
Abstract
Azo dyes are highly recalcitrant, persistent, and toxic compounds, extensively used in the textile industry. The untreated discharge of dye effluents from the textile industry poses severe environmental and health risks. This research aimed to isolate and identify bacterial strains from textile wastewater that can decolorize azo dyes. After the subsequent screening of 89 isolates, 4 novel strains were identified utilizing the 16S rRNA gene sequencing technique that could effectively decolorize and degrade azo dyes, methyl red, direct yellow 12, and acid black 210. A thorough assessment of physicochemical parameters was conducted to optimize for maximum decolorization for all four strains. At pH 7, 37° C, and 50 mg/L dye concentration, the maximum decolorization for methyl red, direct yellow 12, and acid black 210 was 79.09% > 72.20% > 64.76%; 84.45% > 62.59% > 54.29%; 83.12% > 70.22% > 61.42%; and 92.71% > 83.02% > 69.84%, for isolate 1, isolate 2, isolate 3, and isolate 4, respectively. The novel strains belonged to the Sphingomonas, Pseudomonas, Shewanella, and Priestia species. The unique sequences of these bacterial strains have been submitted to the GenBank database under the accession numbers “OQ202071”, “PP708911”, “PP708909”, and “PP086977,” respectively. Further, an enzyme study and statistical optimization of Priestia flexa species was performed. A Central Composite Design and Response Surface Methodology has been applied for synergistic effects of process parameters namely pH (5–9), initial dye concentration (100–250 mg/L), and temperature (25°–45° C) on the decolorization of the model dyes. The regression analysis indicated a strong correlation between the experimental data and the second-order polynomial supported by a high coefficient of determination (R²). For all three dyes analyzed, the difference between the experimental and predicted values was found to be less than 10%. Fourier Transform Infrared spectroscopy was further employed to analyze and confirm the degradation of the three dyes.
Similar content being viewed by others
Introduction
Environmental pollution is one of the grave challenges for the contemporary world. To address the needs of the overgrowing population, perpetual development in agriculture, petroleum, leather, dye, coal, plastic, rubber, coating, pharmaceuticals, and chemical industries is inescapable. Consequently, these industries produce environmental pollutants disseminated into the air, water, or soil that disastrously affect human health, plants, and animals. A substantial number of liquid effluxes are generated from numerous industries that are tainted with hazardous compounds.
The textile industry has a great ascendancy towards the global economy and our daily lives, which significantly employs lakhs of people and encompasses 30% of the nation’s GDP in several countries. Depicting a prominent role in development and industrialization, the industry is one of the fastest-growing sectors. Concurrently it consumes large amounts of water (almost 150 L to dye 1 kg of cotton) and produces a remarkable amount of wastewater1. Worldwide, over 105 dyes have been yielded with the production of over 7 × 105 metric tons, contributing to global sales of approximately US$ 6 billion per annum2. Moreover, such mass production and extensive implementation produce a daily discharge of tons of harmful dye effluents from industries discarded into the nearby water bodies. Before the mid-19th century, natural dyes were preferred and utilized for several centuries3. Their use declined substantially with the breakthrough of synthetic dyes in 1856, driven by industrial advancements and the growing global demand for apparel4,5. Since then, synthetic dyes have been extensively utilized as they are cost-effective in synthesis, stable to temperature and light variation, and stipulate quick coloration6.

Workflow for Azo dyes degradation: A bioremediation approach.
Azo dyes are among the most widely utilized synthetic dyes in the textile industry and are characterized by their strong xenobiotic nature. Azo dyes feature the azo chromophore (–N = N–), with R1 and R2 representing aromatic substituents attached to this azo linkage, which significantly contributes to their chromatic properties7. The untreated disposal of these dyes from industries is one of the foremost concerns of the environment, as it contains a remarkable number of hazardous intermediates that trigger serious environmental and health hazards. Direct disposal of dyes tends to deposit on the surface of the water, which not only increases turbidity and affects the aesthetic water quality but also adversely impacts soil microflora and both marine and terrestrial forms of life8,9. This deposition also recedes the amount of dissolved oxygen in water. The dyes released on the surface of water bodies hinder the sunlight penetration to water which drops the photosynthesis rate that subsequently affects the basis of the food chain10. Besides this, humans are susceptible to the direct and indirect effects of dyes in terms of skin diseases, allergies including dermatitis11, and the formation of tumors and cancer in the worst cases. Earlier studies reported the intermediate compounds of dyes have reported to cause bladder cancer and Central Nervous System ailments.
Various conventional physical and chemical treatments have been applied to treat dyes before their disposal into the water bodies. These treatments include oxidation, coagulation photocatalysis, ozonation, and the Advanced oxidation process (AOP)12,13. Removal of dyes through these processes is effective for only a limited volume of effluents, as they necessitate inordinately expensive reagents and catalysts. Parallelly, it produces a large amount of secondary sludge, comprising several volatile compounds and other byproducts upon incomplete degradation9. However, implying microorganisms for the removal of harmful constituents can be a potential approach in an eco-efficient manner. Numerous studies have been documented involving the decolorization and degradation processes, encompassing a variety of microorganisms such as bacteria, fungi, yeast, actinomycetes, and algae under aerobic and anaerobic conditions. So far, bacteria are considered the most versatile organism for bioremediation14. The phylum portrays its existence in all extreme conditions of varying values of pH, temperatures, and salinity15,16. Due to their significant activity, widespread distribution, and exceptional adaptability, bacteria are widely used in decolorization17.
The present study addresses the necessity for effective bioremediation strategies for treating dye-contaminated textile wastewater. Four novel strains of bacteria capable of decolorizing azo dyes have been isolated from textile wastewater. These are Sphingomonas mali, Pseudomonas leuteola, Shewanella putrefaciens, and Priestia flexa. The study highlights the decolorization of the three toxic azo dyes under aerobic conditions: Methyl Red (MR), Direct Yellow 12 (DY 12), and Acid Black 210 (AB 210) by the four novel bacterial strains17,18,19. The impact of various environmental factors on the decolorization of each has been studied extensively for each dye with the traditional method of single-factor optimization20. This study offers profound insights into the potential application of bacterial strains in large-scale wastewater treatment processes by determining the optimal conditions for the effective degradation of dyes.
Further, the decolorization of model dyes by the strain Priestia flexa AKSKSLAB04 has been meticulously studied using Fourier Transformed Infrared Spectroscopy (FTIR)21. The effect of pH, temperature, and initial concentration on dye decolorization was investigated and optimized using Response Surface Methodology (RSM). Figure 1 briefs the study’s workflow.
Materials and methods
Sample collection and preservation
The textile effluent water samples were collected from the surface of wastewater discharged from a textile processing unit in Tirupur, Tamil Nadu, India. The accurate location recorded with GPS was (11.1085 N 77.3411 E). The samples were collected following a standard procedure using the grab sampling method. 200 mL of wastewater was sampled using sterilized plastic bottles to prevent contamination. Following collection, the samples were immediately placed in an icebox to maintain a low temperature during transportation to the laboratory for further analysis. To ensure clarity and to remove any fine suspended particles, the collected samples were filtered through traditional filter paper with a pore size of 0.45 μm. The resulting filtrate was promptly stored at 4° C to preserve the sample integrity until subsequent laboratory analyses were conducted22.
Dyes and chemicals
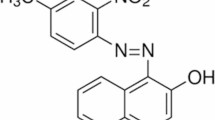
The three dyes were selected based on the number of azo bonds in their chemical structure: i.e., mono-azo dye, i.e. methyl red (MR; λmax 520 nm), di-azo dye, i.e., direct yellow 12 (DY 12; λmax 580 nm), and tri-azo dye, i.e., acid black 210 (AB 210, λmax 604 nm) dyes. The dyes were purchased from High Media India Ltd. The molecular weights of dyes are 269.30 g/mol, 680.66 g/mol, and 938.0 g/mol, corresponding to molecular formulas C15H15N3O2, C30H26N4Na2O8S2, and C34H25K2N11O11S3 respectively. The chemical structures of the dyes are illustrated in Fig. 2. Other properties of the compounds are summarised in Table S1.

Chemical structure of the model dyes (a) Methyl red, (b) Direct yellow12, and (c) Acid black 210.
Isolation, screening and enrichment of bacterial consortium
The filtered textile wastewater sample was used for isolating bacterial strains employing a serial dilution method. Pure cultures were obtained by streaking on Nutrient agar media (NAM). The distinct colonies with bacterial cells (10⁸ CFU/mL) were inoculated in a 250 mL Erlenmeyer flask containing 100 mL sterile Nutrient broth (NB) (comprising 1.0 g/L of yeast extract, 5.0 g/L of peptone, 5.0 g/L of sodium chloride 15.0 g/L of agar) at 37° C for 24 h on a rotary shaker at 120 rpm. After incubating for 24 h, 10% (v/v) inoculum was subcultured in sterile NAM containing 10 mg/L respective dyes. The cultures were further incubated in similar conditions, as described above. The distinct colonies that could utilize dyes as a nutrient source were picked and stored in NB for subsequent analysis23,24.
Decolorization study and quantification
The potential bacterial isolates were examined for their ability to decolorize dye in liquid media. The four strains were subjected to three different toxic and extensively used azo dyes in the textile dyeing industry. A loopful of distinct bacterial strains was grown on a nutrient agar plate for 24 h at 35º C and further inoculated in an Erlenmeyer flask in 100 mL NB. The 48-hour grown cells were supplemented with 50 mg/L of each dye. To prevent biomass interference amidst dye concentration, the determination cells were purged out by centrifugation at 6,000xg for 15 min. Uninoculated samples with dye were taken as a control to calibrate the spectrophotometer. Post incubation 3 mL of aliquot was drawn from each flask at 0 h and 48 h for measurement of absorbance at 495 nm (MR), 580 nm (DY 12), and 500 nm (AB 210), respectively using a UV-vis spectrophotometer. The experiments were carried out in duplicates. Further, based on the following Equation1, the decolorization efficiency is determined as a percentage ratio25.
