
Abstract
Neonicotinoids are a group of neurotoxic insecticides that possess significant threats not only to the environment but also to human health. This underlines the importance of developing efficient and accurate tools to detect neonicotinoids and track their behavior. Aptamers have been widely used as stable, efficient, and specific biorecognition molecules in biosensors. Nonetheless, no aptasensor was reported for the multiplexed detection of neonicotinoids. Herein, a graphene-based electrochemical biosensor was fabricated with three aptamers for the detection of imidacloprid, thiamethoxam, and clothianidin. The imidacloprid-specific aptamer underwent a truncation, which showed strong affinity with KD = 12.8 nM compared to 20.1 nM of the original sequence when studied with differential pulse voltammetry (DPV). Screen printed electrodes were coated with graphene oxide. After electrochemical reduction, 1-pyrenebutyric acid was used to functionalize the electrodes and covalently immobilize the aptamers. The electrodes were characterized by scanning electron microscopy (SEM), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS) to ensure successful aptasensor fabrication. The biosensor displayed excellent sensitivity compared with reported aptasensors and linear ranges from 0.01 ng/mL to 100 ng/mL for imidacloprid, thiamethoxam, and clothianidin. It also demonstrated excellent selectivity to the three analytes. Spiked extracts from tomato and rice samples were analyzed using our aptasensor, and results were validated through conventional chromatography assays. High recovery rates for all three neonicotinoids were obtained, demonstrating excellent agreement between the two methods. This study presents a cost-effective and simple multiplex detection for the sensitive, specific, and accurate on-site analysis of neonicotinoids.
Similar content being viewed by others

Introduction
Neonicotinoids are a class of insecticides that target the central nervous system (CNS) of insects by blocking the transmission of stimuli, making them highly selective and efficient for pest control in agriculture1. Imidacloprid, a commonly used neonicotinoid, disrupts the nicotinergic neuronal pathway by inhibiting nicotinic acetylcholine receptors, which can lead to paralysis and death as it interferes with nerve impulse transmission2,3. Compared to insects, imidacloprid shows significantly less toxicity to mammals due to its reduced affinity for mammalian neuron receptors4. Similarly, clothianidin, another neonicotinoid, operates in the same manner but offers increased durability and neurotoxicity5. Thiamethoxam, a novel neonicotinoid, stands out for its unique mechanism of action and effectiveness against pests that have developed resistance to other insecticides such as carbamates, organophosphates, and pyrethroids6,7.
Despite their agricultural benefits, the extensive use of neonicotinoids has raised significant environmental concerns. These compounds persist in soil and water, adversely affecting non-target species like beneficial insects, birds, and aquatic organisms8,9. These neurotoxins can also end up in the food chain, thus affecting other animals in the ecosystem10. Neonicotinoids effects on the population of bees and production of honey have been under great attention11. In response, regulatory actions have been taken in several countries, including the European Union, which has banned the use of imidacloprid, clothianidin, and thiamethoxam12. In the United Arab Emirates (UAE), commercial pesticides containing imidacloprid are restricted by the Ministry of Climate Change and Environment. Nonetheless, due to their efficacy, neonicotinoids remain difficult to replace, highlighting the need for precise analytical methods to monitor their presence and safeguard public health13.
Various techniques have been developed for detecting neonicotinoids in different matrices. Imidacloprid residues have been detected using liquid chromatography with tandem mass spectrometry (LC-MS/MS) in wheat14, and high-performance liquid chromatography (HPLC) and gas chromatography-mass spectrometry (GC-MS) in tea leaves15. Additionally, thiamethoxam and clothianidin residues were detected with LC-MS/MS in Swiss chard16, and using ultrahigh-performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) in cowpeas17and soil18. While these methods offer low detection limits, high selectivity, and accuracy, they require sophisticated sample pretreatment, complex operations, and the transportation of samples to centralized laboratories, which increases both time and cost.
Therefore, there are ongoing efforts to develop advanced analytical methods for on-site and rapid neonicotinoid detection. Antibodies have been widely used as recognition elements in detection methods such as enzyme-linked immunosorbent assay (ELISA), dual-labeled time-resolved fluoroimmunoassay (TRFIA), and fluorescence polarization immunoassay (FPIA)19,20,21. A gold-lateral flow immunosensor was also developed for imidacloprid, clothianidin, and imidaclothiz detection using antibodies22. Nonetheless, antibodies are limited in stability, reproducibility, and affordability, hence, aptamers were explored as more efficient recognition molecules23. Aptamers are short, single-stranded nucleic acid chains that can specifically bind to target analytes with high affinity and stability24. Produced chemically through the Systematic Evolution of Ligands by Exponential Enrichment (SELEX), aptamers overcome many of the limitations of antibodies, offering advantages like lower cost, easier modification, enhanced scalability and longer shelf life25,26.
Several aptamers were reported for the selective detection of neonicotinoids27. A specific aptamer for imidacloprid has been used in an electrochemical biosensor, demonstrating a limit of detection (LOD) of 0.190 ng/mL without performing any affinity studies28. Another aptamer was screened through capture-SELEX for clothianidin, which was then utilized in a label-free fluorescent biosensor, showing a dissociation constant (KD) of 40.7 nM23. As for thiamethoxam, a total of five aptamer-based biosensors were reported using different aptamers with KDranging between 54.6 nM and 4.93 µM, using fluorescence, colorimetric, electrochemical, and electrochemiluminescence detection methods29,30,31,32,33. Novel nanomaterials, such as carbon quantum dots and ZIF-7-derived porous carbon, have been incorporated to enhance sensitivity30,32.
Among the various detection methods, electrochemical biosensors have attracted significant attention for their portability, low cost, and high sensitivity, making them ideal for rapid, on-site detection34,35,36. However, multiplexed detection in aptamer-based biosensors remains challenging, as aptamers can vary significantly in size and binding conditions. Achieving multiplexing could enhance efficiency, reduce analysis time, and lower production costs37.
