. (b) GO enrichment analysis of OA-related core genes. (c) KEGG enrichment analysis of the core genes related to OA.
Screening of hub genes in the PPI network
The PPI network of the 15 core genes was analysed using the STRING database and Cytoscape software (Fig. 4a). The Degree plugin in Cytoscape was then used to select the top 10 core genes (Fig. 4b).

Protein–protein interaction network related to OA. (a) PPI network of the 15 core genes. (b) PPI network of the top-ranking core genes based on the degree scores (note: darker colours indicate higher ranks).
Screening OA NRGs based on machine learning
The residual box plots of the three machine learning models (SVM, RF and XGB) are shown in Fig. 5a, with the reverse cumulative distribution plots of the residuals shown in Fig. 5b and the gene importance distribution plots in Fig. 5c. The predictive performance of the classifier models was evaluated by plotting receiver operating characteristic (ROC) curves and determining the area under the curve (AUC) values based on GEO data (Fig. 5d). The AUC values for the RF, SVM, and XGB were 0.806, 0.883, and 0.806, respectively. The intersection of genes identified as being related to OA by PPI, RF, SVM, and XGB yielded the final set of OA-related NRGs (Fig. 5e).

Screening for OA NRGs using machine learning. (a) Residual boxplots for machine learning methods including SVM, RF, and XGB. (b) Reverse cumulative distribution plots of the residuals for SVM, RF, and XGB. (c) Gene importance distribution plots for RF, SVM, and XGB models. (d) Receiver operating characteristic curves for the RF, SVM, and XGB models. (e) Venn diagram showing the intersection of OA NRGs identified using PPI, RF, SVM, and XGB analyses (CASP1).
Correlation between CASP1 expression and immune cell infiltration in OA tissues
Using the GEO dataset, we employed the ssGSEA method to analyse immune cell infiltration in OA tissues with higher and lower CASP1 expression (Fig. 6a) and used box plots to identify immune gene sets with significant differences between the higher and lower CASP1 expression groups (Fig. 6b). Subsequently, the CIBERSORT algorithm was used to determine the level of immune cells infiltration in OA tissues (Fig. 6c), and box plots were used to identify immune cell types with significant differences between the higher and lower CASP1 expression groups (Fig. 6d). Finally, we used the xCELL algorithm to evaluate the infiltration levels of 64 types of immune cells in OA tissues (Fig. 6e) and employed box plots to identify immune cells showing significant differences between the higher and lower CASP1 expression groups (Fig. 6f).

Assessment of the immune status in OA tissues. (a) Immune cell infiltration in OA tissues with high versus low CASP1 expression, as determined by the ssGSEA algorithm. (b) Box plots of immune cell infiltration in OA tissues with higher and lower CASP1 expression based on the ssGSEA algorithm. (c) Infiltration levels of 22 varieties of immune cells in OA tissues with higher and lower CASP1 expression, based on the CIBERSORT algorithm. (d) Box plots of immune cell infiltration levels in OA tissues with higher and lower CASP1 expression, based on the CIBERSORT algorithm. (e) Infiltration levels of 64 varieties of immune cells in OA tissues with higher and lower CASP1 expression, based on the xCELL algorithm. (f) Box plots of immune cell infiltration levels in OA tissues with higher and lower CASP1 expression, based on the xCELL algorithm.
Enrichment analysis for higher and lower CASP1 expression groups
After preprocessing the raw data, we identified 2231 differentially expressed genes (Fig. 7a), including 1145 upregulated and 1086 downregulated genes. GO enrichment analysis revealed that these genes were primarily associated with biological functions including cytokine receptor activity, positive regulation of chemotaxis, endoplasmic reticulum lumen, regulation of leukocyte differentiation, and positive regulation of cytokine production (Fig. 7b). KEGG enrichment analysis indicated that these genes were mainly associated with pathways including the Toll-like receptor signalling pathway, neutrophil extracellular trap formation, cell adhesion molecules, chemokine signalling pathway, and PI3K–Akt signalling pathway (Fig. 7c).

Enrichment analysis for higher and lower CASP1 expression groups. (a) Volcano plot of differentially expressed genes between higher and lower CASP1 expression groups based on the sequencing data. (b) GO enrichment analysis of genes differentially expressed between higher and lower CASP1 expression groups. (c) KEGG enrichment analysis for genes differentially expressed between higher and lower CASP1 expression groups.
Validation of CASP1
Using qRT-PCR and WB, we determined that there was a statistically significant difference in CASP1 expression between the normal and OA groups (P < 0.05) (Fig. 8). The expression of CASP1 was markedly higher in OA tissues compared to that in normal tissues.

