
Abstract
To create a more comprehensive genetic analysis panel for peanuts, three high-density SNP panels were exploited. The refined SNP panel, PHR0301_Ah10K, comprises 10,000 SNP sites and demonstrated outstanding performance in sequence data analysis. It recorded the highest proportions of 99.53%, 96.48%, and 59.72% for the SNPs with minor allele frequency thresholds of MAF > 5%, MAF > 10%, and MAF > 20%, respectively. Moderate polymorphic information content (PIC) values were observed, with an average of 0.26, suggesting that the optimized SNP panel is informative. However, the PIC value for the four panels were skewed due to the small population size and limited genetic diversity (GD), as evidenced by the Kinship, PCA, and LD decay analyses. However, PHR0301_Ah10K demonstrated superior performance compared to the others in terms of variance explained in the PCA analysis while the outcomes of the genetic analyses confirmed its genotyping ability in peanut. The putative SNP sites associated with increased oleic acid levels have been integrated into this panel and validated, thus significantly enhancing its breeding potential. Moreover, the cost of genotyping by target sequencing (GBTS) using this panel is less than $9 per sample, making it more affordable. Due to its exceptional informativeness, cost-effectiveness, and breeding potential, we recommend this SNP panel for GBTS in peanut.
Similar content being viewed by others
Introduction
Groundnuts, commonly referred to as peanuts (Arachis hypogaea L.), are a significant agricultural product cultivated in more than 100 countries and consumed in nearly every nation worldwide. Peanuts are not only rich in protein (25–28%) and oil (48–50%), but they also provide essential vitamins and minerals that serve as a valuable energy source for humans1,2. Furthermore, peanuts are abundant in functional components such as fiber, vitamins, and fatty acids like oleic and linoleic acids3,4,5. Over the past decade, the demand for peanuts has consistently risen, driven by rapid population growth in Asia and Africa (http://faostat.fao.org/). The cultivated peanut is an allotetraploid (2n = 4 × = 40) resulting from the hybridization of Arachis duranensis (A subgenome) and Arachis ipaensis (B subgenome), with a genome size of 2.8 Gb6,7,8,9. Peanut subgenomes are significantly related10,11, exhibiting more than 60% of repetition rate, which leads to a notably low level of genetic diversity in cultivated peanuts12. Genome sequences of the two diploid ancestors were completed in 2016, laying the groundwork for a better understanding of the cultivated peanut genome9. Furthermore, the genetic sequencing of the cultivated peanut variety A. hypogaea was completed in 2019, offering essential insights for research in peanut biology, genetics, and breeding13.
Next-generation sequencing (NGS) is a cost-effective method that results in significant progress of biological research12. This method has facilitated the creation of a genome-wide molecular markers such as single-nucleotide polymorphisms (SNPs) for complex genomes through de novo assembly, resequencing and the application of various bioinformatics tools9,14,15,16. In fact, high-density SNP arrays have been developed for accurate mapping, which are widely used in various applications. These applications include high-density genetic profiling, genome-wide association studies (GWAS), and genomic selection17,18,19. Given the relatively large genome size and low genetic diversity in cultivated peanuts, SNP chips is essential for enabling high-throughput genotyping17. Several commercially available peanut SNP chips have been created to address this issue. A 58K Affymetrix SNP array was developed and used to genotype peanut mini core collections in US, which led to the discovery of SNP markers associated with traits related to arachidic and behenic fatty acid compositions20. The 58K SNPs has also been utilized for genetic research and breeding programs in peanuts21. A 48K Axiom Arachis SNP array was designed to detect single nucleotide polymorphisms (SNPs) across two sets of RILs and the two original Nod + parental lines, with the goal of exploring the genetic factors and regions that affect nodulation in peanuts22. Multiple fixed SNP arrays have been developed for many crops, including rice23,24, maize25,26, sunflower27, soybean28, oat29, cotton30, and wheat31,32. However, the DNA probes associated with those SNPs are designed on a standard chip array, which cannot be modified33. Additionally, the high cost of fixed SNP arrays makes them inaccessible to many companies. To address this issue, genotyping by target sequencing (GBTS) has emerged by using liquid chip technology. This approach is notable for its cost-effectiveness, minimal facility requirements, a diverse SNP chips of flexible marker types, shareable and cumulative marker data, and reduced demands for information management and support34. This technology has wide-ranging applications across various domains, including germplasm evaluation, high-density genetic linkage maps development, and safeguarding intellectual property rights related to crop samples34. To date, multiple GenoBaits SNP panels have been developed for both animals and plants. The majority of them were commercially available and utilized for GBTS at Molbreeding Biotechnology Co., Ltd. such as GenoBaits Maize 20K35, GenoBaits Rice 10K36, GenoBaits Soy40K33, GenoBaits Wheat 16K37, and GenoBaits Porcine SNP 50K38. Three SNP panels such as Arachis_hy01 with 40K SNPs, Arachis_hy02 with 40K SNPs, and Arachis_hy03 with 50K SNPs have been extensively utilized for genetic analysis in peanuts. The first two panels are based on the Arachis hypogaea cv. Tifrunner9, while the third panel uses Arachis hypogaea cv. Fuhuasheng as its reference genome39,40. Consequently, there are concerns regarding the analysis of their sequence data, as it can be affected by the genetic background of the plant materials and the reference genome employed in the analysis. Additionally, various genetic and breeding methods, such as genomic selection, require consistent high-resolution genotyping data for the same SNP loci across different germplasm collections, irrespective of the reference genome. To address this challenge and facilitate peanut genotyping, we have refined the three SNP panels. Through this optimization, we effectively merged the top-performing SNPs from these panels to create a cost-efficient 10K SNP panel.