Microbial and biochemical characterization
Four isolates exhibited notable decolorizing efficiencies and proceeded further for the microbial and biochemical tests. All the tests were enacted following the standard protocol according to Bergey’s manual of determinative bacteriology26.
Influence of various parameters on dye degradation
The potential strains were optimized to achieve the highest decolorization rate for the model dyes. An approach utilizing one factor at a time (OFAT) was employed to identify the optimal level of a particular factor. Certain factors that significantly affected decolorization were studied21, and the decolorization percentage was calculated using the Eq1. The sterile media was amended with 50 mg/L of each MR, DY 12, and AB 210 dye and inoculated with 10%(v/v) inoculum. To ascertain the effect of incubation time for maximum decolorization, samples were drawn intermittently at 6, 12, 18, 24, 30, 36, and 48 h from the broth medium and were analyzed for decolorization efficiency. The pH of the medium was amended using dilute acidic and alkaline solutions of 0.1 N hydrochloric acid or 0.1 N sodium hydroxide, respectively. A 48-hour observation was done at 37º C, varying from pH 5–9. To ascertain the optimum temperature, the decolorization was examined by keeping the broth at different temperatures, ranging from 25° C to 45° C27. Lastly, to ascertain the impact of different concentrations of dyes on decolorization, the inoculum was subjected to a 48-hour incubation along with varying concentrations of dye ranging from 50, 100, 150, 200, 250, and 300 mg/L28.
Enzyme study for isolate 4
The decolorization of azo dyes primarily relies on oxidative and reductive enzyme production. The activity of these enzymes determines the ability of microorganisms to adapt to persistent azo-dye pollutants29,30. The bacterial cells of isolate 4 capable of decolorizing all three dyes most efficiently, were cultured aerobically in NB at 35º C, with continuous shaking at 120–160 rpm for 48 h supplemented with the 50 mg/L dye. The cells grown in the absence of dye were treated as a control. After centrifugation at 7000x g for 15 min, the cells were washed thrice. Subsequently, the pellet was resuspended in 5 mL potassium phosphate buffer (50 mM, pH 7), and further sonicated at 4° C, and the resulting supernatant was utilized as the crude enzyme source for further analysis. The following experiments were performed in duplicates to measure the average enzyme activity for each enzyme.
Lignin peroxidase
Lignin peroxidase (EC 1.11.1.14) activity was determined by measuring the increase in absorbance at 310 nm for 270 seconds. The 3 mL reaction mixture constituted of 4mM Veratryl alcohol, 0.2mM H2O2, and 10 µL enzyme in 10 mM sodium tartrate buffer with pH 3.031.
Manganese peroxidase
The Mn-peroxidase (EC 1.11.1.13) activity was assessed by measuring MnSO4 oxidation in a 50 mM sodium malonate buffer with a pH of 4.5, as measured spectrophotometrically at 468 nm for 270 seconds. The reaction mixture, comprising 4 mL, included 1 mM MnSO4, 0.1 mM H2O2, and 100 µL of enzyme32.
Laccase
The activity of laccase (EC 1.10.3.2) was assessed by measuring oxidation at 420 nm over a 270-second duration following the addition of 100 µL of enzyme and 0.4 mL of ABTS (1 mM) into 2.5 mL of sodium succinate buffer (100 mM, pH 4.5)33.
Azoreductases
The azoreductase activity was assessed in a 50 mM potassium buffer (pH 7.5) by monitoring the reduction at 482 nm over 270 s. The assay mixture totals 3 mL and consists of 50 µL of dye, 100 µM NADH, and 100 µL of the crude extract. The assay utilizes respective dyes as a substrate for assessing azoreductases34. One unit of enzyme activity was defined as 1 µg of the particular dye reduced per minute35.
Optimization of decolorization parameters by response surface methodology
RSM is a mathematical and statistical approach used to model and optimize processes influenced by multiple variables36. It generates optimized conditions for a system consisting of multiple variables against response variables by reducing the time and human effort involved in actual experiments and minimizing experimentation errors. RSM with Central Composite Design (CCD) was used to investigate the synergistic impact of various variables to obtain optimum conditions for efficient dye degradation by isolate 4. The design automatically calculates alpha values to ensure rotatability and orthogonality. In this study, pH (X1), temperature (X2), and initial concentration of dye (X3) were chosen as independent variables. The decolorization percentage was considered as a response variable. All statistical analyses were conducted using the R programming language (version 4.2.3), primarily leveraging packages focused on the Design of experiments (DOE) and Response surface methodology (RSM).
Degradation analysis with FTIR
The degradation of the model dyes (initial concentration 50 mg/L) by isolate 4 was characterized by FTIR spectroscopy21,37. The changes in the position and intensity of the peak after 48 h of incubation were studied. The dye-decolorized broth was centrifuged at 4000 g for 20 min. The supernatant was filtered through a 0.02-micron filter. The filtrate was added with an equal volume of ethyl acetate and agitated for 10 min. The aqueous phase and the organic phase were separated in a separating funnel. This organic phase (containing the extracted dye) was harvested and allowed to dry over anhydrous Na2S04. The transmittance of the samples was noted and compared with the changes in the functional group of the corresponding dye, which was taken as the control. The FTIR analysis was in the mid-infrared region of 400–4000 cm− 1.
Genomic DNA extraction and 16S rRNA gene amplification
The genomic DNA was extracted using the protocol outlined by Wright et al.. (2017), which involves the lysis of log-phase bacterial culture followed by PCI (Phenol: Chloroform: Isoamyl alcohol) extraction. The procured pellet was washed with ethanol and kept for air drying. Further, the DNA pellet is resuspended in TE buffer for subsequent use and storage. The extracted DNA was then run on a 1.0% agarose gel to check the presence and concentration38.
The fragment of the genomic DNA was amplified to target the 16S rRNA gene. The universal 16S primers; forward primer “GGATGAGCCCGCGGCCTA” and reverse primer “CGGTGTGTACAAGGCCCGG” were used to amplify the DNA fragments using PCR. The reaction mixture consisted of 14.75 µL of ddH2O,1 µL of genomic DNA template, 2 µL of each forward and reverse primer, 5u/µL of Taq DNA Polymerase, 2.5 µL of 10X buffer with 2 µL of MgCl2, 0.5 µL of dNTPs each. The PCR amplification was performed by thermocycler with initial denaturation at 95° C for 5 min, followed by 35 cycles of 40 s of denaturation at 95° C, annealing at 65° C for 1 min, elongation at 72° C for 2 min, and final extension at 65° C for 1 min. The amplified PCR product was purified using a GeneJet Gel extraction PCR purification kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Sequencing of 16S rRNA and BLAST analysis
The 16S rRNA gene is a highly conserved region in prokaryotes due to its vital role in ribosome assembly, the cell organelle responsible for protein synthesis. The gene encodes for 16S rRNA that integrates along a complex of 19 proteins to form a 30S subunit of bacterial ribosome. For bacterial identification, the nucleotide sequence of the 16S rRNA gene was sequenced by the Sanger di-deoxy chain termination method. The sequence was then deciphered on a genetic analyzer (Applied Biosystems) and the forward and reverse trace files (ABI files) were obtained and later assembled using DNA baser v5.2039,40,41,42,43,44. The assembled sequence was saved in FASTA file format. The acquired sequences were further scrutinized using EMBOSS software for nucleotide composition, GC percentage, etc45,46,47.
Phylogenetic analysis
Gene sequences for 16S rRNA were identified through similarity searching using the NCBI-BLAST (Basic Local Alignment Search Tool) for further comparative analysis with other sequences. Hence, applying a threshold value of > 95%, twenty sequences from the 16S ribosomal RNA sequence database (GenBank) were downloaded48. These sequences were further used to construct a phylogenetic tree using MEGA-XI software (Molecular Evolutionary Genetics Analysis-XI)49. Phylogenetic analysis for evolutionary relationships was conducted employing the Neighbor-joining algorithm to assess the degree of certainty for particular branches50. The bootstrapping value was set to 1000 replications to evaluate the reliability of each node in the phylogenetic tree51. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site52.
rRNA structure prediction
Ribosomal RNA operates as an essential component in ribosomes to regulate protein synthesis, which contain both conserved and hypervariable regions53,54. The secondary structure of rRNA is often used to trace the evolutionary course of an organism and can be effectively predicted from the sequence by thermodynamics. Diverse approaches like absorbance melting curves and microcalorimetry including differential scanning calorimetry and isothermal titration calorimetry have been examined for RNA structure thermodynamics55. The 3-dimensional structure of RNA presumes to evince various shapes that predict the native structure in equilibrium. Consolidation of phylogenetic evidence and thermodynamic principles together are speculated for RNA structure peculiarities for duplex formation which also entails assessment of spectroscopic properties56. The RNA structures, the minimum free energy (MFE), and the entropy of sequences are predicted using the RNA fold web server at 37 ⁰C. UNAfold (Unified Nucleic Acid Folding and Hybridising Package) was operated to predict the secondary structure of 16S rRNA and examine the structural stability57,58. The mountain plot of each sequence is represented which graphically represents the secondary structure of the RNA molecule. It plots the number of base pairs at each position along the sequence, with the height of the plot representing the frequency or abundance of the base pairs at that position. It also gives information about the interaction with the molecule and folding patterns59.
