
Abstract
Altered circulating hormones in ALS patients have been widely reported by previous observational studies, but whether these relationships are causal is unclear. Moreover, the potential therapeutic targets for ALS and the effects of plasma protein fluctuation on ALS progression are not fully understood. Therefore, we conducted a Mendelian randomization (MR) study to evaluate the causal role of 5 hormonal risk factors (insulin-like growth factor-1, IGF-1; sex hormone-binding globulin, SHBG; free testosterone, FT; total testosterone, TT; and estradiol) in ALS risk. Furthermore, we screened up to 90 circulating proteins including cytokines, chemokines, growth factors, and interferons, to identify potential therapeutic targets for ALS. Our MR analysis found genetically predicted higher level of FT was associated with a 23% lowered risk of ALS. Further screening of proteomic traits found that 12 plasma proteins were causally associated with ALS. These findings suggest that higher FT potentially exerts a protective effect on ALS risk. Several proteins may act as potential circulating biomarkers and therapeutic targets for ALS. In the future, high-throughput proteomic analyses and experimental explorations are likely needed to clarify the regulated role and mechanistic pathways.
Similar content being viewed by others


Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disorder with a median survival of about 3 to 5 years after symptom onset. The causes of ALS remain far to be explored and appear heterogeneous. Hitherto, there is no cure or effective medical treatment for ALS, and the standard of disease management remains multidisciplinary care1. Therefore, it is fundamental to identify modifiable risk factors and circulating biomarkers for ALS to develop effective diagnosis, prevention and treatment strategies.
Accumulating evidence has suggested that circulating hormones, including testosterone2,3, estradiol4, and insulin-like growth factor-1 (IGF-1)5,6 may play a role in ALS progression. As a consequence, androgen and IGF-1 therapies were proposed as therapeutic candidates against ALS7. Although observational studies have evaluated the association between the concentrations of these hormones and ALS, the causality and the underlying molecular mechanism remain to be established. On the other hand, biological studies led to the opinion that inflammatory response contributes to the pathogenesis of ALS8. In addition, altered levels of peripheral cytokines and proteins were observed in ALS patients, but the results were inconsistent9,10. Several attempts have been made to identify therapeutic targets; however, few clinical studies are available on human individuals. Thus, the causal nature of these risk factors, and their suitability as intervention targets for ALS prevention, are still unknown.
Observational studies are susceptible to residual confounding and reverse causality, which may bias the association estimates between modifiable risk factors and ALS. Mendelian Randomization (MR) overcomes these defects by using the genetic variants as instrumental variables to investigate the causal effect of exposure (risk factors) on a disease outcome11. As this genetic-based analysis is compliant with Mendel’s law of segregation and independent assortment, the estimates of MR are less likely to be affected by confounding factors and biased by reverse causation.
Given the unclear relevance of circulating hormones and plasma proteins in ALS etiology, we conducted multiple MR analyses to evaluate the causal role of the five known endogenous hormones, including insulin like growth factor (IGF-1), sex hormone-binding globulin (SHBG), free (bioavailable) testosterone (FT), total testosterone (TT), and estradiol, and up to 90 plasma proteins including cytokines, chemokines, growth factors and interferons, in ALS risk.
Materials and methods
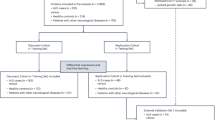
The analytical strategy employed in this study is depicted in Fig. 1. In the initial phase of our study, we examined the causal relationships between five hormones (including IGF-1, FT, TT, SHBG, and estradiol) and the risk of developing ALS. On the other hand, we evaluated the causal effects of circulating proteins on ALS progression.

The study design overview and the conceptual schematic of the Mendelian randomization. ALS, amyotrophic lateral sclerosis; FT, free testosterone; IVW, the inverse-variance weighted method; IGF-1, insulin-like growth factor 1; IVs, instrumental variables; MR, Mendelian randomization; MR-PRESSO, MR-pleiotropy residual sum and outlier; SHBG, sex hormone-binding globulin; TT, total testosterone.