CASP1 expression levels in the normal control (NC) and OA groups. (a) qRT-PCR showing the expression levels of CASP1 in NC and OA groups (n = 3 per group). The expression of CASP1 is significantly higher in the OA group than that in the NC group (*P < 0.05). (b) Western blot (WB) analysis of CASP1 and GAPDH in NC and OA groups (n = 3 per group). CASP1 protein levels were higher in the OA group compared to that in the NC group. The full-length, unprocessed blot is included in Supplementary Fig. 2. (c) Quantification of WB results. Relative protein expression of CASP1 normalised to GAPDH is significantly increased in the OA group compared to that in the NC group (P < 0.01).
Discussion
In this study, we successfully identified key NRGs that influence the development of OA through the integrated application of machine learning methods and bioinformatics analyses. In OA tissues, elevated CASP1 expression is closely associated with immune cell infiltration. These results further develop our understanding of the pathophysiology of OA and offer potential molecular targets for developing new treatment strategies.
Initially, by intersecting the differentially expressed NRGs with the genes positively correlated with OA obtained from the WGCNA analysis, we identified 15 NRGs associated with OA. Furthermore, the GO and KEGG enrichment analyses corroborated previous findings. Necroptosis is a regulated form of cell death characterised by the rupture of the cell membrane and release of contents, potentially triggering inflammatory responses. JNK, a class of stress-activated protein kinases, plays a crucial role in regulating cell survival and death, which can promote necroptosis and subsequently influence the production of inflammatory cytokines. Additionally, the regulation of cytokine secretion is closely related to necroptosis, where the release of cellular contents can stimulate surrounding cells to produce and release more cytokines, thereby exacerbating inflammation12. Furthermore, in immune and inflammatory responses, the activation of the Toll-like receptor signalling pathway is a crucial step, indirectly promoting necroptosis by enhancing the production of inflammatory cytokines13,14. These findings emphasise the complex roles and mechanisms of necroptosis in OA development, identify several potential molecular targets and regulatory points, and provide important clues for future research directions and therapeutic strategies.
Our study reveals significant insights into the functional and pathway-specific roles of differentially expressed genes associated with CASP1 expression levels. The GO enrichment analysis highlights critical biological processes, including cytokine receptor activity, positive regulation of chemotaxis, and leukocyte differentiation regulation. These functions are closely linked to immune responses and inflammation, supporting the pivotal role of CASP1 in mediating immune system activity15. Moreover, the identification of DEGs related to the endoplasmic reticulum lumen suggests a potential involvement in protein folding and stress responses, which are known to influence cytokine production16. The KEGG analysis further emphasizes the importance of CASP1 in inflammatory and immune-regulatory pathways, such as the Toll-like receptor signalling pathway and the chemokine signalling pathway. These findings suggest CASP1 plays a role in enhancing innate immune mechanisms and promoting neutrophil extracellular trap (NET) formation, which is critical in pathogen defence17,18. Additionally, the enrichment of pathways such as PI3K–Akt signalling highlights potential links between CASP1 and cellular processes such as survival, proliferation, and apoptosis19. Collectively, these findings underscore the multifaceted role of CASP1 in immune modulation and inflammation, providing valuable avenues for therapeutic intervention.
In OA, damage and degeneration of the joint cartilage and other tissues are associated with the infiltration of immune cells, particularly macrophages and T cells, which are closely related to disease progression and pain perception20. Our study results indicate that CASP1 expression is significantly elevated in OA tissues compared to normal tissues and is closely associated with immune cell infiltration, particularly B cells, T cells, macrophages, endothelial cells, and others. Previous studies have shown that the high expression of CASP1 may contribute to the formation of an inflammatory microenvironment, attracting a large number of immune cells, thereby exacerbating cartilage destruction and disease progression in OA21. Additionally, evidence indicates that macrophage and T cell infiltration is significantly increased in the affected joints of patients with OA, and the activation of these cells is closely associated with the secretion of inflammatory mediators and degradation of the cartilage matrix22. Endothelial cells also participate in the inflammatory process by promoting the migration of immune cells to synovial tissues. The high expression of CASP1 related to endothelial cell activity may reflect an enhanced vascular response, further exacerbating inflammation and tissue damage in OA23. These findings support our hypothesis that CASP1 plays an important role in OA pathophysiology by regulating immune cell infiltration.