Results
Isolation and screening of samples
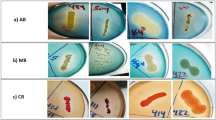
The textile effluents successfully yielded a total of 89 samples. Out of the total samples, four pure cultures were assessed for their ability to utilize azo dyes and preserved in agar slants at 4° C. The colonies from isolate 1 appeared smooth and glistening. They were yellow-colored and mostly circular. The colonies from isolate 2 were also yellowish and appeared wrinkled and sticky. The colonies from isolate 3 had a tint of pink with a round and smooth appearance. The colonies from isolate 4 were cream-colored, round, moderate in size, and exhibited smooth texture. The colonies were round with well-defined edges and appeared convex or raised. The diverse colony morphologies observed among the four isolates suggest the presence of a heterogeneous population of bacteria in textile wastewater capable of potentially degrading the azo dyes.
Decolorization assay
This preliminary screening aimed to identify bacterial isolates, capable of decolorizing dyes under aerobic conditions within 48 h. The decolorization percentage of the four pure cultures is depicted in Table 1. MR, DY 12, and AB 210 dyes are extensively utilized in the textile industry and have diverse molecular structures. These dyes are known to have speculative health risks and resistance to environmental degradation due to their persistence and bioaccumulation. Isolate 1 displayed moderate to high decolorization percentages (65–80%) for all three dyes. Isolate 2 and isolate 3 also showcased efficient decolorization rates, especially for MR and DY 12. Isolate 4 consistently demonstrated the highest decolorization percentage (almost > 70%) among all the isolates for all three dyes. A prominent pattern of decreasing decolorization percentage i.e. Monoazo > Diazo > Triazo was consistently observed across all strains. This owes to the fact that the presence of functional groups also significantly contributes to the decolorization of azo dyes.
Morphological and biochemical characterization of the dye decolorizing strains
Four samples out of the batch yielded a response for the decolorization. Table 2 depicts conclusive results for the biochemical and microbial tests. Figure 3 illustrates the prominent images for isolate 4.

Biochemical tests for the Isolate 4 AKSKSLAB04; (a) pure culture, (b) Citrate utilization test, (c) Indole test
Optimization of environmental conditions and calculation of decolorization percentage by the isolates
The effect of varied physicochemical parameters on model dyes by the 4 potential isolates was conducted extensively. For certain parameters, a consistent trend was observed across all three dyes. Whereas, for other parameters, the observed patterns were distinct and dye-specific. A positive correlation between incubation time and decolorization efficiency was observed in all strains (Fig. S1). Isolate 4 also exhibited greater decolorization with increased incubation time (Fig. 4).
The decolorization experiments were conducted across a pH range of 5 to 9. The results showed that the isolates could effectively decolorize the three model dyes within the alkaline pH range of 7 to 9. A sensitivity to acidic conditions has been seen distinctively for all strains across all dyes. Isolate 1 exhibited effective decolorization performance at pH levels between 8 and 9, while others ranged between pH 6–8 (Fig. S2). Isolate 4 displayed optimal decolorization between pH 7 and 9, showing a range (neutral to alkaline) of decolorization compared to the other isolates (Fig. 4). A range of initial dye concentrations (50–300 mg/L) was employed to ascertain the maximum concentration of each dye for decolorization. Effective decolorization exceeding 50% was observed for all dyes up to 150 mg/L across all isolates (Fig. S3). Beyond this concentration, a decrease in decolorization rates is observed, which proves toxic for the growth of bacteria and enzymatic functions. Compared to other isolates, isolate 4 exhibited a remarkable decolorization efficiency exceeding 70% at a dye concentration of 250 mg/L (Fig. 4), surpassing the optimal decolorization levels of the other three isolates, which ranged within the 100–150 mg/L range (Fig. S3). The effect of incubation temperature on the decolorization efficiency of the isolates was investigated and distinct temperature-dependent decolorization patterns were observed. Isolates 1, 2, and 3 exhibited decolorization within the temperature range of 25° C to 35° C (Fig. S4). Interestingly, isolate 4 exhibited the optimal temperature range, achieving maximum decolorization between 30° C and 40° C (Fig. 4).

Graph depicts the percentage decolorization corresponding to varying physiochemical parameters for Isolate 4 (AKSKSLAB04) (a) Incubation time (b) pH (c) Concentration (d) Temperature.
Enzyme study
The potential of various bacterial enzymes in the degradation of azo dyes has been extensively studied. The cell-free extracellular content of isolate 4 exhibited prominent activities of azoreductase, lignin peroxidase, manganese peroxidase, and laccase. Table 3 shows the activity of the degrading enzymes recorded at 48 h of incubation. The enzyme azoreductases exhibited a significant increase after decolorization, showcasing a fold change of 2.34, 1.97, and 1.57, respectively. Similarly, laccase shows a fold change of 2.08, 1.70, and 1.33 for 3 dyes respectively. This indicated the direct involvement of these enzymes in the cleavage of azo bonds resulting in simpler metabolites owing to efficient oxidative biodegradation. The activity of lignin peroxidase and manganese peroxide remained barely altered compared to the control. The synergistic activities of these enzymes enable the bacteria to break down a wide variety of azo dyes.
Response surface methodology
RSM with CCD was used to study the impact of pH, temperature, and initial dye concentration on decolorization. The range of variables is shown in Table 4, and the CCD matrix and experimental design (20 runs) are presented in Table 5. The experimental and predicted values for the model dyes are shown in Table S2. A second-order polynomial, also known as a quadratic equation, is used to fit the data for the model dyes (MR, DY 12, and AB 210), as it is generally preferred for capturing curvature and providing a more precise depiction of intricate trends. This approach offers enhanced flexibility, maintaining accuracy and minimizing the risk of overfitting.
Methyl red
The decolorization percentage obtained ranges from 10.11 to 82.28% for methyl red, as shown in the design matrix table (Table 5). A regression analysis was conducted to model the response variable and estimate the percentage decolorization. The regression model equation derived is presented in Eq. 2.
The linear, square, and interaction terms were significant as the p-value was less than 0.05. This model exhibited a Multiple R-squared of 0.958 and an adjusted R-squared value of 0.921 indicating a satisfactory fit with the experimental results and model predictions. The closer R2 to infinity the better the model data fits the actual data whereas, a lower R2 value does not ascertain the significance of the dependent variable in elucidating the behavior of the variations. The results from the ANOVA analysis provide a statistical assessment of the model’s ability to predict the decolorization of methyl red (Table 6). The Lack of Fit p-value of 0.41 indicates that the model does not exhibit a significant lack of fit and is adequate for representing the data. The signal-to-noise ratio (SNR) of 13.67 suggests the model had an adequate signal. The 2D contour plots and the 3D surface plots, illustrated in Fig. 5 indicate the regression equation and relationships between the variables. The statistical coefficients and their significant values of methyl red are summarized in Table S3.


Contour plots and corresponding 3D surface plots for decolorization percentage of methyl red. (a) Temperature- pH;(b) Temperature-concentration; (c) pH-concentration.
Direct yellow 12
The decolorization percentage obtained ranges from 14.65 to 79.37% for direct yellow 12 (Table 5). The regression model equation derived is presented in Eq. 3:
The linear and square terms are highly significant and all interaction terms are fairly significant as the p-value was less than 0.05. This model exhibited multiple R-squared values of 0.975 and an adjusted R-squared value of 0.953, suggesting an optimal alignment with the experimental results and model predictions. The Analysis of Variance (ANOVA) of the response surface model for the decolorization of DY 12 is summarized in Table 7. The Lack of Fit (p-value is 0.36) is not statistically significant, suggesting that the model adequately represents the data. Adequate model precision of 11.85 indicating SNR did not affect the prediction of process variables. The 2D contour plots and the 3D surface plots (Fig. 6) signify the regression equation and relationships between the variables. The statistical coefficients and their significant values of DY 12 are summarized in Table S4.

Contour plots and 3D surface plots for decolorization percentage of direct yellow 12. (a) Temperature- pH; (b) Temperature-concentration; (c) pH-concentration.
Acid black 210
The decolorization percentage obtained ranges from 7.39 to 59.74% for acid black 210 (Table 5). The regression model equation derived is presented in Equation4.
All the linear, interactions, and square terms exhibit significant value. This model achieves a multiple R-squared value of 0.995 and an adjusted R-squared value of 0.99 recommending best fit with the experimental results and model predictions. The Analysis of Variance (ANOVA) of the response surface model for the decolorization of acid black 210 is summarized in Table 8. The Lack of Fit p-value of 0.541 does not show a significant value for representing the data. Adequate model precision of 10.77 suggesting SNR did not affect the prediction of process variables. The 2D contour plots and the 3D surface plots are given in Fig. 7. The statistical coefficients and their significant values are summarized in Table S5.

Contour plots and corresponding 3D surface plots for decolorization percentage of acid black 210.(a) Temperature- pH; (b) Temperature-concentration; (c) pH-concentration.