All analyses were based on summary-level data on measures of hormones, ALS, and circulating proteins from published genome-wide association studies (GWASs). Appropriate ethical approval and informed patient consent could be found in the original studies.
Growth and sex hormones instrument selection
Genetic instruments associated with IGF-1 were obtained from a GWAS including 358,072 European-descent participants of the UK Biobank12. Single‐nucleotide polymorphisms (SNPs) predicting levels of FT, TT, and SHBG were extracted from a GWAS conducted by Ruth et al. using UK Biobank data13, which included 425,097, 382,988, and 368,929 European‐descent participants for FT, TT, and SHBG, respectively. Variants selection for estradiol levels was based on a GWAS including 229,966 women of European ancestry from the UK Biobank dataset14. To construct genetic instruments for growth and sex hormones, we obtained independent (r2 < 0.01 and clump distance > 10,000 kb) SNPs that reached genome-wide significance (P < 5 × 10–8). Information on genetic instruments is provided in Table S1.
The data source for ALS
Summary genetic association data on ALS was obtained from a publicly available GWAS conducted by Nicolas et al.15. This large-scale GWAS involved 12,663 ALS patients and 53,439 controls. The data was then incorporated into a meta-analysis with a previous GWAS including 12,577 ALS patients and 23,475 controls16. After imputation and quality control measures, genetic variants from 20,806 cases and 59,804 controls in populations of European ancestry were available for association analyses. ALS patients were diagnosed by neurologists according to the EI Escorial criteria15. In the replication analyses, the summary data of ALS (GWAS ID: finn-b-G6_ALS) was extracted from FinnGen consortium and provided by the IEU database (https://gwas.mrcieu.ac.uk/), which included 219 cases and 111,621 controls.
Circulating proteins study population
The summary statistics and genetic instruments for circulating proteins were derived from a GWAS conducted by Folkersen et al.17. Ninety proteins, comprising cytokines, chemokines, growth factors, and interferons, in up to 30,931 European individuals from 13 cohorts passed quality control, were available for our hormone-protein MR analyses. Details of the included proteins are shown in Table S2. To select genetic instruments, we first extracted SNPs associated with circulating proteins at the genome-wide significance level (P < 5 × 10–8) and then pruned these SNPs according to linkage disequilibrium (r2 < 0.01) based on the 1000 Genomics European reference panel. We excluded proteins that had no or limited (< 2) genetic instruments. After removing 14 proteins, 76 circulating proteins were included in the protein-ALS MR analyses. Detailed information on used SNPs is presented in Table S3.
Statistical analyses
In our primary analyses, we used the Wald ratio to generate effect estimates and used the delta method to approximate standard errors18. For circulating proteins instrumented by only two SNPs, inverse-variance weighted (IVW) fixed-effects models were used to estimate causal effects. For traits instrumented by three or more SNPs, IVW random-effects models were used18. To evaluate the presence of horizontal pleiotropy (i.e. the genetic variant influences an outcome through a biological pathway independent of the exposure), we conducted various sensitivity analyses, including MR Egger regression19, weighted median estimation20, maximum likelihood estimation21, and MR Pleiotropy Residual Sum and Outlier (MR-PRESSO) estimation22. Each of these methods consists of different assumptions regarding the potential horizontal pleiotropy, thus conducting all of them can provide complementary support to the IVW method and examine the robustness of associations. These methods were not performed when instruments consisted of only two SNPs because of the reduced statistical power to detect horizontal pleiotropy in these circumstances.
All statistical analyses were conducted using R (Vienna, Austria) version 4.0.1 together with the R package TwoSampleMR (https://github.com/MRCIEU/TwoSampleMR) and MR-PRESSO (https://github.com/rondolab/MR-PRESSO).