Materials and methods
Plant materials
A panel of 48 peanut recombinant inbred lines (RILs) was utilized to generate sequence data through three commercial peanut SNP panels. Based on our prior knowledge of their pedigrees, the RIL population likely originated from a select number of commonly cultivated commercial peanut hybrids in China. All peanut samples were grown in an open field at the Experimental Demonstration Station of Molbreeding Biotechnology Co., Ltd. in Shijiazhuang, China.
Commercial peanut SNP panel
Three high-density SNP panels used in this work, including Arachis_hy01 and Arachis_hy02 based on the Arachis hypogaea cv. Tifrunner, and the Arachis_hy02, which is relied on the Arachis hypogaea cv. Fuhuasheng for sequence data analysis. Those SNP panels were currently used for peanut genotyping.
DNA isolation, sequencing, and first step of SNP selection
Genomic DNA was extracted from young leaves using the Polysaccharide-Polyphenol Plant Genomic DNA Extraction Kit (GenoPrep v2.0, Molbreeding Biotechnology Co., Ltd., Shijiazhuang, China). The purity and integrity of the extracted DNA were assessed through 1% agarose gel electrophoresis, while the DNA concentration was accurately measured using a Qubit. High-quality DNA samples (ranging from 20 to 300 ng) were then utilized for DNA library preparation with the DNA Library Prep Kit for ILM (GenoBaits v4.0, Molbreeding Biotechnology Co., Ltd., Shijiazhuang, China). Genomic DNA was fragmented using an ultrasonic Crusher (Ultrasonic Crusher Q800R3, Qsonica Co Ltd, USA) to achieve average DNA fragment sizes ranging from 200 to 500 base pairs (bp), with an input of 200 ng per sample. Following fragmentation, the DNA underwent a series of processes including end repair, adenylation, and ligation of Illumina paired-end adapters to the fragmented DNA. The resulting ligated DNA products were selected and amplified, then purified using GenoPrep DNA Clean Beads (Molbreeding Biotechnology Co., Ltd., Shijiazhuang, China) in freshly prepared 80% ethanol, following the manufacturer’s instructions. These fragmented DNA samples were then captured using the Peanut 40K SNP panel and 50K SNP panel based on the Arachis hypogaea cv. Tifrunner reference genome, as well as the Peanut 40K SNP panel based on the Arachis hypogaea cv. Fuhuasheng reference genome, at 65 °C using the DNA Hybridization Kit for ILM (GenoBaits v3.3, Molbreeding Biotechnology Co., Ltd., Shijiazhuang, China). The paired-end libraries were enriched using GenoBaits DNA probe beads (Molbreeding Biotechnology Co., Ltd) and sequenced on an Illumina Hiseq X Ten PE150 sequencer (Molbreeding Biotechnology Co., Ltd., Shijiazhuang, China) with a sequencing depth of 100-fold. The raw data underwent filtering using using FASTQ software41. The quality control process for genotype data included the removal of contaminated adapters and primers, as well as reads with more than 10 missing values (NA > 10) and those where 50% or more of the nucleotides had a quality score of 20 or lower. The cleaned reads were aligned to the reference genome using BWA (version 0.7.10-r789)42, applying a mapping score threshold of 30. The alignment results, initially in SAM format, were converted to Binary Alignment Map (BAM) files with SAMTools (Version 1.9)43. To remove duplicate reads, the Mark Duplicates function from the Picard tool (version 1.102)44 was used, and the resulting BAM files were then employed for further analyses. Single nucleotide polymorphisms (SNPs) for genotyping were identified using the standard pipeline of the Genome Analysis Toolkit (GATK, version 3.1)45.
Selection of SNP and probes
The first SNP batch was selected based on the following filtering criteria: missing rate < 0.2, minor allele frequency (MAF) > 0.05, bed uniformity > 0.1, and target rate > 0.5. The DNA probes associated with these SNP across the three SNP panels were aligned with the draft reference genome of Arachis hypogaea cv. Fuhuasheng to evaluate their matching events. The probes filtering criteria were as follows: a mismatch base pair rate less than 5% between the probes and the targeted DNA sequences. These mismatch rate comprised of the nucleotide length variation and mutated nucleotide. Probes that satisfied these criteria were mixed to develop the preliminary GenoBaits Peanut 10K panel.
Validation of the GenoBaits Peanut 10K panel
After experimental validation of the preliminary products, stringent filtering criteria was applied to ensure the selection of high-quality and representative SNPs. These filtering criteria are as follow: (1) SNP sites should not have missing data, ensuring 100% locus completeness; (2) SNP sites with MAF less than 0.2 will be removed; (3) SNP sites with a heterozygosity ratio greater than 50% must be removed; (4) Redundant probes should be removed; (5) SNPs are expected to be uniformly distributed throughout the genome. To achieve this goal, those SNPs with MAF threshold between 0.1 and 0.2, as well as those with a MAF range of 0.05 to 0.1, could be utilized to fill in the gaps.