To improve the performance of the aptasensor, it is also necessary to optimize the aptamer length. Aptamer truncation enhances the binding affinity and reduces the production costs even further37. Graphene-based materials are of particular interest in electrochemical biosensors due to their unique properties. Their planar sp2carbon atoms offer a large specific surface area, while the highly mobile electrons increase their electrical conductivity and reduce charge transfer resistance. The large surface area enhances detection sensitivity when employed in biosensors due to the high loading capacity of biomolecules. In addition to their cost-effective preparation, the thermal and chemical inertness of graphene materials highlights their suitability for integration into biosensors38. The integration of reduced graphene oxide (rGO) and graphene oxide (GO) with aptamers has been previously employed for the detection of various pesticides39,40,41,42,43,44. Moreover, these materials provide a wide range of functionalization capabilities that enhance the versatility in various applications45.
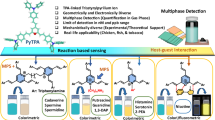
Herein, a rGO-based multiplexed electrochemical, aptamer-based biosensor was developed for simultaneous detection of imidacloprid, clothianidin, and thiamethoxam. To the best of our knowledge, this is the first aptasensor capable of detecting all three neonicotinoids within a single platform. Furthermore, the Imidacloprid-specific aptamer was truncated for the first time and evaluated in affinity studies. The developed biosensor was used to detect the three neonicotinoids in tomato and rice extracts, and its results were validated against the LC/MS/MS assay.
Experimental sections
Materials and reagents
Amine-labeled aptamers for clothianidin (Cloth) (5′-NH2-(CH2)6/GGCCGCCTCTGGGACTCTCGTTCCCTGGAG/−3′)23, thiamethoxam (Thiam) (5′-NH2-(CH2)6/GACGGATCCACCGACCATGCAAAGATGCACAAAAACG/−3′)31, imidacloprid (Imidac) (5′-NH2-(CH2)6/AGGAATTCAGATCTCCCTGCAGTGTCGTCTACGGTTTT GGTTGTTGTTTGTTGGTGGGTGTACTCGAGGAGCTCAGGATCCCG/−3′)28 and the designed truncated imidacloprid aptamers (Imidac T1: 5′-NH2-(CH2)6/AGGAATTCAGATCTCCCTGCAGTGTCGTCTACGGTTTTGG/−3′, Imidac T2: 5′-NH2-(CH2)6/TTGTTGTTTGTTGGTGGGTGTACTCGAGGAGCTCA GGATCCCG/−3′, Imidac T3: 5′-NH2-(CH2)6/TTCAGATCTCCCTGCAGTGTCGTCTACGGTGAGGAGCTCAGGATCCCG/−3′) were synthesized and HPLC purified by Metabion (Munich, Germany). Graphene oxide dispersion (4.00 mg/mL) was obtained from Graphenea (USA). The pesticides clothianidin, thiamethoxam, and imidacloprid, in addition to Tris, sodium chloride, magnesium chloride, 2-aminoethanol, dimethyl sulfoxide (DMSO), and acetonitrile were acquired from Supelco, Merck (Darmstadt, Germany). Graphene oxide (GO), 1-pyrenebutyric acid (Py), Potassium ferrocyanide (K4[Fe(CN)6]), and potassium ferricyanide (K3[Fe(CN)6]), HPLC-grade acetonitrile, methanol, ethanolamine (EA), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), and N-Hydroxy succinimide (NHS), ammonium acetate, magnesium sulfate (MgSO4), sodium citrate dibasic ((CH2COONa)2), and tri-sodium citrate (Na3C6H5O7) were obtained from Sigma-Aldrich (Massachusetts, USA). Dipotassium hydrogen phosphate (K2HPO4) and potassium dihydrogen phosphate (KH2PO4) were purchased from Merck (Darmstadt, Germany). Primary Secondary Amine (PSA) and octadecyl-modified silica (C-18) were supplied by DIKMA Technologies (Beijing, China). Phosphate buffer saline (PBS), 0.100 M, pH of 7.4, prepared from K2HPO4, KH2PO4, and NaCl, was used as a supporting electrolyte for CV and DPV measurements. The electrochemical measurements were performed using 5.00 mM K4[Fe(CN)6]/K3[Fe(CN)6] in 0.100 M PBS, pH 7.4. Binding buffer, pH 7.5, was prepared from 50.0 mM Tris, 2.00 mM magnesium chloride, and 150 mM sodium chloride and used in pesticides and aptamers dilutions. Lastly, spin-X (0.450 μm) cellulose acetate centrifuge tube filters were purchased from Costar (Utah, USA).
Instrumentations
The morphology of the electrodes was characterized using scanning electron microscopy (SEM) by Quanta 250 FEG, FEI (Oregon, USA), and WITEC alpha300 Raman spectroscopy. X-ray photoelectron spectroscopy (XPS) by Thermofisher Scientific was used for surface characterizations. Electrochemical analyses were carried out using CV and DPV with the MultiEmStat4 Potentiostat by PalmSens (Houten, Netherlands), run using the MultiTrace 5.0 software. Screen printed electrodes (SPE) with eight carbon working electrodes (WEs) and an Ag/AgCl reference electrode and cable connectors for SPEs (Ref. CAC) were acquired from DropSens, Metrohm (Herisau, Switzerland). Comparative chromatographic analyses were conducted with an Agilent 6430 LC/MS/MS (Santa Clara, USA) instrument using a C-18 analytical column (Zorbax 2.10 mm inner diameter x 100 mm length, 3.00 μm particle size; Agilent Technologies, Santa Clara, USA).