One limitation of this study was the reliance on publicly available datasets, which may not fully represent the diverse patient populations and varying stages of OA. Additionally, while the power calculation supports the statistical validity of the results within the study, the sample size and dataset origin still pose challenges to the generalizability of the findings. We also reiterate the need for validation in larger, independent, and diverse cohorts to strengthen clinical applicability of these results.
In conclusion, this study advanced OA NRGs research through machine learning techniques, offering fresh insights into the molecular mechanisms of OA and paving the way for future therapeutic strategies. Nevertheless, further experimental validation and detailed mechanistic studies are necessary to apply these findings clinically.
Data availability
Data will be made available by the corresponding author upon reasonable request.
References
Liu, Z., Qian, Z., Wang, Y. & Wang, H. Necroptosis in pathogenesis of osteoarthritis and its therapeutic implications. Zhejiang Da Xue Xue Bao. Yi Xue Ban 51, 261–265 (2022).
Kolasinski, S. L. et al. 2019 American College of Rheumatology/Arthritis Foundation Guideline for the Management of Osteoarthritis of the Hand, Hip, and Knee. Arthritis Care Res. (Hoboken) 72, 149–162 (2020).
Yang, J. et al. Targeting cell death: Pyroptosis, ferroptosis, apoptosis and necroptosis in osteoarthritis. Front. Cell Dev. Biol. 9, 789948 (2021).
Li, X. T. et al. TNFAIP3 derived from skeletal stem cells alleviated rat osteoarthritis by inhibiting the necroptosis of subchondral osteoblasts. Stem Cells 42, 360–373 (2024).
Kokkotis, C., Moustakidis, S., Papageorgiou, E., Giakas, G. & Tsaopoulos, D. E. Machine learning in knee osteoarthritis: A review. Osteoarthr. Cartil. Open 2, 100069 (2020).
Liang, Y., Lin, F. & Huang, Y. Identification of biomarkers associated with diagnosis of osteoarthritis patients based on bioinformatics and machine learning. J. Immunol. Res. 2022, 5600190 (2022).
Qiu, Y. et al. Machine learning identifies ferroptosis-related genes as potential diagnostic biomarkers for osteoarthritis. Front. Endocrinol. (Lausanne) 14, 1198763 (2023).
Li, J. et al. Identification of immune-associated genes in diagnosing osteoarthritis with metabolic syndrome by integrated bioinformatics analysis and machine learning. X. Front. Immunol. 14, 1134412 (2023).
Zhou, J. et al. Identification of aging-related biomarkers and immune infiltration characteristics in osteoarthritis based on bioinformatics analysis and machine learning. Front. Immunol. 14, 1168780 (2023).
Xuan, A. et al. The application of machine learning in early diagnosis of osteoarthritis: A narrative review. Ther. Adv. Musculoskelet. Dis. 15, 1759720X231158198 (2023).
Zhao, Z. et al. Necroptosis-related lncRNAs: Predicting prognosis and the distinction between the cold and hot tumors in gastric cancer. J. Oncol. 2021, 6718443 (2021).
Kearney, C. J. et al. Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ. 22, 1313–1327 (2015).
Hu, Y. H. et al. SPOP negatively regulates toll-like receptor-induced inflammation by disrupting MyD88 self-association. Cell. Mol. Immunol. 18, 1708–1717 (2021).
Wang, S. et al. Overexpression of toll-like receptor 4 affects autophagy, oxidative stress, and inflammatory responses in monocytes of transgenic sheep. Front. Cell Dev. Biol. 8, 248 (2020).
Winkler, S. & Rösen-Wolff, A. Caspase-1: an integral regulator of innate immunity. Semin Immunopathol 37, 419–427 (2015).
Bhattarai, K., Riaz, T., Kim, H. & Chae, H. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp Mol Med 53, 151–167 (2021).
Melbouci, D., Ahmad, A. & Decker, P. Neutrophil extracellular traps (NET): not only antimicrobial but also modulators of innate and adaptive immunities in inflammatory autoimmune diseases. RMD Open 9 (2023).
Yang, H. et al. New Insights into Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Inflammation. Front Immunol 7, 302 (2016).
Manning, B. D. & Toker, A. AKT/PKB signaling: Navigating the network. Cell 169, 381–405 (2017).
Shen, P. C. et al. Lentiviral small hairpin RNA knockdown of macrophage inflammatory protein-1γ ameliorates experimentally induced osteoarthritis in mice. Hum. Gene Ther. 24, 871–882 (2013).
Uible, E. et al. The Zymogen Form of Caspase-1 Is Required to Fine Tune Excessive Cell-Intrinsic Inflammation in Acute Myeloid Leukemia. Blood (2023).
Mathiessen, A. & Conaghan, P. G. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res. Ther. 19, 18 (2017).
Li, Y. et al. SIRT1 inhibits inflammatory response partly through regulation of NLRP3 inflammasome in vascular endothelial cells. Mol. Immunol. 77, 148–156 (2016).
Acknowledgements
We would like to thank Cong Lai for their encouragement and inspiration, as well as Editage (https://www.editage.com) for their writing assistance. This work was supported by the Youth Fund Project of Hainan Provincial Natural Science Foundation of China (grant no. 822QN450), Institutional Project of Hainan General Hospital of China (grant no. 2021QNXM22), and National Natural Science Foundation of China (grant no. 82260439).
Author information
Authors and Affiliations
Contributions
HH and YZ conceived and designed the experiments, analysed the data, and reviewed the drafts of the manuscript. YW, XG, and BW analysed the data, and wrote and reviewed the drafts of the manuscript. KL, JZ and ZZ analysed the data, prepared figures, and reviewed the drafts of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Guo, X., Wang, B. et al. Screening necroptosis genes influencing osteoarthritis development based on machine learning. Sci Rep 15, 9019 (2025). https://doi.org/10.1038/s41598-025-92911-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-92911-y