Phylogenetic and genetic diversity analyses
To determine genetic diversity of the 48 peanut samples, a phylogenetic tree using the neighbor-joining method with the Kimura 2-parameter/p-distance model in MEGA-X software46, with 1000 bootstrap replicates was constructed. The resulting tree is visualized with MEGA-X software (www.megasoftware.net). To understand genetic diversity (GD), observed allele number (Ao), effective allele number (Ae), observed heterozygosity (Ho), expected heterozygosity (He) and polymorphism information content (PIC) were evaluated47,48. PIC values were classified in 3 different ranges as described48,49. Serrote et al.49 established the following classifications for PIC: low PIC (0–0.10), medium PIC (0.10–0.25), high PIC (0.3–0.4), and very high PIC (0.4–0.5). In comparison, Botstein et al.48 divided PIC into three categories: not informative PIC (0–0.25), informative PIC (0.25–0.5), and very informative PIC (> 0.5). The following formula is used to calculate allele frequency and PIC:
which: pi and pj are allele frequencies at allele i and j and n is number of allele, respectively.
Population structure
Principal component analysis (PCA) of the SNPs was conducted using GCTA (version 1.92.4) software, following the methodology outlined50. We calculated the variance explained by each principal component (PC) and created a score matrix for each sample across the PCs. High-confidence SNPs were utilized to analyze the population structure among 48 samples with ADMIXTURE (version 1.22)51, testing K values (the estimated number of populations) from 1 to 15. The number of sub-populations was evaluated through five-fold cross-validation. Stacked assignment bar plots of the Q matrix for each K value were generated using the R package Pophelper (http://royfrancis.github.io/pophelper). The optimal number of clusters was determined by examining the cross-validation error (CV error), with the K value yielding the lowest CV error indicating the most appropriate number of clusters.
Kinship and linkage disequilibrium
In a natural population, kinship refers to the genetic relatedness between two specific accessions, as well as the relative genetic relatedness among any accessions. The GCTA software (version 1.92.1)52 was utilized to estimate kinship among individuals within the population. The mean expected variance of SNPs was employed to adjust the expected variance, resulting in a heatmap of the kinship G matrix. Genetic relatedness of samples was analyzed following the methodologies outlined53,54. PopLDdecay (version 3.41)55 software was applied to calculate LD of two SNPs within 5000 kb on one chromosome. The LD coefficient was represented by r2 value.
Kompetitive Allele-Specific PCR (KASP) analysis
The KASP assay and results visualization were performed using the Automate High-throughput Genotyping System GeneMatrix (HC Scientific, Chengdu, China). For allele amplification, PACE® Genotyping Master Mix 2x (standard ROX) V2.0 (3CR Bioscience, United Kingdom) was utilized. The primer specific to high OA is associated with a HEX dye-labeled sequence, while the primer for low OA is linked to FAM dye-labeled sequences.
Phenotyping
The 48 peanut samples were cultivated at the Experimental Field of Molbreeding Biotechnology Co., Ltd during the late growing seasons of 2024 in Shijiazhuang, China. Seed quality related traits, in occurrence OA and LA were obtained using a near infrared analyzer (DA7250, Perten Instruments, Hägersten,Sweden). About 30 seeds were analyzed for two peanut varieties:冀花6331和冀花086-2-7. The OLR was obtained by calculating a ratio of oleic acid and linoleic acid.
Results
GBTS using the commercial SNP panels and promising SNPs selection
To evaluate the commercial SNP panels, such as Arachis_hy02, Arachis_hy01, and Arachis_hy03, 48 peanut samples were sequenced using GBTS. Here, we referred to the evaluation data as sequencing data performance (SDP). The DNA sequencing results revealed an average of clean bases ranging from 13,396,397.04 bp with a mapped ratio of 0.82 for Arachis_hy01 to 5,706,620.79 bp with a mapped ratio of 0.99 for Arachis_hy03. The uniformity20 varied from 0.87 for both Arachis_hy02 and Arachis_hy03 to 0.88 for Arachis_hy01, while the uniformity at 50 ranged from 0.60 for Arachis_hy01 to 0.63 for Arachis_hy03 (see Table 1 and Supplemental Table 1). The on-target values varied from 0.19 for Arachis_hy02 to 0.48 for Arachis_hy03, while capture efficiency ranged from 25.00% for Arachis_hy01 to 57.78% for Arachis_hy03 (see Table 1 and Supplemental Table 1). Upon analysis, the commercial SNP panel Arachis_hy03 scored the highest SDP values among the commercial panels. To determine promising SNP for the generation of a 10K panel, SNP with MAF > 0.05 were selected from the sequence data of three commercial panels and approximately 20,000 SNPs were prepared for the forthcoming SNP panel, designated PHR0301_Ah10K. Those SNP were experimentally validated with the 48 peanut samples. Hereafter, a more stringent filtering criteria were applied, which resulted in the selection of 10,000 SNP batch with three MAF classes, where SNP with MAF > 0.2 were used as primary criteria, SNP with MAF > 0.1 and MAF > 0.05 were used for gap filling. In this experiment, the average clean base pair count was 3,229,963.29 bp, with a mapping ratio of 0.99. The uniformity metrics were 0.99 for uniformity 20 and 0.85 for uniformity 50. The on-target and capture efficiency values were 0.59 and 65.64%, respectively (Supplemental Table 1). Despite having limited sequence data, PHR0301_Ah10K has demonstrated the highest SDP when compared to the three commercial SNP panels. In summary, the newly created 10 K panel outperformed the existing panels in terms of SDP in the GBTS analysis (see Table 1 and Supplemental Table 1).