Methods
Truncation design study
The secondary structures and free energies of the three aptamers were obtained through the mfold software46 (https://www.unafold.org/mfold/applications/dna-folding-form.php) at 25 ℃ with [Na+]: 100 nM and [Mg2+]: 2.00 nM. Three truncations were then proposed for Imidac aptamer based on its structure that contains three stems and loops. The first truncation (Imidac T1) included two of the stem-and-loop structures, while the second truncation (Imidac T2) contained the remaining stem-and-loop. The final truncation (Imidac T3) was tailored to contain all three loops and exclude the linear region. All sequences are provided in TableS1.
Aptasensor fabrication and characterization
Starting with the multiplexed SPE, 5.00 µL of 1.00 mg/mL GO dispersion was dropcasted and left overnight at room temperature. Electrochemical reduction was then performed using CV in the range from − 0.3 to −1.3 V at a scan rate of 0.05 V/s for 20 cycles. The DPV with E pulse 0.05 V, T pulse 0.02 s, and scan rate of 0.02 V/s was utilized for the measurement. Both CV and DPV were conducted in 0.100 M PBS buffer solution, pH 7.4, containing 5.00 mM K4[Fe(CN)6]/K3[Fe(CN)6]. The GO/SPE and rGO/SPE were characterized using SEM and Raman spectroscopy. Functionalizing was then done by dropping 5.00 µL of 0.100 M Py, incubating for an hour, and rinsing with PBS buffer to remove excess reagents. A solution of 100 mM EDC and 20.0 mM NHS in 0.100 M PBS buffer at pH 5.5 was then incubated on the Py-modified electrodes. The electrodes were then washed and incubated with 1.00 µM of amine-labeled aptamers in PBS (pH 7.4) for 3 h and consequently with 0.100 M ethanolamine (EA) for half an hour to block the remaining binding sites on the electrode surface, while taking CV and DPV measurements throughout. The electrodes were lastly washed with PBS buffer to be utilized in further studies. For multiplexed detection, an 8 WEs SPE was used with two electrodes per analyte. To confirm successful functionalization, XPS spectra, CV and DPV were obtained at each step of the fabrication protocol. The steps are summarized in Fig. 1.
Binding affinity of the truncated aptamers
Four electrodes were then prepared and characterized as described earlier for Imidac parent aptamer and its three truncations (T1-T3). Initially, 1 mM of pesticide stocks were prepared in DMSO. Serial dilutions were performed on the imidacloprid stock solution with binding buffer. 10.0 µL of each concentration was then dropped on the electrode, sequentially, and left for 30 min before washing with PBS. The resulting DPV measurements were recorded with the operating conditions mentioned above. KD were calculated on GraphPad Prism 8 software using nonlinear regression analysis.
Binding optimization
Working solutions of the aptamers for all three analytes were prepared with PBS in a series of solutions with concentrations of 0.50, 1.00, 1.50, 2.00, and 2.50 µM. The aptasensors were then fabricated with the different aptamers and aptamer concentrations. DPV signals were then recorded after the addition of 100 ng/mL of the corresponding analytes to the electrodes. After fixing the aptamer concentration to 1.00 µM, 100 ng/mL of analytes were incubated for different periods of time (10–60 min). The concentrations/ binding time were then plotted against the change in current \(\:\varDelta\:I\) observed in DPV, which was calculated using the following formula:
Where \(\:{I}_{^\circ\:}\) is the peak current of the background signal of the aptasensor and \(\:I\) is the peak current of the solution being tested.
Multiplexed detection
Sensitivity studies were conducted at room temperature by dropping 10.0 µL of each concentration (0.0100–100 ng/mL) of each pesticide on its corresponding electrode and recording the DPV signal after 30 min incubation and washing with PBS. The change in current was plotted against the analyte concentration in the log scale to obtain a linear graph. The LOD values were calculated using formula 2 based on the standard deviations of blank measurements (s) and the slopes of the constructed detection plots.
For selectivity studies, the individual electrodes dedicated to imidacloprid, thiamethoxam, and clothianidin were tested against 10.0 ng/mL of all three analytes, in addition to dinotefuran, sequentially. The multiplexed electrode (8 WEs) was then utilized to test different mixtures of neonicotinoids to determine selectivity using the same conditions mentioned above. The first mixture (Mix 3) was prepared using 10 ng/mL of each imidacloprid, thiamethoxam, and clothianidin. Mix 2 excluded imidacloprid and contained 10 ng/mL of the latter two. Lastly, 10 ng/mL of clothianidin was also tested on the multiplexed electrode.
Real sample analysis
A globe tomato and basmati rice were used for real sample analysis (obtained from a local supplier in Abu Dhabi, UAE). 1.00 mL of distilled water was added to the rice before each sample was blended separately to form a homogenous mixture. One gram of each was mixed with 2.00 mL of distilled water and 4.00 mL of acetonitrile and then vortexed for 3 min. The solutions were centrifuged at 4000 g for 10 min. One milliliter of each of the resulting supernatants was transferred to new tubes and placed in a dry bath set at 60 ℃ to fully evaporate. The residues were dissolved in 1.00 mL of the binding buffer and vortexed to homogenize. The solutions were then filtered in 200 µL aliquots using 0.450 μm cellulose acetate centrifuge filters. The extracts were diluted 1:10 using the binding buffer and were then spiked with the three analytes in the concentrations 10.0, 50.0, and 100 ng/mL. Using the same detection procedure above, the spiked samples were tested using the prepared aptasensors.