Fourier-transform infrared (FTIR) analysis
Methyl red
The FTIR spectra of bacteria-treated methyl red dye obtained after incubation of 48 h exhibited significant changes in the position of the peak when compared with untreated confirming its degradation. The FTIR spectra of control shown in Fig. 8a displayed sharp peaks at 601–611 cm− 1 indicating alkyne C-H bending37. However, the peaks at 719 cm− 1, and 920 cm− 1, represent methylene bending and aromatic C-H bending. The single peak at 1050 cm− 1,1270 cm− 1 and 1492 cm− 1 displayed the cyclohexane ring vibrations, C-N stretch of aromatic amines, and C-C stretch60,61. Moreover, the small peaks at 1593–1623 cm− 1 represented the aromatic ring stretching. The peaks at 2925 cm− 1, 3260 cm− 1, and 3360 cm− 1 resulted in asymmetric CH3 stretch, alkyne C-H stretch, and polymeric OH stretch. On the other hand, the FTIR spectra of bacterial-treated methyl red at 48 h of incubation displayed peaks at different variations. The peaks at 766 cm− 1, 1030 cm− 1, and 1210 cm− 1 depicted a mono substitution phenyl aromatic C-H bend, reflecting changes in the phenyl ring due to bacterial treatment. The minor peaks at 1030 cm− 1 and 1210 cm− 1 corresponded to cyclohexane ring vibrations and aromatic C-H in-plane bend. The minor peaks at 1340 cm− 1, 1640 cm− 1, and 3340 cm− 1 describe methane C-H bend, alkenyl C = C stretch, and an aliphatic primary amine21. The absence of a peak at 1623 cm− 1 indicates the breakdown of the azo bond by the action of azoreductases produced by bacteria62. The appearance of new peaks in the bacterial-treated dye spectrum, along with shifts in existing peaks, indicates the transformation of the dye into different metabolites. These changes reflect the bacterial degradation and detoxification processes that occurred over the 48-hour incubation period (Fig. 8b).

FTIR spectrum of methyl red (a) control; (b) treated sample (48 h).
Direct yellow 12
A significant difference was observed while studying the functional properties of direct yellow 12 before and after treatment. The peak intensities in the direct yellow 12 as control Fig. 9a showed peaks at 1070 cm-1 and 1120 cm-1 revealing the sulfate functional group and C-O stretching21,63. The distinctive peaks at 1575 cm-1 and 1590 cm-1 represented the azo bond and NH bending. The peak at 2750 cm-1 demonstrated a C-H stretch. The major peak at 3650 cm-1 represented N-H stretching vibrations of secondary amines, confirming the presence of secondary amine groups in the dye, and contributing to its chemical properties. Alternatively, the bacteria-treated dye exhibited major variations in peak intensities with the appearance and disappearance of certain peaks after 48 h of incubation, as demonstrated in Fig. 9b.

FTIR spectrum of direct yellow 12 (a) control; (b) treated sample (48 h).
These changes indicate significant modifications resulting from the bacterial treatment. The absorption intensities in the region 600–1280 cm-1 were decreased; hence, suggesting a reduction in ether or alcohol content due to the breakdown of these functional groups. The appearance of a new peak at 1300 cm-1 represented aromatic ring vibrations and -C-O stretching vibrations as intermediates or new products of the bacterial degradation process. The peak at 2770 cm⁻¹ may change in intensity or position, reflecting alterations in the aliphatic hydrocarbon content of the dye molecule. A new peak appeared at 2990 cm-1 which denotes the asymmetric OH stretch. The presence of asymmetric OH stretching vibrations indicates increased hydroxylation, which enhances the dye’s solubility and biodegradability. The alterations in the peaks ranging from 3290 to 3480 cm⁻¹ indicate modifications to the amine groups (N-H stretching)37. This could result from deamination, where amine groups are removed, or the formation of new amine structures. These spectral changes underscore the effectiveness of bacterial treatment in breaking down complex dye structures into simpler, more biodegradable forms.
Acid black 210
The peaks in the acid black 210 (control) spectrum showed multiple variations as described in Fig. 10a. The small peaks at 966–974 cm-1 represented trans-C-H out-of-plane bending indicating the existence of trans-alkene structures within the dye molecule. The sharp peak at 1030 cm-1 depicted cyclohexane ring vibrations representing the cyclic structures, contributing to the overall stability and rigidity of the molecule. The distinctive peaks at 1600 cm-1 and 1770 cm-1 resulted in azo bond formation and N-H bend stretch63,64.The peak at 2850 cm-1 resulted in a C-H stretch indicating that the dye contains alkyl chains or non-aromatic carbon structures. In contrast, the single highest peak at 3430 cm-1 revealed aliphatic primary amine, respectively, indicating that the azo group in the dye contributed to its chemical reactivity and solubility properties37.

FTIR spectrum of acid black 210 (a) control; (b) treated sample (48 h).
Simultaneously, treatment of the dye often involves chemical modifications that can make the dye more susceptible to bacterial degradation. These modifications can include breaking down complex aromatic structures, reducing molecular weight, or introducing functional groups more easily metabolized by bacteria as shown in Fig. 10b. The slightest peak at 608 cm-1 depicted alkyne C-H bend however the peaks at the following wavelength 1100 cm-1, 1260 cm-1, 1410 cm-1, and 1540 cm-1 revealed C = C stretch of aromatic compound forms and S = O stretching of sulphonic acids and sulphonamides, respectively63,65. The peak at 1080 cm− 1 indicated C–OH stretching of primary alcohol while the one at 814 cm− 1 showed C–H deformation of tri-substituted benzene. The peaks at 3290 cm-1 and 3370 cm-1 revealed N-H stretching of amines66. The disappearance of certain peaks in the control spectrum suggests that specific functional groups or structural features have been broken down or altered during the treatment process thus indicating the crucial reduction of dye during the incubation period of 48 h.
DNA isolation and PCR amplification
Genomic DNA was extracted from the selected isolates using the phenol-chloroform method and appropriately labeled. DNA concentration was measured using a Nanodrop (Thermo Fisher Scientific) and a Qubit fluorometer, ensuring optimal purity. The absence of non-specific amplification products or contamination confirms the specificity and reproducibility of the PCR reaction. The DNA was subsequently used for the amplification and sequencing of the partial 16S rRNA gene.
Sequence analysis and alignment
The acquired sequences were then aligned against the nucleotide GenBank database by nucleotide Blast software using a cut-off value of > 95%. The BLASTn result displayed numerous taxonomic sequences with some degree of similarity. The results indicate that the sequences did not exhibit 100% similarity with any existing sequences, suggesting that strains AKSKSLAB01, AKSKSLAB02, AKSKSLAB03, and AKSKSLAB04 belong to a distinct and novel category. The results intimated that the strains AKSKSLAB01, AKSKSLAB02, AKSKSLAB03, and AKSKSLAB04 belonged to Sphingomonas mali, Shewanella putrefaciens, Pseudomonas leuteola, and Priestia flexa, respectively. Priestia flexa is regarded as a basonym for Bacillus flexus. The bioinformatics analysis of the strains revealed that the 16S rRNA sequence of AKSKSLAB01 is 1162 base pairs long, has a molecular weight of 352,127.00 Daltons and has a GC content of 53.61%. Similarly, the 16S rRNA sequence AKSKSLAB02 has 1403 base pairs, a molecular weight of 425,459.00 Daltons, and a GC content of 54.81%. The 16S rRNA sequence AKSKSLAB03 consists of 1403 base pairs exhibiting a molecular weight of 425,516.00 Daltons and having a GC content of 53.88%. The 16S rRNA sequence AKSKSLAB04 has 1423 base pairs long with a molecular weight of 434233.00 Daltons and a GC content of 53.34%. The novel sequences of the bacterial strains have been deposited to the GenBank database with accession numbers “OQ202071”, “PP708911”, “PP708909”, and “PP086977”, respectively.
Phylogenetic analysis
The phylogenetic analysis proceeded by Multiple Sequence Alignment using the Muscle algorithm. The DNA sequences determined were compared to 20 homologous nucleotide sequences > 95% were retrieved in FASTA format from the NCBI GenBank database. These sequences were further used to construct a phylogenetic tree. The evolutionary history was inferred using the Neighbor-Joining method. The bootstrap consensus tree inferred from 1000 replicates represents the evolutionary history of the analyzed taxa. Branches corresponding to partitions reproduced in less than 50% of bootstrap replicates are collapsed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site. All ambiguous positions were removed for each sequence pair (pairwise deletion option). In the final dataset, there was a total of 1162, 1411, 1407, and 1432 positions for Sphingomonas mali strain AKSKSLAB01, Shewanella putrefaciens AKSKSLAB02, Pseudomonas leuteola AKSKSLAB03, and Priestia flexa strain AKSKSLAB04, respectively.
The BLASTn against rRNA/ITS Database search generated a list of hits, revealing the degree of similarity between the query sequence and those in the database. The 20 sequences were selected and had > 95% similarity with the query. The query Sphingomonas mali strain AKSKSLAB01 displayed a significant similarity of 98.28% with Sphingomonas mali strain NBRC 15,500 16S ribosomal RNA, partial sequence, and 98.11% with both Sphingomonas pruni strain NBRC 15,498 16S ribosomal RNA, partial sequence and Sphingomonas mali strain IFO 15,500 16S ribosomal RNA, partial sequence (Fig. S5).
The top two strains with the highest similarity with the query sequence Shewanella putrefaciens strain AKSKSLAB02 were Shewanella seohaensis strain S7-3 16S ribosomal RNA, partial sequence by 97.92% and Shewanella decolorationis strain S12 16S ribosomal RNA, partial sequence by 97.49% (Fig. S6). The top two strains with the highest similarity with the query sequence Pseudomonas leuteola AKSKSLAB03 are Pseudomonas luteola strain NBRC 103,146 16S ribosomal RNA, partial sequence by 98.77% and Pseudomonas luteola strain 4239 16S ribosomal RNA, partial sequence by 98.70% (Fig. S7). The two strains with the highest similarity with the query sequence Priestia flexa strain AKSKSLAB04 are Priestia megaterium strain ATCC 14,581 16S ribosomal RNA, partial sequence by 99.41%, Priestia aryabhattai B8W22 16S ribosomal RNA, partial sequence by 99.38%. The phylogenetic tree of Priestia flexa strain AKSKSLAB04 is depicted in Fig. 11.