Distribution and density of selected SNP across peanut chromosomes
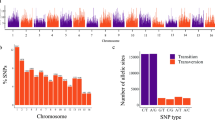
The total SNP number from each panel were utilized to perform a chromosome distribution analysis. The results indicated a high-density SNP distribution across the genome for Arachis_hy01, Arachis_hy02, and Arachis_hy03 panels (Fig. 1a–c). In contrast, The SNP distribution for PHR0301_Ah10K appeared to be sparse, exhibiting noticeable gaps (Fig. 1d). To investigate the polymorphic rate of different SNP panels, the proportions and densities of the SNPs at chromosomal level were calculated. Here, we defined 3 different classes of SNPs based on different minor allele frequency (MAF) threshold levels. Notably, for the MAF > 0.2 threshold level, PHR0301_Ah10K surpassed others in terms of proportion of SNP with high MAF per chromosome. However, it showed the lowest SNP density score, which was attributed to the removal of low-quality SNPs during the optimization process (see Table 2 and Supplemental Table 2). In fact, PHR0301_Ah10K achieved SNP proportions of 99.53%, 96.48%, and 59.72% for the three MAF classes of MAF > 5%, MAF > 10%, and MAF > 20%, respectively, when compared to the results obtained from the commercial SNP panels (Fig. 1e). This result demonstrated that the PHR0301_Ah10K is more informative than others.

Genome density and polymorphism of SNPs using the four different SNP panels. Genome-wide SNP distribution of SNP using panel Arachis_hy01 (a), Arachis_hy02 (b), Arachis_hy03 (c), and PHR0301_Ah10K (d) respectively, and (e) is the Percentage of SNPs with MAF threshold levels (MAF > 5%, MAF > 10%, and MAF > 20% across the various panels.
Genetic diversity analysis using the four SNP panels
This study focused on five key parameters, including PIC, Ao, Ae, Ho, and He to illustrate the potential of SNP panels for conducting genetic diversity analysis in peanuts. The SNP panel PHR0301_Ah10K achieved the highest scores for each parameter compared to the three commercial SNP panels, thus corroborating its high informativeness. In fact, average PIC values varied from 0.11 for Arachis_hy01 to 0.26 for PHR0301_Ah10K while average Ao values varied from 1.58 for Arachis_hy03 to 2.00 for PHR0301_Ah10K (Table 3). Average Ae ranged from 1.23 for Arachis_hy01 to 1.53 for PHR0301_Ah10K while He varied from 0.14 for Arachis_hy01 to 0.32 for PHR0301_Ah10K. Overall, the four SNP panels indicated a value of 0 for Ho, suggesting the lowest level of genetic diversity among the peanut samples analyzed (see Table 3 and Supplemental Table 3). Further, a series of genetic analyses were conducted, encompassing population structure, phylogenetic analysis, principal component analysis (PCA), linkage disequilibrium (LD) decay, and kinship analysis.
Population structure and phylogeny analyses using the four SNP panels
To evaluate the population clustering capabilities of these panels, a population structure analysis was performed. K values ranging from 1 to 15 were assessed for all the four SNP panels. The optimal K value was determined by examining the graphical representation of cross-validation errors (CV error) (Fig. S1 a–d). For the refined SNP panel PHR0301_Ah10K, optimal K values of 6 and 7 were identified (Fig. S1a and Fig. 2a, b). In contrast, the SNP panels Arachis_hy01 and Arachis_hy02 both showed an optimal K value of 6 at the lowest CV error, while Arachis_hy03 was assigned a K value of 7 (Fig. S1b–d and Fig. 2c–e). In conclusion, the three commercial SNP panels successfully classified the samples into either 6 or 7 clusters, depending on the specific panel used. Likewise, the refined SNP panel also grouped the samples into 6 and 7 clusters (Fig. 2a–e). Regarding phylogenetic analysis, all SNP panels classified the 48 samples into three distinct subgroups, although some individuals within specific subgroups exhibited variation (Fig. S2a–d). These results demonstrate that both the commercially available panels and their optimized versions are effective for population clustering analysis. Furthermore, the refined panel acts as an intermediate genotyping tool that reflects the original panels.

Population structure analysis of the peanut varieties using the four different SNP panels. Based on the optimal K values, samples were classified into subgroups 6 and 7 with the optimized SNP panel PHR0301_Ah10K (a, b). In contrast, the SNP panels Arachis_hy02 (c) and Arachis_hy01 (d) grouped the samples into 6 subgroups, while the SNP panel Arachis_hy03 (e) categorized them into 7 subgroups.
Principal component analysis, Linkage disequilibrium, and Kinship using the four SNP panels
Upon PCA analysis, variance explained (VE) values for PC1 were 12.38%, 12.57%, 13.91%, and 19.28% for Arachis_hy02, Arachis_hy01, Arachis_hy03, and PHR0301_Ah10K, respectively while for that of PC2, the values were 7.66%, 8.24%, 8.64%, and 8.83% for Arachis_hy03, Arachis_hy02, Arachis_hy01, and PHR0301_Ah10K, respectively (Fig. S3a–d). The results indicated that PHR0301_Ah10K is more prone to classifying samples, as shown by PC1 and PC2, even though no distinct subgroup was identified. The PCA outcome further demonstrated the closed genetic relation between samples used. To validate this assertion, a kinship analysis was conducted. The results indicated that the kinship values for the 48 samples, assessed with four different SNP panels, ranged from − 1 to 4 (Fig. S4a–h). This finding confirmed the close genetic relationships among the samples and highlighted the strong consistency of the experiments utilizing these SNP panels. On the other hand, the genetic mapping resolution of panels were further investigated through linkage disequilibrium (LD) analysis. The result revealed a notably gradual decline in LD, with the average LD (r2) of more than 0.4 across for all panels. In particular, the LD decay for Arachis_hy01 and PHR0301_Ah10K was relatively slow, showing a decay distance of around 1000 Kb. In contrast, Arachis_hy02 and Arachis_hy03 exhibited a relatively short decay distance of approximately 500 Kb (Fig. S5a–d).