LC-MS/MS analysis
The LC-MS/MS analysis followed a previously reported protocol47. Rice and tomato samples were homogenized thoroughly and then spiked with three different concentrations (10.0, 50.0, and 100 ng/mL) of pesticide standards. 10.0 g of the homogenized tomato was weighed and added to 10.0 mL of acetonitrile before being hand-shaken vigorously for 1 min. Similarly, 5.00 g of homogenized rice was mixed with 5.00 mL of water and 10.0 mL of acetonitrile. MgSO4 (4.00 g), NaCl (1.00 g), (CH2COONa)2 (0.500 g), and Na3C6H5O7 (0.500 g) were then added to each sample and shaken using a vortex mixer for 1 min. The tubes were then centrifuged at 5000 rpm at 4 °C for 5 min. Rice samples were stored at − 20 °C for 2 h. The supernatants (1.00 mL) were transferred into 2.00 mL dispersive solid-phase extraction tubes containing 50.0 mg of PSA, 100 mg of C-18, and 150 mg of anhydrous MgSO4. The tubes were shaken vigorously for 1 min before another centrifugation was conducted at 5000 rpm at 4 °C for 5 min. The supernatants obtained were filtered through 0.450 μm polyvinylidene fluoride (PVDF) filters into vials. The analysis was accomplished with the Agilent LC-MS/MS instrument at 40 °C. The flow rate and injection volume were 0.200 mL/min and 10.0 µL, respectively. The mobile phases used were (A) 5.00 mM ammonium acetate and 20%methanol in water, and (B) 5.00 mM ammonium acetate in methanol. The gradient elution program started with 40%of mobile phase B for 3.5 min, then increased to 55%for 4.5 min, then 95% for 22 min, and then decreased to 15%for 10 min. The final run time of the method was 40 min.

Schematic representation of the prepared multiplexed aptasensor.
Results and discussion
Electrode modification
Reduction of GO generally enhances the electron transfer process, and this high conductivity improves the utility of the material. Besides the excellent electrochemical properties of rGO, its graphitic sites are utilized in π-π stacking in the functionalization with Py48. Carboxylic groups in Py are activated with the addition of EDC/NHS, which allowed the immobilization of amine-labeled aptamers as previously reported49. The morphology of GO casting on a carbon electrode was studied using SEM. In Fig. 1B, it can be observed that GO shows full coverage, indicating good distribution on the electrode surface. The wrinkles and folds on the GO represent the presence of oxygen in functional groups including carboxyl, hydroxyl, and epoxy50. Reduction of GO therefore removed some of the wrinkles. The Raman spectra in Fig. 1B demonstrated typical carbon nanostructure G and D bands at 1579 and 1347 (cm−1), respectively, where the former represents C-C stretching and the latter is generated by aromatic ring breathing mode, associated with defects in the graphene structure51. The intensity ratio of the D and G bands (ID/IG) for rGO (0.940) was higher than GO (0.707). Additionally, the calculated AD/AG ratios for GO and rGO samples were 0.73 and 1.02, respectively. Following reduction, the increase in the ID/IG and AD/AGratios may suggest a decrease in sheet size, which increases edge defects52.
XPS was used to investigate the chemical composition of the functionalized electrode. The C/O ratio evidently increased from 4.08 in GO to 7.15 in rGO, confirming that a reduction took place. Figure 2A shows the carbon 1s peak of GO before reduction where the peaks indicate the presence of C-C, C = C. C-O, and C = O bonds. After reduction, a decrease in the C-O and C = O bonds was confirmed as presented in Fig. 2B. The emergence of COOH bond and the increase in C-O and C = O content in the C 1s spectrum depicted in Fig. 2C indicates the successful Py functionalization on the rGO surface. This acts as a substrate for amide bond formation once the amine labeled aptamers are added. As can be seen in Fig. 2D, the addition of the Imidac aptamer showed the emergence of C-N bond. This step was also tracked using N 1s spectra, where C = N, C-NH peaks confirm the presence of the DNA nitrogen bases and NHC = O confirms the formation of the amide linkage (Fig. 2E). The same trend was observed with Cloth and Thiam aptamers electrode modification, shown in Fig. S1.

XPS C 1s spectra of the imidacloprid electrode modification steps (A) GO, (B) rGO, (C) after functionalization with Py, (D) after aptamer addition, and (E) N 1s spectrum after aptamer addition.
Aptamer Truncation
Aptamers selected through SELEX may contain flanking nucleotides that are not involved in binding. Retaining these non-binding regions can complicate their integration into biosensor platforms due to minimal conformational changes upon target binding and increased synthesis cost31. Previous reports suggest that the main binding sites in aptamers are stem-loop, hairpin, and other structures53. While stems mainly stabilize the structure, the loops undergo conformational changes driven by hydrogen bonds and hydrophobic interactions as the aptamer encounters its target23. The sequence for clothianidin was used after omitting the primer binding sites. However, three truncations were proposed for the aptamer previously selected for imidacloprid28 based on its secondary structure, which showed three stem-loops as in Fig. 3A. Imidac T1 and Imidac T2 aptamers (Fig. 3B and C) did not possess any additional intrastrand interactions when compared to Imidac parent aptamer. Imidac T3 aptamer (Fig. 3D), however, has undergone significant conformational change, resulting in a big stem with two loop structures and almost no linear region. These properties are expected to increase Imidac T3’s binding affinity towards imidacloprid. The affinity is usually expressed by determining KD, where smaller KDvalues indicate stronger affinities54.
It is worth noting that the KD of Imidac parent aptamer was not previously reported. To determine the KD values of the parent and the truncated sequences, different concentrations of imidacloprid were incubated on aptasensors prepared using the long, T1, T2 and T3 aptamer sequences and the signal detected using DPV. A saturation curve was plotted using \(\:\varDelta\:I\) values against imidacloprid concentration, which was then used to calculate KD by nonlinear regression (Fig. 3E). The KD values were found to be 20.1 nM for Imidac aptamer and 12.8 nM for Imidac T3 aptamer. Imidac T1 and Imidac T2 aptamers showed small changes in signal, which did not allow accurate calculation of KD.