Results
Causal estimates of the genetically predicted Circulating hormone in ALS
In the growth hormone analysis, IGF-1 was not associated with ALS (odds ratio (OR) per increase in standard deviation (SD) IGF-1, 1.00, 95% confidence interval (CI) 0.93–1.08, P = 0.901) (Fig. 2). As for sex hormones, a genetically predicted higher level of FT was associated with a 23% lowered risk of ALS (OR per increase in INT nmol/L FT 0.77, 95% CI 0.65–0.91, P = 0.003), corroborated by sensitivity estimates (Weighted median method, OR = 0.73, 95% CI 0.55–0.97, P = 0.029; MR Egger method, OR = 0.62, 95% CI 0.43–0.91, P = 0.015; Maximum Likelihood, OR = 0.77, 95% CI 0.65–0.91, P = 0.002). The MR PRESSO method showed no outliers in IVs. In addition, MR-Egger intercept was not significantly different from zero (P = 0.218). We found that no specific SNP had a decisive role regarding the association between FT and ALS by applying the leave-one-out analysis. Further validation analyses by using ALS data from FinnGen consortium yielded consistent negative association of FT with ALS risk through IVW method (OR = 0.30, 95% CI 0.11–0.80, P = 0.016) (Table S4).

Mendelian randomization association of genetically predicted growth and sex hormones with ALS. Odds ratios are scaled per genetically predicted standard deviation change in IGF-1, inverse-normal transformed nmol/L change in FT and TT, natural log transformed nmol/L change in SHBG, and pmol/L change in estradiol. ALS, amyotrophic lateral sclerosis; FT, free testosterone; IVW, the inverse-variance weighted method; IGF-1, insulin-like growth factor 1; MR, Mendelian randomization; OR, odds ratio; SHBG, sex hormone-binding globulin; TT, total testosterone.
In contrast, the IVW MR estimates for TT (OR per increase in inverse-normal transformed [INT] nmol/L TT 0.95, 95% CI 0.79 to 1.14, P = 0.600), SHBG (OR per increase in natural log transformed nmol/L SHBG 1.08, 95% CI 0.94 to 1.24, P = 0.280), and estradiol (OR per increase in pmol/L estradiol 1.02, 95% CI 0.60 to 1.72, P = 0.953) were not significant (Fig. 2).
Identification of causal risk proteins for ALS
After removing the instruments that did not satisfy the inclusion criteria, seventy-six plasma proteins were tested for the causal relationships with ALS (Table S5). The MR estimates identified twelve potential causal risk proteins for ALS: HSP-27, CA-125, MMP-1, U-PAR, CTSD, IL-1ra, PECAM-1, Dkk-1, MMP-7, ST2, IL-27, and PSGL-1. Among of which, Genetic predispositions to 1-SD increase in HSP-27 (OR 1.08, 95% CI 1.03 to 1.09, P = 1.1 × 10–4), CA-125 (1.06, 95% CI 1.02 to 1.11, P = 0.007), MMP-1 (1.09, 95% CI 1.01 to 1.17, P = 0.020), U-PAR (1.16, 95% CI 1.02 to 1.31, P = 0.023), and CTSD (1.06, 95% CI 1.00 to 1.13, P = 0.049) were associated with higher risk of ALS (Fig. 3). In contrast, the results from the IVW method suggested the protective effects of IL-1ra (0.92, 95% CI 0.87 to 0.97, P = 0.001), PECAM-1 (0.92, 95% CI 0.86 to 0.97, P = 0.003), Dkk-1 (0.91, 95% CI 0.84 to 0.98, P = 0.011), MMP-7 (0.90, 95% CI 0.84 to 0.98, P = 0.014), ST2 (0.96, 95% CI 0.93 to 0.99, P = 0.017), IL-27 (0.93, 95% CI 0.88 to 0.99, P = 0.022), and PSGL-1 (0.94, 95% CI 0.90 to 0.99, P = 0.022) on ALS risk (Fig. 3). The causal estimates were broadly consistent, when using the additional methods for sensitivity analysis (Table S5). Horizontal pleiotropies were not observed in the MR-Egger intercept test.

Mendelian randomization association of genetically predicted circulating proteins with ALS. Odds ratios are scaled per increase in standard-deviations of proteins. Detailed information for the proteins are provided in Supplemental Material. ALS, amyotrophic lateral sclerosis; CI, confidence interval; IVW, the inverse-variance weighted method; MR, Mendelian randomization; OR, odds ratio; SNP, single nucleotide polymorphism.