Effectiveness and distinctiveness of the refined SNP panel
Cost-effectiveness of the optimized 10 K SNP panel was assessed through evaluation of costs associated with GBTS analysis using different panels. The costs per sample were found to be 77 RMB (10.70 USD) for Arachis_hy01, 70 RMB (9.73 USD) for Arachis_hy02 or Arachis_hy03, and 60 RMB (8.39 USD) for PHR0301_Ah10K (see Supplemental Table 4). These data suggest that the choice of the optimized SNP panel can significantly reduce GBTS costs in peanuts. Furthermore, the refined panel included functional SNP sites (A/G, A/-) identified in the FAD2A and FAD2B genes, which are associated with increased oleic acid (OA) levels. Specifically, DNA probes for GBTS and primers for KASP analysis were created (see Supplemental Table 5). The OA and linoleic acid (LA) content of two representative varieties from the 48 samples was assessed. The OA content ranged from 45% to 81.1%, while the LA content varied from 34.40% to 3.14%. The oleic-linoleic ratio (OLR) was found to be between 1.31 and 25.83 for 冀花6331 and 冀花086-2-7, respectively (see Fig. 3a and Supplemental Table 6). GBTS and KASP assays were further conducted for validation. The GBTS results indicated the presence of AA alleles at both targeted sites in FAD2A and FAD2B for 冀花086-2-7, while alleles GG and -/- were found for 冀花6331, respectively (Data not shown). KASP assay has clustered samples into three distinct groups: group 1, group 2, and group 3. The high OA content variety 冀花086-2-7 was grouped with samples in group 1 with HEX-dye due to the presence of AA alleles at the targeted sites in both FAD2A and FAD2B genes. In contrast, 冀花6331 was classified among individuals in group 2 with FAM-dye as a result of GG and -/- alleles in FAD2A and FAD2B genes (Fig. 3b, c).

Validation of putative SNP sites associated with high OA content in peanut. OA and LA content as well as OLR value of two representative peanut samples used in this study (a). KASP assay results for the SNP on peanut chromosome A09 (b) and that for SNP on chromosome B09 (c). The P-value indicates highly significant difference between the two samples. OA, LA and OLR indicate oleic acid, linoleic acid, oleic and linoleic acid ratio, respectively. The notation A/A represents homozygous alleles associated with high OA content, while G/G and -/- denotes homozygous alleles linked to low OA content. A/G and A/- indicate heterozygous alleles correspond to low OA content while the NTC indicates No template control.
Discussion
Creation of a high-efficiency SNP panel utilizing existing commercial SNP panels
In recent decades, significant advancements have been achieved in the genotyping of living organisms through genotyping by target sequencing (GBTS). This progress is largely attributed to the decreasing costs of next-generation sequencing (NGS) technology and the emergence of hybridization capture techniques. Numerous SNP liquid chips are now commercially available, offering a high-throughput genotyping solution that overcomes the higher costs and inflexibility associated with traditional SNP arrays. Molbreeding Biotechnology Co., Ltd. is a research and technical service company that specializes in the development of liquid chips and their application in genotyping. We utilize our proprietary hybridization capture method, known as the Genobaits tool, along with a high-throughput genotyping platform. Peanut is one of the most widely cultivated oil crops, serving as a significant energy source for people around the world, particularly in China1,2. At Molbreeding Biotechnology Co., Ltd, three liquid chip panels are utilized for commercial genotyping of peanut. The GenoBaits Peanut 40K (Arachis_hy01) and 50K (Arachis_hy03) panels are based on the Arachis hypogaea cv. Tifrunner, while the GenoBaits Peanut 40K (Arachis_hy02) is derived from Arachis hypogaea cv. Fuhuasheng. Despite their dependence on genome references, these panels exhibit suboptimal polymorphism, and they also require a relatively high volume of sequencing data. To address these challenges, an optimization process was initiated, resulting in the creation of a new SNP panel that demonstrates exceptional sequencing data performance during GBTS analysis, particularly in terms of uniformity at 20 and 50, as well as on-target values. This panel, designated PHR0301_Ah10K, consolidates all high-resolution SNPs from three separate panels and features 10,000 SNP sites with higher minor allele frequency (MAF) values compared to them. To ensure the panel’s reproducibility and to standardize the reference genome sequence, we aligned all probes to a single reference genome, specifically Arachis hypogaea cv. Fuhuasheng, achieving a mismatch base pair rate of under 5%. It is demonstrated that probes with up to 10% mismatches could still effectively capture the targeted sequence56, suggesting that the filtering criteria applied for probe selection are quite reasonable.