Figure S2 and Fig. 4 show CV and DPV measurements of Imidac aptamer and its three truncations after incubation with imidacloprid. The voltammograms revealed that Imidac T3 aptamer exhibits the largest change in peak current when compared to Imidac, Imidac T1, and Imidac T2 aptamers. This indicates that Imidac T3 aptamer has the highest affinity towards its target analyte, which was evidently greater than the original sequence. Imidac parent aptamer was shortened by 35 bases, starting with an 83 nucleotide-long sequence and ending with onl y 48. This showed improved affinity and conformational changes towards imidacloprid indicated by the strong change in signal. Since T3 includes all three stem loops and excludes the linear region of the original aptamer, it can be confirmed that the complex structures are the core binding sites.

Secondary structures of the aptamers (A) Imidac, (B) Imidac T1, (C) Imidac T2, (D) Imidac T3, and (E) KD fitting of Imidac and Imidac T3 aptamers.

DPV characterization of electrodes immobilized with Imidac sequences. (A) Imidac electrode, (B) Imidac T1 electrode, (C) Imidac T2 electrode, and (D) Imidac T3 electrode.
Electrode characterization
The individual steps of electrode modification are examined by CV in Fig. S2 (A-D) for Imidac aptamer and its truncations. All the observed curves are owed to the redox reaction of K4[Fe(CN)6]/K3[Fe(CN)6]. The peak intensities decreased with each step, as expected, since the buildup of material on the electrode surface limits the redox process30. This was observed starting with the decrease in peak current with the addition of Py. The coupling agents EDC and NHS were then added to activate the carboxylic groups in Py to facilitate the formation of amide bonds with the amine-labeled aptamers55. This was confirmed by the additional decrease in peak current with the aptamer due to the negative charge on the DNA which repels the anions of the redox probe. This suggests that the aptamer immobilization is successful. After occupying the non-specific sites with EA, the peak current decreased even more.
The same trend was also observed with DPV with a sequential decrease in cathodic peak intensities. Generally, DPV has a relatively higher resolution and sensitivity compared to CV56. It can be seen in Fig. 4 (A-D) that the DPV curves show better quality peaks demonstrated in a significant change in current. This suggests that the electrochemical behavior of the assay is better observed using DPV, and it is therefore the technique used moving forward. DPV experiments were also conducted to examine the electrode modification for the three pesticides and the results are shown in Fig. S3. The feasibility of the prepared electrodes to detect the corresponding analytes was observed after incubating the pesticides in question. All three electrodes showed a significant decrease in peak current, suggesting that the analyte-aptamer complex remarkably hinders electron transfer as the aptamer conformation changes upon binding. This indicates that the immobilized aptamers can retain their targets well.
Binding optimization
The assay conditions (binding time and aptamer concentration) were optimized to obtain the ideal performance of the aptasensor. These parameters were plotted against the change in peak current\(\:\:(\varDelta\:I)\). Fig. S4A shows the increase in \(\:\varDelta\:I\) with increased binding time of clothianidin on its corresponding electrode. The change is significant during the first 30 min, while further incubation showed little to no change, suggesting that the electrode has reached equilibrium. Similar patterns can be seen in Fig. S4B for imidacloprid and Fig. S4C for thiamethoxam detection, all showing the maximum \(\:\varDelta\:I\) around 30%. For imidacloprid, however, the curve did not reach a plateau at 30 min, but rather continued to increase at a much slower rate. Thus, 30 min was selected as the optimum binding time for the subsequent aptasensing experiments.
Additionally, aptamer concentration was optimized in the range of 0.50 to 2.50 µM. For Cloth aptamer (Fig. S4D), the increase in \(\:\varDelta\:I\) was abrupt until 1.00 µM but then continued to slowly increase until 2.00 µM. Imidac aptamer (Fig. S4E) reached the maximum value at 1.00 µM, after which the \(\:\varDelta\:I\:\)started to decrease. As for Thiam aptamer (Fig. S4F), maximum peak current change was reached at 1.50 µM and the values decreased beyond that. We attribute the decrease in the aptasensor response at higher aptamer loading to the steric hindrance effect which may negatively impact the binding of the aptamers to the analytes. Taking all three cases into consideration, the optimum aptamer concentration is found to be 1.00 µM.
Multiplexed detection of Imidacloprid, Clothianidin, and Thiamethoxam
With the optimized conditions, the aptasensor performance was evaluated by measuring the electrochemical response with changes in analyte concentration. The DPV curves of imidacloprid binding given in Fig. 5A indicate that higher analyte concentration leads to lower peak current as the aptamer-imidacloprid complex hinders the electron transfer. Moreover, the aptamer conformational changes upon binding have a contribution to the change in signal. A linear plot with the regression equation \(\:\varDelta\:I=35.2+11.1\:Log\left[C\right]\) and \(\:{R}^{2}=0.983\) was achieved when the change in peak current was plotted against the logarithm of imidacloprid concentrations from 0.0100 ng/mL to 100 ng/mL (Fig. 5B). Similar results were achieved with clothianidin (Fig. 5C and D) giving \(\:\varDelta\:I=31.0+9.83Log\left[C\right]\), \(\:{R}^{2}=0.986\) and thiamethoxam (Fig. 5E and F) showing \(\:\varDelta\:I=38.6+11.0\:Log\left[C\right]\), \(\:\:{R}^{2}=0.989\). The LOD for the three biosensors were 6.30, 6.80, and 7.10 pg/mL for imidacloprid, clothianidin, and thiamethoxam, respectively. These values were significantly lower than the LODs for other electrochemical, colorimetric, and fluorescence aptasensors reported previously for the detection of neonicotinoids, as presented in Table S2. Truncation of Imidac aptamer showed a significant decrease in the LOD when compared to the previously reported aptasensors. The LODs obtained for clothianidin and thiamethoxam were also lower than other reported methods likely due to the enhanced electrochemical behavior of the rGO-modified electrode and the efficient aptamer immobilization. Moreover, with the sample extraction method of 1 g in 6 mL of solvent, as described in the experimental section, the LODs in µg/kg are significantly lower than the maximum residue levels (MRLs) established by the European Commission. This demonstrates that our method is highly sensitive and capable of detecting residues well below regulatory limits. Specifically, for clothianidin and thiamethoxam, our LOD (0.0408–0.0426 µg/kg) is far below the lowest MRL (10 µg/kg). Similarly, for imidacloprid, the MRL is 600 µg/kg, which greatly exceeds our LOD (0.0378 µg/kg). These findings confirm that the developed aptasensor exhibits exceptional sensitivity toward all three analytes, making it highly suitable for trace-level detection.