Discussion
Since the incidence of ALS has been reported to be sex-related, there raised the hypothesis that sex hormones may be involved in the etiopathogenesis of this disorder23. Our systematic MR analysis of endogenous hormones in 20,806 ALS cases and 59,804 controls provided genetic evidence for the protective role of free (bioavailable) testosterone (FT) in the risk of ALS. Although other hormones including IGF-1, SHBG, and total testosterone (TT) have been investigated in previous observational and laboratory studies, little evidence from our study suggested that they may be causally implicated in ALS risk. Moreover, our screening of molecular traits found that genetic predisposition to higher levels of IL27, IL-1ra, Dkk-1, PSGL-1, MMP7, PECAM-1, and ST2 were persistently associated with lower odds of ALS, whereas genetic liability to HSP-27, CA-125, cathepsin D, MMP1, and U-PAR were positively associated with ALS risk.
Previously, the impact of androgens, including testosterone and its reduced metabolites, on the morphology and viability of spinal and brainstem motoneurons has been well-documented. Several studies have demonstrated that the administration of androgens, especially testosterone, promotes the restoration of motoneurons, leading to the regeneration of both axons and dendrites7. Nevertheless, despite the safety and low cost of androgen therapy, few clinical studies are available on human individuals. Although previous observational study observed that the serum level of FT was reduced in ALS patients, and significantly lowered CSF testosterone in ALS patients compared to healthy controls was found in a recent study3,24, discrepancy was reported that female ALS patients showed higher testosterone levels and lower progesterone/FT ratio presented a more rapid worsening of the monthly forced vital capacity25. Our primary and validated analyses in different ALS populations both revealed inverse association of genetically predicted plasma FT with ALS risk, suggested a potential protected effect of FT on ALS progression. Similarly, a previous study by using different dataset reported that bioavailable testosterone was negatively associated with the risk of developing ALS26, further supported the confidence of the findings. In contrast, we found no significant difference in the relationship between TT and ALS. It is accepted that the distribution of circulating testosterone is constituted by bound and unbound forms (the latter represents FT). 60–70% of circulating testosterone is tightly bound to SHBG, whereas the remaining 30–40% binds loosely to albumin; only 0.5–2% is free and in this form the hormone can cross the blood–brain barrier (BBB)27. Given the BBB permeability of FT, we proposed that this form of hormone is potentially involved in the pathophysiology of ALS, which may partially explain the inconsistent effects of TT and SHBG. Notably, testosterone has also been reported to be negatively associated with other neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease and cognitive functions26,28,29,30, suggesting that the genetic effect and therapeutic potential of testosterone should be generalized to more neurodegenerative diseases not only for motor neuron dysfunctions. Although dysregulation of IGF-1 was suggested to play a significant role in the progression of ALS5, and elevated IGF-1 was reported to delay disease progression, extend survival time, and decrease disease progression in the ALS mouse model6, we found little evidence to support a protective role of IGF-1 in ALS risk.
Mechanistically, testosterone treatment of ALS mice demonstrated inhibition of astrogliosis in the spinal cord, leading to the reduction of proinflammatory factors2. Moreover, testosterone increased the expression of TGFβ1, a factor well-known for its anti-inflammatory properties31. Besides, testosterone was reported to improve the survival of human neurons and astrocytes, acting directly on the mitochondrial membrane, and inhibiting the generation of reactive oxygen and nitrogen species, both of which are clear triggers for ALS32. On the other hand, since testosterone treatment demonstrated improvement on muscle atrophy and physical function of ALS mouse models2, and clinical trials of testosterone therapy in low-testosterone-level populations exhibited benefit effects on muscle strength and recovery33,34,35, it is reasonable to infer the possibility of increased muscle reserve as a potential driver of the protective effects for testosterone in ALS progression. This finding may redefine our understanding of the disease as it being a male-predominant disease and also affecting high-functioning athletes who presumably have higher levels of testosterone. Due to the etiological complexity and heterogeneity of the disease, causes of which including genetic, environment, and gene–environment interactions, disease genetics can help guide further insights that challenge our current understanding and provide starting point for targeted drug development and personalized medicine36.