Evaluation of the refined SNP panel for genetic analysis in peanut
The genetic analysis of these panels is performed through a series of methods, including genetic diversity (GD), principal component analysis (PCA), genetic structure assessment, linkage disequilibrium (LD), and kinship analyses. Two key parameters for evaluating the polymorphism status of markers and the genetic diversity of plant materials are polymorphism information content (PIC) and expected heterozygosity (He). The Arachis_hy01 panel has a PIC value of 0.09, while the other three panels, including PHR0301_Ah10K, have PIC values ranging from 0.10 to 0.25. Based on the classification of Serrote et al.49, Arachis_hy01 is categorized as having low PIC, whereas the other three panels are classified as having medium PIC, including PHR0301_Ah10K. On the other hand, a PIC value below 0.25 is considered not very informative, values between 0.25 and 0.5 are informative, and values above 0.5 are very informative48. Based on the classification by Botstein et al.48, the three commercial panels are considered to be less informative than the optimized panel, which has an average PIC value of 0.26. This average PIC value of 0.26 is consistent with the results reported by Liu et al.57, Afolayan et al.58, Enyew et al.59, and Yahaya et al.60, all of whom employed molecular markers like SSR and SNPs to analyze collections of soybean and sorghum germplasm. While PHR0301_Ah10K performs better than the other panels in terms of PIC value, all panels exhibit relatively low PIC values. Additionally, all panels show reduced He values, which are often linked to low genetic diversity among the samples analyzed. To explore the genetic relationships among the samples, a kinship analysis was conducted. Kinship refers to the genetic relationships between individuals who share common ancestry. The kinship values for all four panels across the samples ranged from -1 to 4, indicating full or half-sibling relationships as defined by Kristen et al.54. This analysis reveals a strong paternal relationship among the samples used in this study, as evidenced by the PIC, He, and kinship values. These results further elucidate the limited genetic background of the RIL population, supporting the assertion that these RILs were derived from a small number of commercial peanut varieties. To further validate our findings, we conducted a Principal Component Analysis (PCA) to evaluate the level of genetic diversity and background differences among the samples. As anticipated, the PCA did not reveal any distinct subgroupings among the samples analyzed with the SNP panels. However, the PHR0301_Ah10K panel exhibited the highest variance explained (VE) values for both PC1 and PC2, indicating that it is more polymorphic than the other panels. Linkage Disequilibrium (LD) and LD decay values provide insights into the population’s type, size, and genetic diversity, as well as the number of markers used in the genetic analysis. Notably, our results showed a gradual decline in LD with increasing distance for LD decay across all four panels. It has been observed that inbred cultivars generally display a greater LD decay distance than diverse germplasm accessions and collections61,62,63,64. In our study, we employed inbred lines and similarly identified increased LD decay distances across all SNP panels. Additionally, Yan et al.65 highlight that both the number of markers and the population size play a crucial role in estimating LD values. They demonstrate that a smaller number of markers is associated with higher LD values and longer LD decay distances, which is attributed to a smaller sample size65. Our results align with these findings; however, the number of markers does not seem to correlate with LD decay distance, as shown in the Arachis_hy01 and PHR0301_Ah10K panels. Given the limited sample population in this study, the observed high LD decay distances for the SNP panels are primarily influenced by the population size. Conversely, the reduced He values are typically linked to a greater distance in linkage disequilibrium decay66. The reduced He value observed in the four panels supports this assertion. Overall, the relatively low levels of polymorphism and clustering ability seen in these SNP panels may be attributed to the high genetic similarities and the small sample sizes. Nevertheless, the limited genetic diversity of the samples can serve as an additional filtering criterion, aiding in the identification of highly polymorphic SNP panels. In terms of genetic analysis applications, PHR0301_Ah10K functions as a bridge panel, capable of substituting the three commercial SNP panels for peanut genotyping and various genetic analyses using a unique standardized reference genome sequence. This includes assessments of population structure, phylogenetic analysis, and genetic diversity, among others.
Breeding potential and cost efficiency of the enhanced SNP panel
High oleic peanut oils are highly valued for their superior nutritional benefits and improved thermo-oxidative stability, making them suitable for both health and industrial uses. In this study, we have introduced SNP sites associated with high oleic acid (OA) genes into a new panel to enhance breeding value. The SNPs included in this panel have been experimentally validated using KASP analysis, demonstrating a strong correlation between OA content and SNP genotyping results. As a result, these newly developed SNP panel can be effectively employed for marker-assisted breeding of high OA peanut. Currently, this panel is being utilized in high OA breeding by Molbreeding Company. Additionally, we have assessed the GBTS costs associated with four different panels and found that the newly developed SNP panel is more cost-effective for genetic analysis. Furthermore, costs can decrease even further as the volume of materials for genotyping increases. We believe this panel will be accessible to breeding units and laboratories, as cost-effective genotyping is essential for molecular breeding in small to medium enterprises and for breeding initiatives in developing nations.
Data availability
The data supporting these findings and the Peanut genotype services are available upon request. Please reach out to the corresponding authors for more information. The datasets exploited in this study can be accessed in the public repository of the National Center for Biotechnology Information (NCBI). The sequence reads for Arachis hypogaea cv. Tifrunner can be found under BioProject accession number PRJNA419393, while the sequence data for Arachis hypogaea cv. Fuhuasheng is available in the NCBI Sequence Read Archive under accession numbers SRP167797 and SRP033292.
References
Dhillon, S. S. et al. Reassociation kinetics and cytophotometric characterization of peanut (Arachis hypogaea L.) DNA. Plant Physiol. 65, 1121–1127 (1980).
Win, M. M. et al. Phenolic compounds and antioxidant activity of peanut’s skin, hull, raw kernel and roasted kernel flour. Pak. J. Bot. 43, 1635–1642 (2011).
Pasupuleti, J. et al. Groundnut improvement: use of genetic and genomic tools. Front. Plant Sci. 4, 785. https://doi.org/10.3389/fpls.2013.00023 (2013).