The average pairwise distances of the four novel strains to the 20 BLAST hits were 0.02679 (AKSKSLAB01), 0.021165 (AKSKSLAB02), 0.02209 (AKSKSLAB03), and 0.025344 (AKSKSLAB04). Divergence, a measure of genetic difference, assesses the evolutionary relationship between strains, with lower divergence values indicating closer relationships and higher values suggesting greater genetic separation. These strains exhibit corresponding divergences of 2.68%, 2.12%, 2.21%, and 2.53%, reflecting their close phylogenetic relationship to known sequences and indicating potential novelty at the species level.

Phylogenetic analysis of Priestia flexa strain AKSKSLAB04 against other species of Priestia.
rRNA structure prediction
The 16S rRNA genes contain both conserved and hypervariable regions. These regions provide a vital insight into understanding the basic structure, translational functions, and their interaction with ribosomal protein. These regions are also used for the primer designing, to ascertain whether the primer binds to the target sequence. On the other hand, hypervariable regions are crucial for bacterial phylogenetic analysis and taxonomic identification to scrutinize the bacterial community and closely related species. The hypervariable region is also concerned with the structural roles of small ribosomal subunits and ribosomal proteins. The conserved and the variable regions of the four novel strains are listed in Table S6.
The secondary structure of 16S rRNA was detailed using the RNA-UNAFOLD software UNAFOLD (Unified Nucleic Acid Folding and Hybridization), which testifies the helical regions that may bind to 23S rRNA. Various structures including interior hairpin, bulge, and multi-branched loops are annotated via colored base characters or dots. The minimum free energies for the optimal secondary strains AKSKSLAB01 AKSKSLAB02, AKSKSLAB03, and AKSKSLAB04 were calculated to be -428.10 kcal/mol, 522.90 kcal/mol, -508.80 kcal/mol, and − 515.50 kcal/mol, respectively. The thermodynamics result of each strain is depicted in Table S7 which delineates the entropy of the sequences. The Thermodynamics of Priestia flexa strain AKSKSLAB04 is shown in Table 9. The mountain plot was further used to visualize the secondary structure of the RNA molecule. Mountain plots and rRNA structure of AKSKSLAB01, AKSKSLAB02, and AKSKSLAB03 are shown in Fig. S8-S13. The mountain plot and the rRNA structure for the AKSKSLAB04 are shown in Figs. 12 and 13.

Hill plot -Sequence-wise energy and entropy distribution of 16S rRNA for Priestia flexa strain AKSKSLAB04.

16S rRNA secondary structure of Priestia flexa strain AKSKSLAB04.
Discussion
The study’s principal objective was to scrutinize the decolorization potential of the novel bacterial isolates from the untreated textile wastewater from industrial locations at Tirupur. This study cumulatively reports the isolation of four novel bacterial strains; Sphingomonas mali, Pseudomonas leuteola, Shewanella putrefaciens, and Priestia flexa. Since these strains efficiently degrade a monoazo, diazo, and, triazo dye, the study can be extended to treat a wide range of other dyes.
Decolorization is considered a crucial parameter for evaluating the degradation of dyes. It indicates the initial breakdown of dye molecules, which decreases color intensity67. A consistent pattern is observed across decolorization for all isolates (monoazo > diazo > triazo). Therefore, decolorization can be attributed to the structural complexity of the dye molecules. Additionally, the functional groups of the dye influence the electron density around azo bonds, directly affecting decolorization rates. Upon oxidative degradation, dye loses an electron leaving a positively charged intermediate. With the phenomenon of resonance, electron-releasing groups such as amino, hydroxyl, or alkyl groups get stably oxidized to be positively charged and provide favorable conditions for degradation. The highest percentage of decolorization is seen for MR, which contains a single -OH group, this can be attributed to the dye’s structural simplicity. Contrary to this, electron-withdrawing groups such as sulphonyl or halo groups add to the steric hindrance around the azo bond which suppresses the oxidative decolorization of dye68. This is evident in the decolorization of DY 12 and AB 210. These dyes contain sulphonyl groups, which are stable and resistant to degradation. Further, the sulphonyl group adds to the solubility of the dye and significantly hinders decolorization. The current findings of our study align with the research conducted by Emadi et al., where mono-azo dyes (MR; methyl red and MO: methyl orange) were decolorized at a faster rate than di-azo dye (AB-113: acid blue 113). The presence of the sulphonate group in AB-113 is an additional reason for the lower decolorization of AB-11317.
The decolorization of model dyes has been widely studied, focusing on optimizing various parameters such as temperature, pH, dye concentration, and incubation time69. With increased incubation time, the decolorization intensity of all three dyes across all isolates also increased. This suggests that longer incubation allows more extensive microbial and enzymatic processes, enhancing degradation. Similar results have been reported by a study that shows an increase in decolorization from 8.33% to almost 100% of Crystal Violet dye by Enterobacter sp. CV–S1 with the increase in incubation time from 12 to 72 h70. pH is also a crucial factor in the decolorization process. Effective decolorization is observed at neutral to alkaline pH in the optimum range between 6 and 871. Extreme acidic or alkaline conditions decrease dye removal efficiencies72. pH impacts dye removal efficiencies, by altering the dye molecules in an aqueous solution, which directly alters the dye absorption process. Additionally, the permeation potency through the cell membrane is also affected. This further restricts bacterial decolorization and degradation16. For all isolates, MR decolorization increased with an increase in the initial dye concentration, subsequently, it began to diminish beyond which dye concentration proved toxic to the growth of bacteria and enzymatic functions67,73,74. On the contrary, the increase in dye concentration leads to a decreased decolorization efficiency, especially noticeable with dyes DY 12 and AB 210 due to one or more sulfonic groups. These sulfonic acid (SO3H) groups within the dye’s structure act as to deterrent microbial growth, particularly evident at higher dye concentrations25. Incubation temperature significantly impacts the bacterial ability to decolorize dye. Extreme temperature disrupts bacterial reproduction, impedes enzymatic activity because of the thermal inactivation of proteins, and may also damage cell membranes. High temperatures can even lead to mortality16. An increase in temperature beyond optimal level results in a drastic decrease in decolorization efficiency. This concord with the study on the decolorization of Acid Orange dye at 35° C, by Staphylococcus hominis RMLRT03 showed 92.38% decolorization. Similarly, significantly lower temperature also results in decreased decolorization. The decolorization of Methyl Orange (MO) dye at 25° C was much lower (8.9%), while at 37° C, nearly complete decolorization (∼100%) was achieved75.
The rationale for studying the lignolytic is to gain additional information and understand the mechanism of decolorization and degradation. Apart from azoreductases, enzymatic properties and the role of laccase, MnP, and LiP have been extensively studied and reported in azo dye decolorization76,77,78. These enzymes oxidize various complex compounds, including dyes, thereby contributing to decolorization. In alignment with this, these enzymes were investigated in the novel bacterial strain, Prestia flexa AKSKSLAB04, which had not been characterized for such enzyme production before, making it a novel candidate for dye decolorization studies. The variation in enzyme activities observed towards different dyes is due to the difference in their molecular structures. Isolate 4 effectively shows the role of azoreductases and laccases in decolorizing the model dyes under aerobic conditions.
The process of decolorization can take place under aerobic and anaerobic conditions. Under anaerobic conditions, azoreductases play an initial role in cleaving the N = N bonds by the ping-pong mechanism. This leads to the formation of highly toxic aromatic amines69. On the contrary, aerobic degradation enables the complete mineralization of dyes and produces fewer toxic intermediates. This was also confirmed by a novel bacterial strain Bacillus cereus strain ROC, that efficiently degraded Reactive Orange 16 and Reactive black 579. Subsequently, oxidative enzymes like laccases act upon aromatic amines like aniline, phenol, and polyphenols to completely degrade the dye molecule. A prominent fold change in the activity of laccases shows its involvement in decolorization. This shows similar results where laccase is the key enzyme produced by Enterobacter aerogenes ES014 and has been utilized for large-scale decolorization in a batch bioreactor76. Laccase demethylates the compounds that aid in the breakdown of dye molecules80. A docking study justifies the binding of different dyes with enzymes azoreductases and laccase deciphers the enzyme mechanisms for efficient biodegradation of various azo dyes81. Further, laccases oxidize the phenolic group of azo dyes, forming phenoxy radicals, which are further oxidized to carbonium ions, and then undergo nucleophilic attack by water to cleave the azo bond82. Although bacterial extracellular peroxidases have been characterized in our study, no significant LiP and MnP activity was observed.
Studying the impact of individual factors fails to elucidate the synergistic interaction between the factors affecting decolorization. Hence, this optimization technique doesn’t accurately reflect the complete effects of variables on the decolorization83. The effects of the three process variables pH, initial dye concentration, and temperature considerably governed the decolorization of the three model dyes at p < 0.05, as depicted by ANOVA of response surface methodology. The outward-decreasing contour rings indicate a reduction in decolorization in that direction. The 2D and 3D response surface graphs display cross-section slices at fixed conditions: pH 7 (ph_actual), temperature 35° C (temp_actual), and concentration 175 mg/L (conc_actual). These fixed slices illustrate the interactions between the other two variables (Figs. 5, 6 and 7). The R2 value of the model for three dyes is close to 1. It demonstrates a better fit to the actual data, whereas a smaller value does not provide certainty regarding the relevance of dependent variables in explaining variation in the model84. Also, an insignificant lack of fit positively indicates that the model is a good fit for the data85. Adequate precision ascertains the signal-to-noise ratio. It signifies the differences observed between predicted results and their related errors with a recommended threshold of greater than 4, while values exceeding 10 are desirable for a strong model. Adequate precision of the model predicting the decolorization of MR, DY 12, and AB 210 by isolate 4 is 12.06, 12.59, and 10.61, respectively, indicating the statistical models demonstrated a sufficient signal-to-noise ratio86. Analyzing 16S rRNA gene sequences precisely identifies and classifies bacterial strains. They are extensively used in microbiome studies to assess microbial diversity and community structure87. They play a crucial role in the functional dynamics of rRNA during protein synthesis. Since secondary structures are more conserved than primary sequences across species, they are valuable for understanding phylogeny and evolutionary relationships88. FTIR spectroscopy was employed to analyze the decolorization and confirm the degradation of the dyes into various intermediate compounds.