The screening of circulating proteome in our study identified several risk proteins for ALS. Similar to previously reported results, higher levels of IL-1ra were causally associated with lower odds of ALS, suggesting the robustness of our findings37. Evidence is accumulating that IL-27 has neuroprotective activities as it promotes neuronal survival by regulating pro- and anti-inflammatory cytokines, neuroinflammatory pathways, oxidative stress, apoptosis, autophagy, and epigenetic modifications38. We found a causal inverse association of IL-27 levels with ALS, suggesting a potential protective effect. Abnormal angiogenesis may occur in ALS due to increased microvascular density and accumulation of collagen fibers in the spinal cord perivascular space. Dkk-1 and U-PAR are pro-angiogenic factors, both of which have been demonstrated to increase in ALS patients from prior observational studies9,39. However, our findings suggested inverse directions of these two proteins for ALS risk, the underlying mechanism needs further investigation. Previous studies on the association between HSP-27 and ALS have been contradictory, one study reported that HSP-27 from peripheral blood was elevated in patients10, whereas another one found serum levels of HSP-27 did not differ significantly between the patient and control groups40. By conducting MR analysis, our result suggested that HSP-27 was causally and positively associated with ALS risk, supported by the experimental study that HSP-27 regulates p62 condensates and promotes lysophagy by forming platforms for autophagosome biogenesis at damaged lysosomes thus may contribute to neurodegeneration41. Cathepsins are intracellular proteases located within the lysosomes and are critical for normal protein turnover. The upregulation of cathepsin D was observed in the spinal cord of ALS mice, accompanied by alterations in the localization and associations of cathepsin D with lysosomes42. We further observed a negative association between cathepsin D and ALS, supporting its detrimental effect. MMPs are zinc-dependent enzymes that respond to inflammation, they are known to play a role in brain development and physiology43. MMP1 was suggested as a disease progression marker for ALS before44, which was also identified by our MR analysis. Apart from the above, the function of other potentially risk proteins identified by our study, including PSGL-1, MMP7, PECAM-1, ST2, and CA-125, need to be explored in future studies.
There are several limitations to our analysis. First, we could not completely rule out pleiotropy. However, several aspects of our results indicated the presence of pleiotropy was minimal, including little indication from the MR-Egger intercept analyses, no outlier detected in the MR-PRESSO analyses, and consistent results from sensitivity models. Second, although we have identified potential risk hormone and proteins for ALS, rigorous experimental verification is essential, and the underlying mechanisms demand further exploration. Third, this analysis was restricted to individuals of European ancestry to minimize bias from population stratification, which may constrain the generalizability of our findings to other ethnicities. Fourth, this study only looked at ALS risk, limiting the application of the results to other neurodegenerative diseases. Moreover, as a clinically, genetically and pathologically heterogenous disease, the results may not be indicative of subgroups within the ALS risk group, which could explain heterogeneity within some of the data. Fifth, the dynamic changes in hormone levels during life stages may also exert an influence on ALS development. Future investigations could explore time-dependent effects with the availability of relevant GWAS data.
Conclusion
Our results based on genetic MR analyses implicated a protective role of circulating free testosterone in the development of ALS. Moreover, ALS progression was also affected by several plasma protein alterations. In further studies, high-throughput proteomic measurements are needed to clarify the underlying mediated and mechanistic pathways.
Data availability
Data generated or analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- IGF-1:
-
Insulin-like growth factor-1
- MR:
-
Mendelian Randomization
- SHBG:
-
Sex hormone-binding globulin
- FT:
-
Free testosterone
- TT:
-
Total testosterone
- GWASs:
-
Genome-wide association studies
- SNPs:
-
Single-nucleotide polymorphisms
- IVW:
-
Inverse-variance weighted
- OR:
-
Odds ratio
- SD:
-
Standard deviation
- CI:
-
Confidence interval
References
Beers, D. R. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. 18, 211–220 (2019).
Lara, A. et al. Neuroprotective effects of testosterone in male wobbler mouse, a model of amyotrophic lateral sclerosis. Mol. Neurobiol. 58, 2088–2106 (2021).