Radhakrishnan, R. et al. Evaluation of luteolin from shells of Korean peanut cultivars for industrial utilization. Afr. J. Biotechnol. 12, 4477–4480 (2013).
Musa, Ö. M. et al. Some nutritional characteristics of kernel and oil of peanut (Arachis hypogaea L.). J. Oleo Sci. 59, 1–5 (2010).
Smartt, J. et al. The genomes of Arachis hypogaea L. Cytogenetic studies of putative genome donors. Euphytica 27, 665–675 (1978).
Seijo, G. et al. Genomic relationships between the cultivated peanut (Arachis hypogaea, L.) and its close relatives revealed by double GISH. Am. J. Bot. 94, 1963–1971 (2007).
Robledo, G. et al. Species relations among wild Arachis species with the A genome as revealed by FISH mapping of rDNA loci and heterochromatin detection. Theor. Appl. Genet. 118, 1295–1307 (2009).
Bertioli, D. J. et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 48, 438–446 (2016).
Moretzsohn, M. C. et al. A study of the relationships of cultivated peanut (Arachis hypogaea) and its most closely related wild species using intron sequences and microsatellite markers. Ann. Bot. 111, 113–126 (2012).
Nielen, S. et al. Matita, a new retroelement from peanut: characterization and evolutionary context in the light of the Arachis A-B genome divergence. Mol. Genet. Genom. 287, 21–38 (2012).
Kim, K. S. et al. Development of SNP-based molecular markers by re-sequencing strategy in peanut. Plant Breed. Biotechnol. 5, 325–333 (2017).
Chen, X. et al. Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proc. Natl. Acad. Sci. 113, 6785–6790 (2016).
Yang, H. et al. Application of next-generation sequencing for rapid marker development in molecular plant breeding: a case study on anthracnose disease resistance in Lupinus angustifolius L.. BMC Genom. 13, 318 (2012).
Lee, J. et al. Genome-wide SNP identification and QTL mapping for black rot resistance in cabbage. BMC Plant Biol. 15, 32 (2015).
Kang, Y. J. et al. Resequencing of Capsicum annuum parental lines (YCM334 and Taean) for the genetic analysis of bacterial wilt resistance. BMC Plant Biol. 16, 235 (2016).
Pandey, M. K. et al. Advances in Arachis genomics for peanut improvement. Biotechnol. Adv. 30, 639–651 (2012).
Varshney, R. K. et al. Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics. Biotechnol. Adv. 31, 1120–1134 (2013).
Pandey, M. K. et al. Emerging genomic tools for legume breeding: current status and future prospects. Front. Plant Sci. 7, 455 (2016).
Otyama, P. I. et al. Evaluation of linkage disequilibrium, population structure, and genetic diversity in the US peanut mini core collection. BMC Genom. 20, 481 (2019).
Peng, Z. et al. Comparison of SNP calling pipelines and NGS platforms to predict the genomic regions harboring candidate genes for nodulation in cultivated peanut. Front. Genet. 11, 222 (2020).
Zou, K. et al. Genetic diversity and genome-wide association study of seed aspect ratio using a high-density SNP array in peanut (Arachis hypogaea L.). Genes 12, 1. https://doi.org/10.3390/genes12010002 (2021).
McCouch, S. R. et al. Development of genome-wide SNP assays for rice. Breed. Sci. 60, 524–535 (2010).
Chen, H. et al. A high density SNP genotyping array for rice biology and molecular breeding. Mol. Plant. 7, 541–553 (2014).
Ganal, M. W. et al. A large maize (Zea mays L.) SNP genotyping array: development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS One 6, e28334 (2011).
Unterseer, S. et al. A powerful tool for genome analysis in maize: development and evaluation of the high density 600 k SNP genotyping array. BMC Genomics 15, 823 (2014).
Bachlava, E. et al. SNP discovery and development of a high-density genotyping array for sunflower. PLoS One 7, e29814 (2012).
Song, Q. et al. Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS One 8, e54985 (2013).
Tinker, N. A. et al. A SNP genotyping array for hexaploid oat. Plant Genome 7, 3. https://doi.org/10.3835/plantgenome2014.03.0010 (2014).
Hulse-Kemp, A. et al. Development of a 63K SNP array for cotton and high-density mapping of intraspecific and interspecific populations of Gossypium spp. G3 (Bethesda) 5, 1187–1209 (2015).
Wang, S. et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 12, 787–796 (2014).
Winfield, M. O. et al. High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnol. J. 14, 1195–1206 (2016).
Liu, Y. et al. GenoBaits Soy40K: a highly fexible and lowcost SNP array for soybean studies. Sci. China Life Sci. 65, 1898–1901 (2022).
Xu, Y. B. et al. Genotyping by target sequencing (GBTS) and its applications (in Chinese). Sci. Agric. Sin. 53, 2983–3004 (2020).
Guo, Z. et al. Development of multiple SNP marker panels afordable to breeders through genotyping by target sequencing (GBTS) in maize. Mol. Breed. 39, 3. https://doi.org/10.1007/s11032-019-0940-4 (2019).
Hussain, I. et al. Identifcation of heterotic groups and patterns based on genotypic and phenotypic characteristics among rice accessions of diverse origins. Front. Genet. 13, 78. https://doi.org/10.3389/fgene.2022.811124 (2022).
Huang, S. et al. Epistatic interaction efect between chromosome 1BL (Yr29) and a novel locus on 2AL facilitating resistance to stripe rust in Chinese wheat Changwu 357–9. Theor. Appl. Genet. 135, 2501–2513 (2022).