The results highlight the potential of bacterial species for the bioremediation of textile effluents contaminated with azo dyes, suggesting that these strains can also be employed to treat other azo dyes, as their degradation mechanisms targeting the azo bond may be effective across various azo dye structures. However, it is too early to propose the exact mechanism behind dye degradation and the formation of secondary metabolites.
Conclusion
This is the first comprehensive report on the novel bacterial strains’ effective decolorization of monoazo, diazo, and triazo dyes. This highlights the bacterial potency for eco-friendly and economical remediation technologies. The analysis of enzymes from Priestia flexa AKSKSLAB04 indicated that azo reductase and laccase are key contributors to the breakdown of the model dyes. FTIR analysis illustrated the structural changes in the dyes, revealing the degradation and transformation of the dyes into less harmful metabolites. Further research on decolorization and enhancement of enzyme activity is required to improve the degradation capabilities of the bacteria. Harnessing the potential of specialized bacterial strains in bioreactors for large-scale decolorization of untreated effluents before discharging them into water bodies. This can prove effective in treating large volumes of dye-contaminated effluents. Additionally, fostering collaboration between academia and industry is essential for the effective application in the bioremediation of dye-contaminated sites. Applying these strains for textile dye decolorization may significantly benefit biological systems. Such benefits encompass improved water quality, enhanced soil health, favorable effects on plant life, and better human health due to decreased toxicity in water resources. This represents a significant step towards sustainable and eco-friendly wastewater management, safeguarding our precious water resources for future generations.
Data availability
Most of the data generated or analyzed during this study are included in this article. We have included additional data in the supplementary material. The 16S rRNA sequence data that support the findings of this study are available in GenBank database with IDs. Moreover, queries can be directed to the corresponding author for any clarifications about the study if needed.https://www.ncbi.nlm.nih.gov/nuccore/OQ202071https://www.ncbi.nlm.nih.gov/nuccore/PP708911https://www.ncbi.nlm.nih.gov/nuccore/PP708909https://www.ncbi.nlm.nih.gov/nuccore/PP086977.
References
Castillo-Suárez, L. A., Sierra-Sánchez, A. G., Linares-Hernández, I., Martínez-Miranda, V. & Teutli-Sequeira, E. A. A critical review of textile industry wastewater: green technologies for the removal of Indigo dyes. Int. J. Environ. Sci. Technol. 20 (9), 10553–10590. https://doi.org/10.1007/s13762-023-04810-2 (2023).
McMullan, G. et al. Jul., Microbial decolourisation and degradation of textile dyes, Appl. Microbiol. Biotechnol., 56 1–2 . 81–87, (2001). https://doi.org/10.1007/s002530000587
Sadeghi, M. et al. /+//Biodecolorization of reactive Black5 and reactive Red120 Azo dyes using bacterial strains isolated from dairy effluents. Int. J. Environ. Sci. Technol. 16 (7), 3615–3624. https://doi.org/10.1007/s13762-018-1750-7 (2019).
Siva, R. Status of natural dyes and dye-yielding plants in India. Curr. Sci. 92 (7), 916–925 (2007).
Abdulkadir, A. A. R. S., Dyeing of textiles with eco-friendly natural dyes: A review, 1, 5, pp. 76–81, (2015).
Rodríguez Couto, S. Dye removal by immobilised fungi. Biotechnol. Adv. 27 (3), 227–235. https://doi.org/10.1016/j.biotechadv.2008.12.001 (2009).
Maljaei, A., Arami, M. & Mahmoodi, N. M. Decolorization and aromatic ring degradation of colored textile wastewater using indirect electrochemical oxidation method. Desalination 249 (3), 1074–1078. https://doi.org/10.1016/j.desal.2009.05.016 (2009).
Kandelbauer, A. & Guebitz, G. M. Bioremediation for the decolorization of textile Dyes — A review, in environmental chemistry, (eds Lichtfouse, E., Schwarzbauer, J. & Robert, D.) Berlin/Heidelberg: Springer-, 269–288. https://doi.org/10.1007/3-540-26531-7_26. (2005).
Kaur, A. et al. Physico-chemical analysis of the industrial effluents and their impact on the soil microflora. Procedia Environ. Sci. 2, 595–599. https://doi.org/10.1016/j.proenv.2010.10.065 (2010).
Lellis, B., Fávaro-Polonio, C. Z., Pamphile, J. A. & Polonio, J. C. Effects of textile dyes on health and the environment and bioremediation potential of living organisms. Biotechnol. Res. Innov. 3 (2), 275–290. https://doi.org/10.1016/j.biori.2019.09.001 (2019).
Pratt, M. Disperse blue dyes 106 and 124 are common causes of textile dermatitis and should serve as screening allergens for this condition. Am. J. Contact Dermat. 11 (1), 30–41. https://doi.org/10.1016/S1046-199X(00)90030-7 (2000).
Kebede, G., Tafese, T., Abda, E. M., Kamaraj, M. & Assefa, F. Factors Influencing the bacterial bioremediation of hydrocarbon contaminants in the soil: mechanisms and impacts, J. Chem., vol. pp. 1–17, 2021, (2021). https://doi.org/10.1155/2021/9823362
Singh, K. & Arora, S. Removal of synthetic textile dyes from wastewaters: A critical review on present treatment technologies. Crit. Rev. Environ. Sci. Technol. 41 (9), 807–878. https://doi.org/10.1080/10643380903218376 (2011).
Nadh, A. G. et al. Identification of Azo dye degrading Sphingomonas strain EMBS022 and EMBS023 using 16S rRNA gene sequencing. http://www.eurekaselect.com, Accessed: Nov. 08, 2024. [Online]. Available: https://www.eurekaselect.com/article/70929
Manjarrez Paba, G., Baldiris, R., Ávila & Baena Baldiris, D. Application of environmental bacteria as potential methods of Azo dye degradation systems. Glob J. Environ. Sci. Manag. 7 (1). https://doi.org/10.22034/gjesm.2021.01.10 (Jan. 2021).
Mishra, A. et al. An integrative approach to study bacterial enzymatic degradation of toxic dyes. Front. Microbiol. 12 https://doi.org/10.3389/fmicb.2021.802544 (2022).
Emadi, Z. et al. Simultaneous decolorization/degradation of AB-113 and chromium(VI) removal by a salt-tolerant Klebsiella Sp. AB-PR and detoxification of biotransformed-metabolites. Int. J. Environ. Sci. Technol. 19 (3), 2007–2024. https://doi.org/10.1007/s13762-021-03360-9 (2022).
El Nemr, A., Hassaan, M. A. & Madkour, F. F. HPLC-MS/MS mechanistic study of direct yellow 12 dye degradation using ultraviolet assisted Ozone process. J. Water Environ. Nanotechnol. 3 (1). https://doi.org/10.22090/jwent.2018.01.001 (2018).
Rocha, O. P. et al. Ecotoxicological risk assessment of the ‘acid black 210’ dye. Toxicology 376, 113–119. https://doi.org/10.1016/j.tox.2016.04.002 (2017).
Ikram, M. et al. Biodegradation of Azo dye Methyl red by Pseudomonas aeruginosa: optimization of process conditions. Int. J. Environ. Res. Public. Health. 19 (16), 9962. https://doi.org/10.3390/ijerph19169962 (2022).
Khan, S. & Joshi, N. Molecular identification of dye degrading bacterial isolates and FT-IR analysis of degraded products. Environ. Eng. Res. 25 (4), 561–570. https://doi.org/10.4491/eer.2019.224 (2019).
Moyo, S., Makhanya, B. P. & Zwane, P. E. Use of bacterial isolates in the treatment of textile dye wastewater: A review. Heliyon 8 (6), e09632. https://doi.org/10.1016/j.heliyon.2022.e09632 (2022).
Sakpal, S. B. & Tarfe, K. S. Screening, isolation and characterization of dye degrading bacteria from textile dye effluents,. 20, (2021). https://doi.org/10.1101/2021.12.20.473465
Srinivasan, V., Bhavan, P. S. & Krishnakumar, J. Bioremediation of textile dye effluent by bacillus and pseudomonas spp.
Chen, K. C., Wu, J. Y., Liou, D. J. & Hwang, S. C. J. Decolorization of the textile dyes by newly isolated bacterial strains. J. Biotechnol. 101 (1), 57–68. https://doi.org/10.1016/s0168-1656(02)00303-6 (2003).
American Society for Microbiology, for Microbiology, A. S., Bergey, D. H. & Breed, R. S. Bergey’s Manual of Determinative Bacteriology 7th edn pp. 1–1130 (Williams & Wilkins Co, 1957). https://doi.org/10.5962/bhl.title.10728
Pinontoan, R. et al. Trypan blue dye decolorization by Aeromonas caviae isolated from water sewage in Jakarta, Indonesia, Biodiversitas J. Biol. Divers., vol. 25, no. 4, Art. no. 4, Apr. 2024, Accessed: 13, 2024. [Online]. Available: https://smujo.id/biodiv/article/view/17470
Al-Tohamy, R., Sun, J., Fareed, M. F., Kenawy, E. R. & Ali, S. S. Ecofriendly biodegradation of reactive black 5 by newly isolated sterigmatomyces halophilus SSA1575, valued for textile Azo dye wastewater processing and detoxification. Sci. Rep. 10 (1), 12370. https://doi.org/10.1038/s41598-020-69304-4 (2020).