Militello, A. et al. The serum level of free testosterone is reduced in amyotrophic lateral sclerosis. J. Neurol. Sci. 195, 67–70 (2002).
Heitzer, M. et al. Administration of 17beta-Estradiol improves motoneuron survival and down-regulates inflammasome activation in male SOD1(G93A) ALS mice. Mol. Neurobiol. 54, 8429–8443 (2017).
Kaspar, B. K., Llado, J., Sherkat, N. & Rothstein, J. D. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 301, 839–842 (2003).
Wen, D. et al. The role of insulin-like growth factor 1 in ALS cell and mouse models: a mitochondrial protector. Brain Res. Bull. 144, 1–13 (2019).
Bianchi, V. E., Rizzi, L., Bresciani, E. & Omeljaniuk, R. J. &Torsello, A. Androgen therapy in neurodegenerative diseases. J. Endocr. Soc. 4, bvaa120 (2020).
McCauley, M. E. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 137, 715–730 (2019).
Cao, M. C. et al. Serum biomarkers of neuroinflammation and blood-brain barrier leakage in amyotrophic lateral sclerosis. BMC Neurol. 22, 216 (2022).
Vats, A. et al. Expression analysis of protein homeostasis pathways in the peripheral blood mononuclear cells of sporadic amyotrophic lateral sclerosis patients. J. Neurol. Sci. 387, 85–91 (2018).
Yarmolinsky, J. et al. Causal inference in cancer epidemiology: what is the role of Mendelian randomization?? Cancer Epidemiol. Biomark. Prev. 27, 995–1010 (2018).
Sinnott-Armstrong, N. et al. Genetics of 35 blood and urine biomarkers in the UK biobank. Nat. Genet. 53, 185–194 (2021).
Ruth, K. S. et al. Using human genetics to understand the disease impacts of testosterone in men and women. Nat. Med. 26, 252–258 (2020).
Haas, C. B., Hsu, L., Lampe, J. W. & Wernli, K. J. Cross-ancestry Genome-wide association studies of sex hormone concentrations in Pre- and postmenopausal women. Endocrinology 163, (2022).
Nicolas, A. et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1283e1266 (2018).
van Rheenen, W. et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048 (2016).
Folkersen, L. et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat. Metab. 2, 1135–1148 (2020).
Hemani, G., Bowden, J. & Davey Smith, G. Evaluating the potential role of Pleiotropy in Mendelian randomization studies. Hum. Mol. Genet. 27, R195–R208 (2018).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Bowden, J., Davey Smith, G. & Haycock, P. C. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, 745 (2018).
Verbanck, M., Chen, C. Y. & Neale, B. Detection of widespread horizontal Pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Piemonte&Valle d’Aosta Register for Amyotrophic Lateral. Incidence of ALS in Italy: evidence for a uniform frequency in Western countries. Neurology 56, 239–244 (2001).
Lucchi, C. et al. Reduced levels of neurosteroids in cerebrospinal fluid of amyotrophic lateral sclerosis patients. Biomolecules 14, 478 (2024).
Gargiulo-Monachelli, G. M. et al. Circulating gonadal and adrenal steroids in amyotrophic lateral sclerosis: possible markers of susceptibility and outcome. Horm. Metab. Res. 46, 433–439 (2014).
Huang, Q. & Li, Q. Causal relationship between sex hormones and risk of developing common neurodegenerative diseases: a Mendelian randomization study. J. Integr. Neurosci. 23, 78 (2024).
Goldman, A. L. et al. A reappraisal of Testosterone’s binding in circulation: physiological and clinical implications. Endocr. Rev. 38, 302–324 (2017).
Yeung, C. H. C., Au Yeung, S. L., Kwok, M. K. & Zhao, J. V. The influence of growth and sex hormones on risk of Alzheimer’s disease: a Mendelian randomization study. Eur. J. Epidemiol. 38, 745–755 (2023).
Xu, Q. et al. Causal effects of genetically predicted testosterone on Alzheimer’s disease: a two-sample Mendelian randomization study. Acta Neurol. Belg. 124, 591–601 (2024).