Change in current of the aptasensors after incubation with different concentrations of the corresponding analytes. (A, B) imidacloprid, (C, D) clothianidin, and (E, F) thiamethoxam.
Selectivity is an important factor in evaluating a biosensor’s performance. The cross-reactivity studies included clothianidin, imidacloprid, thiamethoxam, as well as dinotefuran. First, 10.0 ng/mL of each pesticide was independently incubated on each electrode and the corresponding \(\:\varDelta\:I\) values are plotted in Fig. 6A. Signals of the interferents were not significant when compared to the analyte in question on its corresponding electrode. Another test was performed using mixtures of the analytes incubated on the multiplexed electrode containing all three aptamers. The results in Fig. 6B indicate the biosensor’s ability to distinguish between the three analytes, even in scenarios where not all three are present.
Although this Cloth aptamer was previously tested for cross-reactivity, to our knowledge, this is the first selectivity test for the given Imidac and Thiam aptamers. Moreover, the label-free fluorescent aptasensor developed for clothianidin was not tested against thiamethoxam nor dinotefuran, however, it showed good selectivity against other pesticides like imidacloprid and acetamiprid23. In the reported work, the signals obtained were slightly higher with imidacloprid and acetamiprid than our results, suggesting improved selectivity of our platform, likely due to the elimination of primer binding sites in our design. The biosensor’s ability to distinguish between imidacloprid and thiamethoxam despite their structural similarities is outstanding. The results, hence, support the high selectivity of the aptasensor towards each analyte, which is attributed to the high specificity and affinity of each aptamer to its target.

Selectivity of the biosensor. (A) incubation with neonicotinoids and (B) incubation with mixtures of analytes.
Real sample analysis and validation of the aptasensor against LC-MS/MS method
The viability of the developed biosensor in detecting the three analytes in real samples was tested with tomatoes and rice extracts. Aliquots were individually spiked with clothianidin, imidacloprid, and thiamethoxam in three concentrations 10.0, 50.0, and 100 ng/mL. All solutions were analyzed using the aptasensors and were then converted into concentration values as displayed in Table 1. The spiked samples were also analyzed using LC-MS/MS as shown in Table 1. The recoveries using the aptasensor were found to be in the range 99.0–100.8% with relative standard deviation values (RSD) from 1.01 to 3.39%. On the other hand, the recovery range from LC-MS/MS results was much wider (76.0–118.7%) and the RSD varied greatly (0.79–8.16%). The recovery and RSD values of the aptasensor demonstrate good repeatability, consistency, and stability suggesting the capability of the prepared aptasensor to simultaneously detect three neonicotinoids in food samples. This also indicates its potential as a tool for rapid on-site analysis of these pesticides.
Conclusion
In summary, an electrochemical aptasensor allowing multiplexed yet selective detection of imidacloprid, thiamethoxam, and clothianidin was developed. The Imidac aptamer was truncated and analyzed by CV and DPV, giving the lowest KD value of 12.8 nM compared to 20.1 of the original aptamer. The truncation design study demonstrated that imidacloprid specifically binds to the stems and loops in the aptamer, indicated by the largest change in peak current with the third truncation. This sequence, in addition to Thiam and Cloth aptamers, were immobilized on an array of electrochemically reduced graphene oxide modified electrodes to construct a multiplexed electrochemical biosensor. Good sensitivity and selectivity were observed with all three analytes. The biosensor was tested on real samples of tomato and rice, and the percentage recoveries were in the range of 99.0–100.8%. The aptasensor results were validated by comparison with LC-MS/MS method in the detection of spiked samples with the three neonicotinoids, showing excellent agreement. These results show that the prepared aptasensor can be utilized for the rapid and sensitive detection of the three neonicotinoids in environmental and food samples.
Data availability
Data is provided within the manuscript and supplementary information files.
References
Cicero, N. et al. Monitoring of neonicotinoid pesticides in beekeeping. Nat. Prod. Res. 31, 1258–1262 (2017).
Jeschke, P., Nauen, R., Schindler, M. & Elbert, A. Overview of the status and global strategy for neonicotinoids. J. Agric. Food Chem. 59, 2897–2908 (2011).
Buszewski, B., Bukowska, M., Ligor, M. & Staneczko-Baranowska I. A holistic study of neonicotinoids neuroactive insecticides—properties, applications, occurrence, and analysis. Environ. Sci. Pollut. Res. 26, 34723–34740 (2019).
Loser, D. et al. Acute effects of the Imidacloprid metabolite desnitro-imidacloprid on human nACh receptors relevant for neuronal signaling. Arch. Toxicol. 95, 3695–3716 (2021).
Basley, K. & Goulson, D. Neonicotinoids Thiamethoxam and Clothianidin adversely affect the colonisation of invertebrate populations in aquatic microcosms. Environ. Sci. Pollut. Res. 25, 9593–9599 (2018).
Ramanathan, S., Kumar, M., Sanjeevi, S., Narayanan, G., Kurien, A. A. & B. & Thiamethoxam, a neonicotinoid poisoning causing acute kidney injury via a novel mechanism. Kidney Int. Rep. 5, 1111–1113 (2020).