Lade, H., Govindwar, S. & Paul, D. Mineralization and detoxification of the carcinogenic Azo dye congo red and real textile effluent by a polyurethane foam immobilized microbial consortium in an upflow column bioreactor. Int. J. Environ. Res. Public. Health. 12 (6,. ). https://doi.org/10.3390/ijerph120606894 (2015). Art. 6.
Telke, A. A., Kadam, A. A. & Govindwar, S. P. Bacterial enzymes and their role in decolorization of Azo Dyes, in Microbial Degradation of Synthetic Dyes in Wastewaters, S. N. Singh, Ed., in Environmental science and engineering., Cham: Springer International Publishing, 149–168. (2015). https://doi.org/10.1007/978-3-319-10942-8_7
Tien, M. & Kirk, T. K. Lignin peroxidase of phanerochaete Chrysosporium, in methods in Enzymology, vol. 161, Elsevier, 238–249. doi: https://doi.org/10.1016/0076-6879(88)61025-1. (1988).
Bermek, H., Gülseren, İ., Li, K., Jung, H. & Tamerler, C. The effect of fungal morphology on ligninolytic enzyme production by a recently isolated wood-degrading fungus trichophyton rubrum LSK-27. World J. Microbiol. Biotechnol. 20 (4), 345–349. https://doi.org/10.1023/B:WIBI.0000033055.52660.03 (2004).
Zavarzina, A. G., Leontievsky, A. A., Golovleva, L. A. & Trofimov, S. Y. Biotransformation of soil humic acids by blue laccase of Panus Tigrinus 8/18: an in vitro study. Soil. Biol. Biochem. 36 (2), 359–369. https://doi.org/10.1016/j.soilbio.2003.10.010 (2004).
Zhao, M. et al. Biodegradation of Methyl red by Bacillus Sp. strain UN2: decolorization capacity, metabolites characterization, and enzyme analysis. Environ. Sci. Pollut Res. 21 (9), 6136–6145. https://doi.org/10.1007/s11356-014-2579-3 (2014).
Du, L. N. et al. Biodegradation of malachite green by Pseudomonas Sp. strain DY1 under aerobic condition: characteristics, degradation products, enzyme analysis and phytotoxicity. Ecotoxicology 20 (2), 438–446. https://doi.org/10.1007/s10646-011-0595-3 (2011).
Hafshejani, M. K., Ogugbue, C. J. & Morad, N. Application of response surface methodology for optimization of decolorization and mineralization of triazo dye Direct Blue 71 by Pseudomonas aeruginosa, 3 Biotech 4 6 605–619, (2014). https://doi.org/10.1007/s13205-013-0192-7
Nandiyanto, A. B. D., Ragadhita, R. & Fiandini, M. Interpretation of Fourier Transform Infrared Spectra (FTIR): A Practical Approach in the Polymer/Plastic Thermal Decomposition, Indones. J. Sci. Technol. 8 1, Art. no. 1, (2023).
Wright, M. H., Adelskov, J. & Greene, A. C. Bacterial DNA extraction using individual enzymes and phenol/Chloroform separation, J. Microbiol. Biol. Educ. 18 2 18.2.60. (2017). https://doi.org/10.1128/jmbe.v18i2.1348
Nayarisseri, A., Suppahia, A., Nadh, A. G. & Nair, A. S. Identification and characterization of a pesticide degrading flavobacterium species EMBS0145 by 16S rRNA gene sequencing, Interdiscip. Sci. Comput. Life Sci. 7 2 93–99 (2015). https://doi.org/10.1007/s12539-015-0016-z
Bhatia, M., Girdhar, A., Tiwari, A. & Nayarisseri, A. Implications of a novel Pseudomonas species on low density polyethylene biodegradation: an in vitro to in Silico approach. SpringerPlus 3 (1), 497. https://doi.org/10.1186/2193-1801-3-497 (2014).
Jaiswal, M. et al. Identification and characterization of alkaline protease producing Bacillus firmus species EMBS023 by 16S rRNA gene sequencing. Interdiscip Sci. Comput. Life Sci. 6 (4), 271–278. https://doi.org/10.1007/s12539-014-0187-z (2014).
Nayarisseri, A., Singh, P. & Singh, S. K. Screening, isolation and characterization of biosurfactant producing Bacillus subtilis strain ANSKLAB03. Bioinformation 14 (6), 304–314. https://doi.org/10.6026/97320630014304 (2018).
Chandok, H. et al. Screening, isolation and identification of probiotic producing Lactobacillus acidophilus strains EMBS081 & EMBS082 by 16S rRNA gene sequencing. Interdiscip Sci. Comput. Life Sci. 7 (3), 242–248. https://doi.org/10.1007/s12539-015-0002-5 (2015).
Nayarisseri, A., Khandelwal, R. & Singh, S. K. Identification and characterization of lipopeptide biosurfactant producing Microbacterium Sp isolated from brackish river water. Curr. Top. Med. Chem. 20 (24), 2221–2234. https://doi.org/10.2174/1568026620666200628144716 (2020).
Phanse, N., Rathore, P., Patel, B. & Nayarisseri, A. Characterization of an industrially important alkalophilic bacterium, Bacillus Agaradhaerens strain nandiniphanse5. J. Pharm. Res. 6 (5), 543–550. https://doi.org/10.1016/j.jopr.2013.04.035 (2013).
Pyde, A. N. et al. Identification and characterization of foodborne pathogen Listeria monocytogenes strain Pyde1 and Pyde2 using 16S rRNA gene sequencing. J. Pharm. Res. 6 (7), 736–741. https://doi.org/10.1016/j.jopr.2013.07.009 (2013).
Amareshwari, P. et al. Isolation and characterization of a novel Chlorpyrifos degrading flavobacterium species EMBS0145 by 16S rRNA gene sequencing. Interdiscip Sci. Comput. Life Sci. 7 (1), 1–6. https://doi.org/10.1007/s12539-012-0207-9 (2015).
Sharma, K., Nayarisseri, A. & Singh, S. K. Biodegradation of plasticizers by novel strains of bacteria isolated from plastic waste near Juhu beach, Mumbai, India. Sci. Rep. 14 (1), 30824 (2024).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. https://doi.org/10.1093/molbev/msab120 (2021).
Nayarisseri, A. & Singh, S. K. Genome analysis of biosurfactant producing bacterium, Bacillus tequilensis. Plos One, 18(6), e0285994. (2023).
Felsenstein, J. & Confidence limits on phylogenies: an approach using the bootstrap. Evol. Int. J. Org. Evol., 39 4 783–791 https://doi.org/10.1111/j.1558-5646.1985.tb00420.x (1985).
Nayarisseri, A. et al. Promoter–motif extraction from co-regulated genes and their relevance to co-expression using E. coli as a model. Brief. Funct. Genomics. 22 (2), 204–216 (2023).
Wang, Y. & Qian, P. Y. Conserved regions in 16S ribosome RNA sequences and primer design for studies of environmental microbes, in Encyclopedia of Metagenomics, (ed Nelson, K. E.) New York, NY: Springer, 1–6. doi: https://doi.org/10.1007/978-1-4614-6418-1_772-1. (2013).
Nayarisseri, A. & Hood, E. A. Advancement in microbial cheminformatics. Curr. Top. Med. Chem. 18 (29), 2459–2461 (2018).
Nayarisseri, A., Singh, P. & Singh, S. K. Screening, isolation and characterization of biosurfactant-producing Bacillus tequilensis strain ANSKLAB04 from brackish river water. Int. J. Environ. Sci. Technol. 16 (11), 7103–7112 (2019).
Pasternak, A. & Wengel, J. Thermodynamics of RNA duplexes modified with unlocked nucleic acid nucleotides. Nucleic Acids Res. 38, 6697–6706. https://doi.org/10.1093/nar/gkq561 (Oct. 2010).
Zuker, M. On finding all suboptimal foldings of an RNA molecule. Science 244 (4900), 48–52. https://doi.org/10.1126/science.2468181 (1989).
Gruber, A. R., Lorenz, R., Bernhart, S. H., Neuböck, R. & Hofacker, I. L. The Vienna RNA websuite, Nucleic Acids Res. 36, no. Web Server issue, pp. W70-74, (2008). https://doi.org/10.1093/nar/gkn188
Aarti, C., Khusro, A., Agastian, P., Darwish, N. M. & Al Farraj, D. A. Molecular diversity and hydrolytic enzymes production abilities of soil bacteria. Saudi J. Biol. Sci. 27 (12), 3235–3248. https://doi.org/10.1016/j.sjbs.2020.09.049 (2020).
Vijayalakshmi, D., Sivaprasad, B. V., Veera Brahmma Chari, P., Reddy, M. K. & Prasad, D. V. R. Microbial consortia for effective degradation and decolorization of textile effluents, in recent trends in mycological research: volume 2: environmental and industrial perspective, (ed Yadav, A. N.) Cham: Springer International Publishing, 167–193. doi: https://doi.org/10.1007/978-3-030-68260-6_7. (2021).
Takkar, S. et al. Biodegradation of Methyl red dye by a novel actinobacterium Zhihengliuella Sp. ISTPL4: kinetic studies, isotherm and biodegradation pathway. Environ. Technol. Innov. 26, 102348. https://doi.org/10.1016/j.eti.2022.102348 (2022).
Patil, N., Bholay, A., Kapadnis, B. & Gaikwad, V. Biodegradation of model Azo dye Methyl red and other textile dyes by isolate Bacillus circulans NPP1. J. Pure Appl. Microbiol. 10 (4), 2793–2800. https://doi.org/10.22207/JPAM.10.4.38 (2016).