Giannos, P. et al. Associations of bioavailable serum testosterone with cognitive function in older men: results from the National health and nutrition examination survey. J. Gerontol. Biol. Sci. Med. Sci. 78, 151–157 (2023).
Caruso, G. et al. Carnosine prevents Abeta-Induced oxidative stress and inflammation in microglial cells: a key role of TGF-beta1. Cells 2019, 8 (2019).
Atallah, A., Mhaouty-Kodja, S. & Grange-Messent, V. Chronic depletion of gonadal testosterone leads to blood-brain barrier dysfunction and inflammation in male mice. J. Cereb. Blood Flow. Metab. 37, 3161–3175 (2017).
Mauvais-Jarvis, F. Metabolic benefits afforded by estradiol and testosterone in both sexes: clinical considerations. J. Clin. Invest. 134, 1456 (2024).
Alexander, S. E. et al. Bioavailable testosterone and androgen receptor activation, but not total testosterone, are associated with muscle mass and strength in females. J. Physiol. (2024).
Howard, E. E. et al. Effects of testosterone on Mixed-Muscle protein synthesis and proteome dynamics during energy deficit. J. Clin. Endocrinol. Metab. 107, e3254–e3263 (2022).
Akcimen, F. et al. Amyotrophic lateral sclerosis: translating genetic discoveries into therapies. Nat. Rev. Genet. 24, 642–658 (2023).
Yuan, S. & Roos, P. M. Interleukin-1 receptor antagonist, interleukin-2 receptor alpha subunit and amyotrophic lateral sclerosis. Eur. J. Neurol. 27, 1913–1917 (2020).
Nortey, A. N. & Garces, K. N. Exploring the role of interleukin-27 as a regulator of neuronal survival in central nervous system diseases. Neural Regen. Res. 17, 2149–2152 (2022).
Glas, M. et al. A role for the urokinase-type plasminogen activator system in amyotrophic lateral sclerosis. Exp. Neurol. 207, 350–356 (2007).
Miyazaki, D. et al. Elevation of serum heat-shock protein levels in amyotrophic lateral sclerosis. Neurol. Sci. 37, 1277–1281 (2016).
Gallagher, E. R. The selective autophagy adaptor p62/SQSTM1 forms phase condensates regulated by HSP27 that facilitate the clearance of damaged lysosomes via lysophagy. Cell. Rep. 42, 112037 (2023).
Wootz, H., Weber, E., Korhonen, L. & Lindholm, D. Altered distribution and levels of cathepsind and cystatins in amyotrophic lateral sclerosis Transgenic mice: possible roles in motor neuron survival. Neuroscience 143, 419–430 (2006).
Dai, X. Y. et al. Matrix metalloproteinases as attractive therapeutic targets for chronic pain: a narrative review. Int. J. Biol. Macromol. 261, 129619 (2024).
Sanchez-Torres, J. L. et al. Matrix metalloproteinases deregulation in amyotrophic lateral sclerosis. J. Neurol. Sci. 419, 117175 (2020).
Acknowledgements
The authors thank all investigators for sharing these data.
Funding
This study was supported by research grants from the National Natural Science Foundation of China (82470903 and 32000820 to LJ, 82201242 to KW), and the Natural Science Foundation of Zhejiang Province (LY24C110001 to LJ).
Author information
Authors and Affiliations
Contributions
LJ, KW, XYF, YTZ and FFZ were responsible for the writing of the original draft; LJ, KW, XYF, YTZ, FFZ, YX, QXD and STL for reviewing and editing the manuscript; LJ, KW, XYF, YTZ, FFZ, YX, QXD, STL, and YML participated in the revision of the manuscript; LJ, KW, XYF, YTZ, and FFZ for acquisition, analysis, or interpretation of data; LJ and KW for the conceptualization and statistical analysis of the study; and LJ for supervision. LJ and KW accessed the database and raw data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fan, X., Zeng, Y., Zhang, F. et al. Genetic effects of circulating hormone and proteome on amyotrophic lateral sclerosis identified by Mendelian randomization. Sci Rep 15, 10782 (2025). https://doi.org/10.1038/s41598-025-95151-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-95151-2