Wang, X. et al. Using machine learning to improve the accuracy of genomic prediction of reproduction traits in pigs. J. Anim. Sci. Biotechnol. 13, 1. https://doi.org/10.1186/s40104-022-00708-0 (2022).
Chen, X. et al. Sequencing of cultivated peanut, Arachis hypogaea, yields insights into genome evolution and oil improvement. Mol. Plant. 12, 920–934 (2019).
Sun, Z. et al. Development and evaluation of the utility of GenoBaits Peanut 40K for a peanut MAGIC population. Mol. Breed. 43, 10. https://doi.org/10.1007/s11032-023-01417-w (2023).
Chen, S. F. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 145. https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, H. et al. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754. https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. et al. Te Sequence Alignment/Map (SAM) Format and SAMtools. Bioinformatics 25(2078–2079), 45 (2009).
Picard tools. https://broadinstitute.githu.bio/picard/ (2016).
Poplin, R. et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 36, 983. https://doi.org/10.1038/nbt.4235 (2018).
Kumar, S. et al. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Nei, M. et al. Sampling variances of heterozygosity and genetic distance. Genetics 76, 379. https://doi.org/10.1093/genetics/76.2.379 (1974).
Botstein, et al. Construction of a genetic linkage map in man using restriction fragment length polymorphism. Am. J. Hum. Genet.32, 314–331 (1980)
Serrote, C. M. L. et al. Determining the polymorphism information content of a molecular marker. Gene 726, 14415. https://doi.org/10.1016/j.gene.2019.144175 (2020).
Lu, Q. et al. A genomic variation map provides insights into peanut diversity in China and associations with 28 agronomic traits. Nat. Genet. 56, 530–540 (2024).
Alexander, D. H. et al. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Yang, et al. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Weir, B. et al. Genetic relatedness analysis: modern data and new challenges. Nat. Rev. Genet. 7, 771–780 (2006).
Kristen L. et al. Interpretation of DNA typing results for Kinship analysis. In National Institute of Standards and Technology USCIS Working Group on DNA Policy. http://www.cstl.nist.gov/strbase/NISTpub.htm. (2011)
Zhang, C. et al. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788 (2019).
Guo, et al. Development of high-resolution multiple SNP arrays for genetic analyses and molecular breeding through genotyping by target sequencing and liquid chip. Plant Commun. 2, 61. https://doi.org/10.1016/j.xplc.2021.100230 (2001).
Liu, M. J. et al. Genetic diversity of Shaanxi soybean landraces based on agronomic traits and SSR markers. Afr. J. Biotechnol. 10, 4823–4837 (2011).
Afolayan, G. et al. Genetic diversity assessment of sorghum (Sorghum bicolor (L.) Moench) accessions using single nucleotide polymorphism markers. Plant Genet. Resour. 17, 412–420 (2019).
Enyew, M. et al. Genetic diversity and population structure of sorghum [Sorghum bicolor (L.) Moench] accessions as revealed by single nucleotide polymorphism markers. Front. Plant Sci. 2, 799482. https://doi.org/10.3389/fpls.2021.799482 (2022).
Yahaya, M. A. et al. Genetic diversity and population structure of African Sorghum (Sorghum bicolor L. Moench) accessions assessed through single nucleotide polymorphisms markers. Genes 14, 1480. https://doi.org/10.3390/genes14071480 (2023).
Remington, D. L. et al. Structure of link age disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. 98, 11479–11484 (2001).
Flint-Garcia, S. A. et al. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 54, 357–374 (2003).
Van Inghelandt, D. et al. Extent and genome-wide distribution of linkage disequilibrium in commercial maize germplasm. Theor. Appl. Genet. 123, 11–20 (2011).
Beckett, T. J. et al. Genetic relatedness of previously plant-variety protected commercial maize inbreds. PLoS One. 12, 12. https://doi.org/10.1371/journal.pone (2017).
Yan, J. et al. Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS One. 4, 52. https://doi.org/10.1371/journal.pone (2009).
Smith, A. V. et al. Sequence features in regions of weak and strong linkage disequilibrium. Genome Res. 15, 1519–1534 (2005).
Acknowledgements
This work was supported by the MolBreeding Biotechnology Co., Ltd independent Peanut Research project MYF240014 entitled Construction of Peanut Rapid Backcross Breeding System.
Author information
Authors and Affiliations
Contributions
The concept was initially proposed by J.N.Z. and Y.F.X., while Y.R.Z. and Y.M.N.A. worked on developing the projects and the technical approach. The technical strategy received approval from J.N.Z., Y.F.X., and Y.R.L., who also generously supplied the peanut materials along with their pedigree information. Y.R.Z., Y.M.N.A., and S.L. raised the peanut seedlings, with Y.R.Z. be responsible for collecting leaf samples and conducting all GBTS analyses in collaboration with H.F.L., H.F.L. and S.A.Z. have performed DNA sequencing and provided accurate data, Finally, Y.M.N.A., Y.R.Z., J.H.D., R.X.Z. and H.F.L. carried out the data analysis and interpretations, while Y.M.N.A. drafted the manuscript and J.N. Z. provided critical revisions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, Y., Adedze, Y.M.N., Dong, J. et al. Optimization of commercial SNP arrays and the generation of a high-efficiency GenoBaits Peanut 10K panel. Sci Rep 15, 9995 (2025). https://doi.org/10.1038/s41598-025-93445-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-93445-z