Sahasrabudhe, M. M., Saratale, R. G., Saratale, G. D. & Pathade, G. R. Decolorization and detoxification of sulfonated toxic Diazo dye C.I. Direct red 81 by Enterococcus faecalis YZ 66. J. Environ. Health Sci. Eng. 12, 151. https://doi.org/10.1186/s40201-014-0151-1 (2014).
Bedekar, P. A., Saratale, R. G., Saratale, G. D. & Govindwar, S. P. Oxidative stress response in dye degrading bacterium Lysinibacillus Sp. RGS exposed to reactive orange 16, degradation of RO16 and evaluation of toxicity. Environ. Sci. Pollut Res. Int. 21 (18), 11075–11085. https://doi.org/10.1007/s11356-014-3041-2 (2014).
Ghodake, G., Jadhav, U., Tamboli, D., Kagalkar, A. & Govindwar, S. Decolorization of textile dyes and degradation of Mono-Azo dye Amaranth by acinetobacter calcoaceticus NCIM 2890. Indian J. Microbiol. 51 (4), 501–508. https://doi.org/10.1007/s12088-011-0131-4 (2011).
Saratale, R. G., Saratale, G. D., Chang, J. S. & Govindwar, S. P. Ecofriendly degradation of sulfonated Diazo dye C.I. Reactive green 19A using micrococcus glutamicus NCIM-2168. Bioresour Technol. 100 (17), 3897–3905. https://doi.org/10.1016/j.biortech.2009.03.051 (2009).
Pinheiro, L. R. S., Gradíssimo, D. G., Xavier, L. P. & Santos, A. V. Degradation of Azo dyes: Bacterial potential for bioremediation, Sustainability, vol. 14, no. 3, Art. no. 3. (2022). https://doi.org/10.3390/su14031510
Zhang, Q. et al. Deciphering effects of chemical structure on Azo dye decolorization/degradation characteristics: bacterial vs. photocatalytic method. J. Taiwan. Inst. Chem. Eng. 43 (5), 760–766. https://doi.org/10.1016/j.jtice.2012.03.001 (2012).
Fazeliyan, E. et al. Decolorization mechanism, identification of an FMN-dependent NADH-azoreductase from a moderately halotolerant Staphylococcus Sp. MEH038S, and toxicity assessment of biotransformed metabolites. Water Environ. Res. Res. Publ Water Environ. Fed. 93 (10), 2072–2083. https://doi.org/10.1002/wer.1580 (2021).
Roy, D. C. et al. Biodegradation of crystal Violet dye by bacteria isolated from textile industry effluents. PeerJ 6, e5015. https://doi.org/10.7717/peerj.5015 (2018).
Khan, Z., Jain, K., Soni, A. & Madamwar, D. Microaerophilic degradation of sulphonated Azo dye – Reactive red 195 by bacterial consortium AR1 through co-metabolism. Int. Biodeterior. Biodegrad. 94, 167–175. https://doi.org/10.1016/j.ibiod.2014.07.002 (2014).
Chittal, V., Gracias, M., Anu, A., Saha, P. & Bhaskara Rao, K. V. Biodecolorization and biodegradation of Azo dye reactive Orange-16 by marine nocardiopsis Sp. Iran. J. Biotechnol. 17 (3), 18–26. https://doi.org/10.29252/ijb.1551 (2019).
Bera, S. P. & Tank, S. K. Microbial degradation of Procion red by Pseudomonas stutzeri. Sci. Rep. 11 (1), 3075. https://doi.org/10.1038/s41598-021-82494-9 (2021).
Emadi, Z. et al. Decolorization of reactive Black-5 high concentration by vermicompost microflora and detoxification of By-Products by UV-C/H2O2 Post-Treatment. Pollution 6, 3. https://doi.org/10.22059/poll.2019.288500.675 (2020).
Singh, R. P., Singh, P. K. & Singh, R. L. Bacterial decolorization of textile Azo dye acid orange by Staphylococcus hominis RMLRT03. Toxicol. Int. 21 (2), 160–166. https://doi.org/10.4103/0971-6580.139797 (2014).
Al-Ansari, M. M., Li, Z., Masood, A. & Rajaselvam, J. Decolourization of Azo dye using a batch bioreactor by an Indigenous bacterium Enterobacter aerogenes ES014 from the waste water dye effluent and toxicity analysis. Environ. Res. 205, 112189. https://doi.org/10.1016/j.envres.2021.112189 (2022).
Chang, Y., Yang, D., Li, R., Wang, T. & Zhu, Y. Textile dye biodecolorization by manganese peroxidase: A review. Molecules 26 (15), 4403. https://doi.org/10.3390/molecules26154403 (2021).
Parshetti, G. K., Parshetti, S., Kalyani, D. C., Doong, R. & Govindwar, S. P. Industrial dye decolorizing lignin peroxidase from Kocuria rosea MTCC 1532, Ann. Microbiol., vol. 62, no. 1, Art. no. 1, (2012). https://doi.org/10.1007/s13213-011-0249-y
Fareed, A. et al. Decolorization of Azo dyes by a novel aerobic bacterial strain Bacillus cereus strain ROC. PLOS ONE. 17 (6), e0269559. https://doi.org/10.1371/journal.pone.0269559 (2022).
Abadulla, E. et al. Decolorization and detoxification of textile dyes with a laccase from trametes hirsuta. Appl. Environ. Microbiol. 66 (8), 3357–3362. https://doi.org/10.1128/AEM.66.8.3357-3362.2000 (2000).
Srinivasan, S., Sadasivam, S. K., Gunalan, S., Shanmugam, G. & Kothandan, G. Application of Docking and active site analysis for enzyme linked biodegradation of textile dyes. Environ. Pollut. 248, 599–608. https://doi.org/10.1016/j.envpol.2019.02.080 (2019).
Chivukula, M. & Renganathan, V. Phenolic Azo dye oxidation by laccase from pyricularia oryzae. Appl. Environ. Microbiol. 61 (12), 4374–4377. https://doi.org/10.1128/aem.61.12.4374-4377.1995 (1995).
Gautam, P., Kumar, S., Vishwakarma, S. & Gautam, A. Synergistic optimization of electrocoagulation process parameters using response surface methodology for treatment of hazardous waste landfill leachate. Chemosphere 290, 133255. https://doi.org/10.1016/j.chemosphere.2021.133255 (2022).
Cao, W., Zhang, C., Hong, P. & Ji, H. Response surface methodology for autolysis parameters optimization of shrimp head and amino acids released during autolysis. Food Chem. 109 (1), 176–183. https://doi.org/10.1016/j.foodchem.2007.11.080 (2008).
Zin, K. M. et al. Microbial Decolorization of Triazo Dye, Direct Blue 71: An Optimization Approach Using Response Surface Methodology (RSM) and Artificial Neural Network (ANN), BioMed Res. Int., vol. pp. 1–16. 2020, (2020). https://doi.org/10.1155/2020/2734135
Ikram, M. et al. Bacillus subtilis: as an efficient bacterial strain for the reclamation of water loaded with textile Azo dye, orange II. Int. J. Mol. Sci. 23 (18), 10637. https://doi.org/10.3390/ijms231810637 (2022).
Petrov, A. S. et al. Secondary structures of rRNAs from all three domains of life. Plos ONE. 9 (2), e88222. https://doi.org/10.1371/journal.pone.0088222 (2014).
Brimacombe, R. & Stiege, W. Structure and function of ribosomal RNA. Biochem. J. 229 (1), 1–17. https://doi.org/10.1042/bj2290001 (1985).
Acknowledgements
The authors are thankful to LeGene Biosciences Pvt for 16S rRNA sequencing of the bacterium. Sanjeev Kumar Singh thanks DST-PURSE 2nd Phase Programme grant [No. SR/PURSE Phase 2/38 (G); DST-FIST Grant [(SR/FST/LSI—667/2016)]; MHRD RUSA-Phase 2.0 grant sanctioned vide Letter no. [F.24‐51/2014‐U, Policy (TN Multi‐Gen), Department of Education, Govt of India]; Tamil Nadu State Council for Higher Education (TANSCHE) under [No. AU: S.O. (P&D): TANSCHE Projects: 117/ 202, File No. RGP/2019‐20/ALU/ HECP‐0048]; DBT-BIC, New Delhi, under Grant/Award [No. BT/PR40154/BTIS/137/ 34/2021, dated 31.12.2021]; and DBT-NNP Project, New Delhi, under Grant/Award [No. BT/PR40156/BTIS/54/2023 dated 06.02.2023] for providing the research grant and infrastructure facilities in the lab.
Author information
Authors and Affiliations
Contributions
A.K.: Involved in in Conceptualization, Sample Collection, Experimental analysis, Methodology, Data curation and analysis, Formal analysis, Validation, Visualization, Writing – review & editing of the MS. A.N. Involved in Conceptualization, Investigation, Methodology, Project administration, Supervision, Writing – review & editing. S.K.S.: Involved in Investigation, Project administration, Supervision, Writing – review & editing of the MS.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khan, A., Nayarisseri, A. & Singh, S.K. Characterization and optimization of azo dyes degrading microbes isolated from textile effluent. Sci Rep 15, 11241 (2025). https://doi.org/10.1038/s41598-025-95359-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-95359-2
Keywords
This article is cited by
-
Characterization of natural therapeutic compounds producing novel bacterial strains isolated from Hyderabad, India
Scientific Reports (2025)
-
Whole genome sequence of petroleum hydrocarbon degrading novel strain Microbacter sp. EMBS2025 isolated from Chilika Lake, Odisha, India
Scientific Reports (2025)
-
Decontamination of crystal violet and auramine O dyes from aqueous media using pullulan@ sodium alginate based hydrogel
Journal of Porous Materials (2025)
-
Characterization of azo dye-degrading Tistrella mobilis isolated from industrial effluents: a molecular perspective
Biologia (2025)