Khan, H. A. A., Akram, W., Iqbal, J. & Naeem-Ullah, U. Thiamethoxam resistance in the house fly, musca domestica L.: current status, resistance selection, Cross-Resistance potential and possible biochemical mechanisms. PLoS One. 10, 125850 (2015).
Bhandari, G., Atreya, K., Scheepers, P. T. J. & Geissen, V. Concentration and distribution of pesticide residues in soil: Non-dietary human health risk assessment. Chemosphere 253, 126594 (2020).
Teklu, B. M., Haileslassie, A. & Mekuria, W. Pesticides as water pollutants and level of risks to environment and people: an example from central rift Valley of Ethiopia. Environ. Dev. Sustain. 24, 5275–5294 (2022).
Lindenmayer, D. B., Wood, J., MacGregor, C., Hobbs, R. J. & Catford, J. A. Non-target impacts of weed control on birds, mammals, and reptiles. Ecosphere 8, 1804 (2017).
Xu, M. et al. Neonicotinoid microsphere Immunosensing for profiling applications in honeybees and Bee-Related matrices. Biosens. (Basel). 12, 792 (2022).
Azpiazu, C., Bosch, J., Martins, C. & Sgolastra, F. Effects of chronic exposure to the new insecticide sulfoxaflor in combination with a SDHI fungicide in a solitary bee. Sci. Total Environ. 850, 157822 (2022).
Alsafran, M. et al. Neonicotinoid insecticides in the environment: A critical review of their distribution, transport, fate, and toxic effects. J. Environ. Chem. Eng. 10, 108485 (2022).
Huang, M. et al. A liquid chromatography with tandem mass spectrometry method to simultaneously determinate Chlorpyrifos, Imidacloprid and Imidacloprid metabolites in wheat. J. Sep. Sci. 42, 1210–1221 (2019).
Gao, N., Guo, X., Zhang, K. & Hu, D. High-Performance liquid chromatography and gas chromatography — Mass spectrometry methods for the determination of Imidacloprid, Chlorpyrifos, and bifenthrin residues in tea leaves. Instrum. Sci. Technol. 42, 267–277 (2014).
Rahman, M. M. et al. Dynamic behaviour and residual pattern of Thiamethoxam and its metabolite Clothianidin in Swiss Chard using liquid chromatography–tandem mass spectrometry. Food Chem. 174, 248–255 (2015).
Chen, L. et al. Determination of Thiamethoxam and its metabolite Clothianidin residue and dissipation in Cowpea by quechers combining with ultrahigh-performance liquid chromatography–tandem mass spectrometry. Environ. Sci. Pollut. Res. 28, 8844–8852 (2021).
Valverde, S., Ares, A. M., Bernal, J. L., Nozal, M. J. & Bernal, J. Simultaneous determination of Thiamethoxam, Clothianidin, and Metazachlor residues in soil by ultrahigh performance liquid chromatography coupled to quadrupole time-of-flight mass spectrometry. J. Sep. Sci. 40, 1083–1090 (2017).
Sheng, E. et al. Dual-labeled time-resolved fluoroimmunoassay for simultaneous detection of Clothianidin and Diniconazole in agricultural samples. Food Chem. 192, 525–530 (2016).
Li, M. et al. Fluorescence polarization immunoassay for highly efficient detection of Clothianidin in agricultural samples. Anal. Methods. 6, 6541 (2014).
Kumaravel, A. Silver nanoparticle-modified electrodes for the electrochemical detection of neonicotinoid pesticide: Clothianidin. Anal. Methods. 14, 4643–4648 (2022).
Jiao, S. et al. Trace Immunosensing of multiple neonicotinoid insecticides by a novel Broad-Specific antibody obtained from a rational screening strategy. Biosens. (Basel). 12, 716 (2022).
Yang, H. et al. Screening and identification of a DNA aptamer to construct the label-free fluorescent aptasensor for ultrasensitive and selective detection of Clothianidin residue in agricultural products. Talanta 262, 124712 (2023).
Zhang, H. & Liu, J. DNA-based biosensors. TrAC - Trends Anal. Chem. 166, 117164 (2023).
Huang, C. J., Lin, H. I., Shiesh, S. C., Lee, G. & Bin Integrated microfluidic system for rapid screening of CRP aptamers utilizing systematic evolution of ligands by exponential enrichment (SELEX). Biosens. Bioelectron. 25, 1761–1766 (2010).
Chen, A. & Yang, S. Replacing antibodies with aptamers in lateral flow immunoassay. Biosens. Bioelectron. 71, 230–242 (2015).
Almenhali, A. Z. & Eissa, S. Aptamer-based biosensors for the detection of neonicotinoid insecticides in environmental samples: A systematic review. Talanta 275, 126190 (2024).
Bor, G. et al. In vitro selection of aptamer for Imidacloprid recognition as model analyte and construction of a water analysis platform. Electroanalysis 32, 1922–1929 (2020).
Luo, Y., Jin, Z., Wang, J., Ding, P. & Pei, R. The isolation of a DNA aptamer to develop a fluorescent aptasensor for the Thiamethoxam pesticide. Analyst 146, 1986–1995 (2021).
Shi, Q., Tao, H., Wu, Y., Chen, J. & Wang, X. An ultrasensitive label-free electrochemical aptasensing platform for Thiamethoxam detection based on ZIF-67 derived Co-N doped porous carbon. Bioelectrochemistry 149, 108317 (2023).
Kong, Q. et al. Non-immobilized GO-SELEX of aptamers for label-free detection of Thiamethoxam in vegetables. Anal. Chim. Acta. 1202, 339677 (2022).
Li, S. et al. An electrochemiluminescence sensor based on Fe/Zn-BTC@C-dots sensitisation for Thiamethoxam detection. Sens. Actuators B Chem. 394, 134415 (2023).
Yue, Y. et al. Screening and evaluation of Thiamethoxam aptamer based on pressurized GO-SELEX and its sensor application. Biosens. (Basel). 13, 155 (2023).
Rotariu, L., Lagarde, F., Jaffrezic-Renault, N. & Bala, C. Electrochemical biosensors for fast detection of food contaminants - trends and perspective. TrAC - Trends Anal. Chem. 79, 80–87 (2016).
Divya et al. Ultrasensitive aptasensors for the detection of viruses based on Opto-Electrochemical readout systems. Biosens. (Basel). 12, 81 (2022).
Zhu, Y., Chandra, P., Ban, C. & Shim, Y. Electrochemical evaluation of binding affinity for aptamer selection using the microarray chip. Electroanalysis 24, 1057–1064 (2012).
Jia, M., Zhongbo, E., Zhai, F. & Bing, X. Rapid multi-residue detection methods for pesticides and veterinary drugs. Molecules 25, 3590 (2020).
Vashist, S. K. & Luong, J. H. T. Recent advances in electrochemical biosensing schemes using graphene and graphene-based nanocomposites. Carbon N Y. 84, 519–550 (2015).
Fei, A. et al. Label-free impedimetric aptasensor for detection of femtomole level Acetamiprid using gold nanoparticles decorated multiwalled carbon nanotube-reduced graphene oxide nanoribbon composites. Biosens. Bioelectron. 70, 122–129 (2015).
Fu, J., An, X., Yao, Y., Guo, Y. & Sun, X. Electrochemical aptasensor based on one step co-electrodeposition of aptamer and GO-CuNPs nanocomposite for organophosphorus pesticide detection. Sens. Actuators B Chem. 287, 503–509 (2019).
Fu, J. et al. Voltammetric determination of organophosphorus pesticides using a hairpin aptamer immobilized in a graphene oxide-chitosan composite. Microchim. Acta. 187, 36 (2020).
Wang, G. et al. Interference-resistant aptasensor with tetrahedral DNA nanostructure for Profenofos detection based on the composites of graphene oxide and polyaniline. Bioelectrochemistry 148, 108227 (2022).
Zhang, Y. et al. An aptasensor based on the fluorescence resonance energy transfer of nitrogen-doped carbon quantum Dots and graphene oxide to detect fipronil in eggs. Eur. Food Res. Technol. 249, 2887–2895 (2023).
Zhao, Y. et al. A low-background fluorescent aptasensor for Acetamiprid detection based on DNA three-way junction-formed G-quadruplexes and graphene oxide. Anal. Bioanal Chem. 413, 2071–2079 (2021).
Pandey, R. K. et al. Elsevier,. Recent progress in the graphene functionalized nanomaterial-based electrochemical sensors. in Functionalized Nanomaterial-Based Electrochemical Sensors 27–38 (2022). https://doi.org/10.1016/B978-0-12-823788-5.00007-7
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
Khazaal, S., El Darra, N., Kobeissi, A., Jammoul, R. & Jammoul, A. Risk assessment of pesticide residues from foods of plant origin in Lebanon. Food Chem. 374, 131676 (2022).
Suranshe, S. S. & Patil, A. Strategically improving electrical conductivity of reduced graphene oxide through a series of reduction processes. Mater. Lett. 333, 133648 (2023).
Venegas, C. J., Rodríguez, L. & Sierra-Rosales, P. Selective Label-Free electrochemical aptasensor based on carbon nanotubes for carbendazim detection. Chemosensors 11, 117 (2023).
Gunasekaran, B. M. et al. Covalently grafted 4-Aminopyridine-Reduced graphene Oxide-Modified Screen-Printed carbon electrode for electrochemical sensing of lead ions. Arab. J. Sci. Eng. 48, 7721–7738 (2023).
Scardaci, V. & Compagnini, G. Raman spectroscopy investigation of graphene oxide reduction by laser scribing. C (Basel). 7, 48 (2021).
Chadha, N., Sharma, R. & Saini, P. A new insight into the structural modulation of graphene oxide upon chemical reduction probed by Raman spectroscopy and X-ray diffraction. Carbon Lett. 31, 1125–1131 (2021).
Qi, S. et al. High-affinity aptamer of allergen β-lactoglobulin: selection, recognition mechanism and application. Sens. Actuators B Chem. 340, 129956 (2021).
Wang, M. et al. Selection and identification of diethylstilbestrol-specific aptamers based on magnetic-bead SELEX. Microchem. J. 159, 105354 (2020).
Hermanson, G. Bioconjugate Techniques (Academic, 2013).
Arvand, M. & Hassannezhad, M. Magnetic core–shell Fe3O4@SiO2/MWCNT nanocomposite modified carbon paste electrode for amplified electrochemical sensing of uric acid. Mater. Sci. Engineering: C. 36, 160–167 (2014).
Acknowledgements
S. Eissa acknowledge Khalifa University of Science and Technology, United Arab Emirates, provided the financial support for this research through grant number 8474000678/DSUF-2024-004. This research project was also implemented with the support of the Arab-German Young Academy of Sciences and Humanities (AGYA). AGYA is funded by the German Federal Ministry of Education and Research (BMBF) grant 01DL20003.
Author information
Authors and Affiliations
Contributions
A. A.: Data curation, Formal analysis, Methodology, Writing—original draft. P. K.: Formal analysis, Methodology, Writing—review & editing. M. A.: Writing—review & editing. S.K.: Formal analysis, Writing—review & editing, N. E.: Formal analysis, Writing—review & editing, S. E.: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing—review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Almenhali, A.Z., Kanagavalli, P., Abd-Ellah, M. et al. Reduced graphene oxide-based electrochemical aptasensor for the multiplexed detection of imidacloprid, thiamethoxam, and clothianidin in food samples. Sci Rep 15, 10329 (2025). https://doi.org/10.1038/s41598-025-94313-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-94313-6
